
Abstract
Until recently, the only known condition in which complement could mediate transplant injury was the rare occurrence of antibody-mediated rejection, in which the original concept of antibody immunity against the transplant was supported by complementary proteins present in the serum. This has changed within the last two decades because of evidence that the processes of ischaemia–reperfusion injury followed by T cell–mediated rejection are also critically dependent on components generated by the complement system. We now have a clearer understanding of the complement triggers and effectors that mediate injury, and a detailed map of their local sites of production and activation in the kidney. This is providing helpful guidelines as to how these harmful processes that restrict transplant outcomes can be targeted for therapeutic benefit. Here we review some of the recent advances highlighting relevant therapeutic targets.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The complement system is an essential part of the innate immune system, acting both as a first line of defence through pathogen and cell debris removal as well as recruiting components of the adaptive immune system [1]. Recognition of specific patterns on the surface of invasive pathogens or damaged internal cells by pattern recognition molecules (PRMs) activates the complement cascade [2]. Two main pattern recognition pathways are involved: the classical and the lectin complement activation pathways, both of which converge on C3, the pivotal component of complement. In addition, C3 can be cleaved by the alternative pathway, which can start up spontaneously by hydrolysis or can follow the cleavage of C3 to C3b by the classical or lectin pathways. The lectin pathway has a number of PRMs including collectins and ficolins (reviewed in 3, 4), while the classical pathway has only one, C1q [5]. These PRMs initiate the cleavage of early complement components (C4 and C2) to form a C3 convertase, which leads to the cleavage of C3 with subsequent C5 cleavage and generation of the membrane attack complex (MAC), as well as other complement effectors such as C3a and C5a, which mediate inflammation and engage the adaptive immune system [6]. In comparison, the alternative pathway generates a C3 convertase from C3b and factor B (fB), which rapidly increases the amount of cleaved C3 through an amplification loop [7]. While the complement system is an elegant rapid response system and recruiter of the adaptive immune system against invasive organisms, aberrant activation of complement can have a number of detrimental effects on the host. This contributes to inflammatory diseases such as rheumatoid arthritis, age-related macular degeneration and ischaemia–reperfusion injury (IRI) (reviewed in 8). In this review, we will discuss the influence of complement on the pathogenesis of inflammatory disease, particularly in the context of organ transplantation.
Complement in IRI
Ischaemia–reperfusion injury (IRI) arises in a number of traumatic injuries and diseases, as well as during transplantation. It is characterized by a restriction in blood supply to an organ (ischaemia) followed by the restoration of blood supply and reoxygenation (reperfusion) [9]. While ischaemic damage is related to tissue hypoxia, subsequent reperfusion mediates further tissue damage compounded by an inflammatory response [8]. The role of complement effectors in renal IRI has been extensively described [10, 11]. In addition, more recent studies have identified a primary role for the lectin pathway of complement activation, in particular the contribution of the PRM known as collectin-11 (CL-11) and the potential for blocking its downstream effects [12, 13]. Evidence points to a major fucosylated ligand recognized by CL-11 on hypoxic tissue [13] and has highlighted the feasibility to block CL-11 binding to this ligand by treatment with high concentrations of soluble L-fucose [12]. The alternative pathway has also been implicated because fB−/− mice are resistant to renal IRI [14]. It therefore seems likely that a local tissue ligand recognized by CL-11 leads to complement deposition on the tubules, with further amplification of complement deposition by the alternative pathway. Important to this concept is that complement components (notably CL-11 and C3) produced locally in the kidney at the site of tissue damage have a far greater impact on immune pathology than the corresponding circulating components, produced by the liver and other tissues.
In myocardial IRI, all three complement pathways (classical, lectin and alternative) have been implicated. Myocardial infarction remains a major cause of death in modern societies with the pivotal treatment step being early reperfusion but this can also promote cardiomyocyte death and an increase in the final infarct size [15]. The role of the classical pathway has been investigated through the manipulation of C-reactive protein (CRP), an inflammatory protein that significantly increases at the sites of inflammation during infection. CRP activates C1q, the PRM of the classical pathway [16] and consequently the complement system. Addition of human CRP to rats in a myocardial IRI model increased the resulting infarct size [17] and the addition of a CRP inhibitor resulted in the inverse [18, 19], demonstrating a role for C1q and by inference the classical pathway. The lectin pathway has been also implicated, through observed co-localization of mannose-binding lectin (MBL; a PRM of the lectin pathway) and C3 in rat myocardial IRI models [20]. In addition, the inhibition of MBL led to a reduction of infarct size [21]. Furthermore, loss of MBL associated serum protease-2 (MASP-2) in mice significantly protected against IRI consistent with a role of lectin pathway activation in this process [22]. Therefore, both the lectin and classical pathways of complement activation are considered potential therapeutic targets in this condition (reviewed in 15).
The liver too is susceptible to complement-mediated IRI, unsurprisingly because hepatic synthesis is the major source of circulating complement components such as C3, C4 and MBL [23]. In experimental models of liver IRI involving treatment with C3 inhibitor [24] or C5aR1 inhibitor [25] or induction in C6-deficient rats [26], animals showed decreased post-ischaemic damage (reviewed in 27). Additional data for C6-deficient mice suggest that MAC is essential for tissue damage in the liver, confirmed by further work using CR2–CD59 which binds C8 and C9 to block formation of MAC downstream of C3 and C5 cleavage [27]. Interestingly, the treatment with CD59 not only reduced IRI damage but also increased the rate of hepatocyte regeneration after a partial hepatectomy (in which IRI is part of the surgical procedure) [28]. Furthermore, patients undergoing a partial hepatectomy show increased levels of C3d and C4d, indicating activation of the complement system. In the rat IRI model, IgM and CRP binding to damaged tissue also implicate classical pathway activation via C1q [29].
Complement links to the adaptive immune response and coagulation
There is extensive crosstalk between the innate and adaptive arms of the immune system. This principally involves an influence on the function of antigen-presenting cells (APCs), such as a dendritic cell (DC), but can also involve direct interaction of innate immune components with B cells and T cells. APCs take up foreign material and present the digested fragments on MHC molecules, which in turn activate specific T cells [30]. On B cells, complement receptor 2 (CR2) is important in cell activation. CR2 is predominantly expressed on follicular dendritic cells (FDCs) and B cells in humans and mice and detects C3-opsonized antigen [31]. As detailed earlier, activation of complement pathways by pattern recognition results in the cleavage of C3 to C3a and C3b. A proportion of the C3b will remain bound to the target antigen surface via protein amine groups. Factor I, in conjunction with complement regulators such as factor H and complement receptor 1 (CR1), converts C3b to C3i and C3d(g). Interaction of C3d-opsonized antigen with CR2 both promotes retention of the target antigen by FDCs in the B-cell areas of lymphoid tissue and augments the stimulation of B cells by the specific antigen [32]. Links between complement and T cell activity have been confirmed in decay-accelerating factor (DAF)–deficient mice. DAF is a regulator of the complement system and mice lacking DAF have increased complement activation and enhanced T cell responses to the reintroduction of antigen [33]. In addition to opsonization by C3b, the complement activation fragment C5a can directly engage C5aR on APC and T cells, to influence T cell immunity. Mice with reduced C5aR activity, induced by inhibitor or targeted deficiency, show a reduction in CD8 + T cell responses to infection [34, 35]. These examples combine to illustrate extensive regulation of the adaptive immune system by complement and complement control proteins [30, 31, 36].
The complement system not only mediates cell and pathogen injury by lysis and recruitment of immune cells, but it also signals to other pathways, including the coagulation pathways. Briefly, two coagulation pathways, the intrinsic and the extrinsic, converge on factor X (FX). The pathways are initiated by tissue factor (TF) exposure (extrinsic) or an activation cascade of factors XII, XI and IX. These ultimately result in the activation of FX to FXa. In association with FVa, FXa then cleaves prothrombin to thrombin which ultimately generates insoluble fibrin and consequently a blood clot [3, 37]. Because MASP-1 resembles thrombin in the crystal structure of its catalytic domain [38], it can cleave fibrinogen amongst other coagulation factors (such as factor VIII) [39]. Both MBL-MASP and L-ficolin-MASP complexes bind to their targets, thus activating the coagulation pathway and resulting in the formation of fibrin blood clots [39, 40]. MASP-2 can cleave prothrombin to generate thrombin which in turn can cleave fibrinogen and factor VIII, thereby forming fibrin clots. Interestingly, the fibrin generated is known to be covalently bound to the same bacterial surfaces that the MBL-MASP2 complex is bound to and can attract phagocytes and/or act as adhesion points for immune system cells. Therefore, this link to the clotting pathway could have direct functional relevance to the innate immune response [41].
Complement and transplantation
Some of the earliest steps in allograft rejection are mediated by the innate immune system, which plays an additional role enhancing and directing the adaptive immune response to transplantation. There are two peaks of complement activation in transplantation, IRI, as described above, and acute rejection [42]. This complement activation is detrimental in itself as described above but also leads to recruitment of the adaptive immune system, for example by enhancing T-cell activity (reviewed in 43). These varying roles of complement within the transplant process are important in the transplant outcome and will be discussed below.
The first and most direct effect of complement in a transplanted organ is the inflammatory response, which is mediated via generation of biologically active complement fragments. These mediate chemotaxis and neutrophil and macrophage activation. The biological effects depend on the site and trigger of complement activation. For example, in renal IRI, the primary location of complement activation is the tubulo-interstitial compartment of the kidney, which appears to be triggered by locally released CL-11 [14]. In the case of IRI, C3a/C3aR interaction contributes to glomerular and tubular injury [44, 45] and C3a has also been shown to stimulate secondary epithelial cell chemokine production contributing to local inflammation [46]. In native organ studies, from which we can infer information relevant to transplants, it has been shown that C5a is a strong chemoattractant, with C5aR deficiency showing a protective effect in models of ischemia–reperfusion injury [47]. Further evidence of complement in IRI comes from studies using C5 inhibition, as well as lectin blockade to protect from IRI in kidney and cardiac models [22, 47, 48]. Once MAC is formed, cells are induced to produce IL-1α and IL-8, further increasing local tissue inflammation [49].
Complement activation is also critical to the development of adaptive immunity. In an alloimmune response, interactions of antigen-presenting cells (APCs) and T cells are associated with the release of alternative pathway complement components C3, factors B and D, along with C5 [50–52]. There is a corresponding upregulation in the expression of C3aR and C5aR on T cells [53, 54], associated with reduced expression of the complement regulator, DAF. These changes favour local complement activation and enhanced T cell proliferation [53, 55]. Furthermore, studies have identified direct stimulation of B cell priming by opsonized antigens [56, 57], while studies in skin-grafted mice confirm that donor-specific antibody (DSA) production is markedly impaired in the absence of complement activation [58]. Antibody-mediated rejection (ABMR) is due to development of DSAs combined with histologic changes on graft biopsy, with concomitant deterioration in graft function [59]. The role of complement in this setting is well established. ABMR is mediated by the classical pathway, when C1q binds to DSA at the site of endothelial antibody attachment. This mechanism has been described in both acute and chronic transplant injury [60]. Furthermore, it has been shown that complement depletion impairs antibody production via its role in T cell presentation of antigen to B cells [61]. Particularly, there is evidence that DSA interaction with C1q could determine cytotoxic potential of these antibodies and could be used to risk stratify and diagnose ABMR [62]. C5a-C5aR interaction is another important part of ABMR, as shown in mouse allograft studies which have shown that lack of C5aR in both donor and recipient reduced allospecific T cell responses (and therefore improved outcomes). This lack of C5aR also reduced the function of APCs, cellular infiltration and inflammation [63]. Linked to this, recent work has shown that C5 inhibition in highly sensitized mice is protective [64], and importantly, C5 inhibition led to long-term allograft survival despite present DSA [65–68]. Therefore, complement control has both a direct and indirect role to play in the successful outcome of transplants and is an emerging target for therapies.
It should be noted that while IRI and T cell-mediated rejection involve complement activation in the tublointersitial compartment of the kidney, ABMR takes place within the vascular compartment where complement is deposited at the site of antibody binding [69]. Potential therapeutic agents should therefore be appropriately selected for delivery to the appropriate compartment for maximal therapeutic benefit.
Therapeutics
The effectors of the complement cascade are potential therapeutic targets for the treatment of certain inflammatory conditions. A recent review of these targets illustrates the breadth and indications for such approaches [70]. Here, we focus on recent updates with relevance to kidney transplantation.
The component of choice is partly determined by the severity of the complement-mediated inflammatory condition and the degree to which intervention at this point will interfere with the critical anti-microbial effector functions of the complement system, such as immune complex clearance, opsonization and chemotactic movement of immune cells such as polymorphonuclear neutrophils. Prolonged inhibition of complement activation will lead to individuals becoming susceptible to recurrent infections. To this end, tailored therapeutic strategies aimed to interrupt complement activity in an organ-specific fashion are likely to lead to more favourable outcomes for patients [2].
In the setting of whole organ transplant rejection, there is active research into the application of reagents that can target the complement system, aimed at taming ischaemic damage and subsequent allograft rejection. Eculizumab (anti-C5), marketed by Alexion as Soliris, is licensed as a treatment for paroxysmal nocturnal haemoglobinuria (PNH) [71]. Its application in other pathologies is also currently being investigated, particularly as a method of treating IRI injury associated with kidney transplantation [72]. In a recent review, eculizumab has been shown to protect renal allografts in recipients suffering from post-transplant atypical haemolytic uremic syndrome (aHUS) and ABMR, with positive outcomes over the short term for those patients who are not highly sensitized (reviewed in 73). In a more recent study, with an end point follow-up of 9 weeks post-transplant, in patients subjected to a desensitization protocol, Eculizumab was determined to be effective as a treatment for renal allograft rejection. However, the outcome was heavily dependent upon the method of analysis employed, highlighting the complexity of pathological assessment of patient renal biopsies [74]. In a study focusing on longer term outcomes in paediatric renal transplant recipients, those that were treated with Eculizumab presented with significantly improved early graft function and a significant reduction in chronic glomerulopathy up to 3 years after transplantation. However, an unexpected number of Eculizumab-treated patients had graft rejection following flu-like symptoms suggesting more study is required [72].
Alternative approaches recently employed anti-complement therapeutics have been met with promising results, utilizing a method whereby complement inhibitor can be directly administered to the donor kidney ex vivo prior to transplantation. This approach has the advantage of inhibiting complement activity locally within the kidney, thereby having less impact on the recipient’s systemic pool of complement. Mirococept (derived from human CR1; [75]) was administered at 10 mg per volunteer in a trial (EMPIRIKAL) to assess whether treatment would have a positive impact on the length of delayed graft function (DGF; [76]). The first EMPIRIKAL trial successfully showed the feasibility and safety of the ex vivo delivery of Mirococept to the kidney allograft. It also showed the need for a dose calibration study which was subsequently completed in pig kidney. These dosing studies have demonstrated strong localization to the tubular epithelium and capillaries of the treated kidney up to 80 mg, with minimal release into the recipient [77]. The original EMPIRIKAL trial is now undergoing a revised study protocol for human subjects based on this new higher optimal dose achieved in pigs (EMPIRIKAL-2).
There is scope for therapeutic intervention at the ‘trigger’ stage of complement activation in the setting of inflammatory pathologies. We have recently investigated the principle of sugar blockade to obstruct the carbohydrate recognition site on CL-11 and this therapy inhibits the lectin pathway in a model of renal IRI. Following the intraperitoneal administration of L-fucose in doses sufficient to raise the intrarenal concentration of the soluble sugar, mice were protected against the induction of renal IRI injury as shown by improved renal function when compared to control-treated animals [12]. This is the first demonstration of using a simple sugar to inhibit complement activation in the kidney with beneficial effect, early data in transplanted murine kidney (unpublished) suggesting similar results. This approach is simple and cheap, theoretically leaving anti-microbial functions of the other activation pathways intact.
Complement inhibitors as promising future therapeutics
A number of complement inhibitors have recently been approved for therapy or are in the final stages of clinical trials and could have an important role in the future treatment of IRI and transplant complications. Firstly, C1 inhibitors (C1-INH) as general serine protease inhibitors, would target the classical and lectin pathways, and although one clinical trial employing a C1-INH has been evaluated as a treatment for ABMR in renal transplant recipients (CINRYZE; Shire ViroPharma Incorporated). The drug was well tolerated, and no demonstrable difference between groups with respect to graft survival was observed, though the C1-INH group trended toward an improvement in renal function. This study was ultimately terminated [83]. However, a separate C1-INH inhibitor (Berinert; CSL Behring) is being tested in a phase 2 study to test the safety and efficacy of this reagent in renal transplant recipients with refractory ABMR [84].
To target complement more generally, work has been undertaken on C3 inhibitors. The non-PEGylated C3 inhibitor compstatin analogue AMY-101 has been developed by Amyndas Pharmaceuticals and they have already announced plans to develop this C3-targeted inhibitor as a treatment option for prevention of organ rejection during kidney transplantation and treatment of patients with end-stage renal disease (ESRD) [85]. Apellis Pharmaceuticals’ complement protein C3 inhibitor Pegcetacoplan has been approved for the treatment of paroxysmal nocturnal hemoglobinuria (PNH) and in a trial has been shown to inhibit both intravascular and extravascular haemolysis, as reflected by increased baseline haemoglobin levels in treated subjects [78]. It is a cyclic peptide and with a small size (43.5 kDa), so application of this technology is pertinent to therapeutic delivery within the kidney, as molecules of this size may penetrate to the corticomedullary junction, the hypoxia-sensitive region of the kidney.
Factor B inhibitors target the alternative pathway as a therapeutic strategy. A novel-specific antisense oligonucleotide (ASO) targeting human complement factor B gene has been developed with the specific aim of reducing circulating levels of this fluid-phase component of the AP, thereby decrease AP activity. The therapeutic, named IONIS-FB-LRX, was evaluated in a phase 1 safety study of healthy volunteers (Clinical trial ref: ACTRN12616000335493). IONIS-FB-LRX reduced plasma factor B levels in a dose-dependent manner in this study. Based on these findings, a phase 2 trial has since been initiated to assess the ability of IONIS-FB-LRX to diminish the growth of age-related macular degeneration (AMD)-associated lesions [79]. This antisense technology could be employed in other inflammatory settings, such as IRI, to temporarily ‘switch off’ the generation of complement intermediates during the acute injury phase. The kidney itself has the capacity to synthesize complement proteins which makes this therapeutic approach an attractive option to pursue further.
Finally, factor D inhibitors would also target the AP. A small-molecule inhibitor of factor D has been developed by Achillion (now part of Alexion/AstraZeneca). This therapeutic is administered orally and has shown promising results in a phase 2 study of PNH, with inhibition of complement-mediated intravascular haemolysis and extravascular haemolysis curtailed significantly [80]. There may be scope for use of small molecules, such as this that could be developed for transplant recipients. The nature of the drug would qualify it for easy penetrance of the renal interstitium and by its design, and its ease of delivery to patients is highly advantageous. These examples indicate an exciting future for the use of complement inhibitors in combating IRI and the detrimental consequences of transplantation.
Conclusion
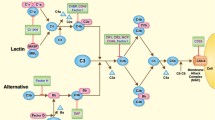
The role of complement in transplantation has grown as a field of study in recent years as it has become clear how great an effect it has on the success or failure of the procedure, at least in experimental models. IRI is a key driver of complement activation at the onset of the transplant procedure, as has been demonstrated in a number of organs but is not the whole story, as the crosstalk between the complement system and the greater immune system and the coagulation system also has roles to play (summarized in Fig. 1). This broader perspective of the role of complement in transplantation has led to emerging therapeutic strategies, which we anticipate will take several more years to validate for clinical use.

A summary of how complement is activated in IRI and transplantation alongside the downstream consequences. IRI causes stressed epithelial tissue to expose fucosylated ligands (black triangles) which are bound by lectin pathway pattern recognition molecules (PRMs). Antibody-mediated rejection (ABMR) is mediated by donor-specific antibody (DSA) which binds donor endothelial cells (ECs), at sites of inflammation—DSA is then bound by C1q, the PRM of the classical pathway. Both the classical and lectin pathway PRMs activate their respective complement pathways, ultimately producing C3 convertases that cleave C3 to C3a and C3b. C3a, amongst other roles contributes to T cell activation and proliferation. C3b causes activation of the alternative pathway which creates more C3 convertase and therefore a further increase in C3a and C3b. C3b combines with other complement pathway components to create C5 convertases and cleave C5. The resulting products of C5 cleavage are C5a which contributes to inflammation, T cell activation and proliferation and antigen presentation, as well as contributing to ABMR. Another product C5b, alongside complement components C6 to 9 form the terminal complement component, membrane attack complex (MAC). Another downstream product of C3 cleavage, C3d also contributes to retention of antigen in FDCs, and increased stimulation of B cells. Alongside the traditional complement pathway activation, the PRMs of the lectin pathway are also able to crosstalk with the coagulation pathway through the MBL-associated serine proteases (MASPs) 1 and 2 which ultimately results in the attraction of phagocytes, as well as providing adhesion points for immune cells. Epithelial and endothelial injury induced by these processes drives the cycle of complement activation and coagulation feeding back into the pathways described above
Unsurprisingly in the context of COVID-19, complement has emerged as a likely contributory factor given the constellation of hyperinflammation, vasculitis and microthrombosis (reviewed in [81]). Furthermore, a recent study showed 46% of patients with COVID-19 had acute kidney injury (AKI) [82]. It remains unclear whether the immune pathology of the AKI is secondary to viral infection of the kidney or is part of the systemic inflammatory response that associates with the acute lung injury. Nonetheless, it is possible that the mechanism of AKI reviewed here may add understanding of how renal involvement is a complication of this condition, to which transplant patients are also vulnerable.
Data availability
N/A.
Code availability
N/A.
Change history
18 March 2022
A Correction to this paper has been published: https://doi.org/10.1007/s00281-022-00924-w
References
Walport MJ (2001) Complement. First of two parts. N Engl J Med 344:1058–1066
Sacks SH, Zhou W (2012) The role of complement in the early immune response to transplantation. Nat Rev Immunol 12:431–442
Howard M, Farrar CA, Sacks SH (2018) Structural and functional diversity of collectins and ficolins and their relationship to disease. Semin Immunopathol 40:75–85
Garred P, Genster N, Pilely K, Bayarri-Olmos R, Rosbjerg A, Ma YJ, Skjoedt MO (2016) A journey through the lectin pathway of complement-MBL and beyond. Immunol Rev 274:74–97
Kang YH, Tan LA, Carroll MV, Gentle ME, Sim RB (2009) Target pattern recognition by complement proteins of the classical and alternative pathways. Adv Exp Med Biol 653:117–128
Morgan BP, Walters D, Serna M, Bubeck D (2016) Terminal complexes of the complement system: new structural insights and their relevance to function. Immunol Rev 274:141–151
Lachmann PJ (2009) The amplification loop of the complement pathways. Adv Immunol 104:115–149
Howard MC, Nauser CL, Vizitiu DA, Sacks SH. 2020. Fucose as a new therapeutic target in renal transplantation. Pediatr Nephrol
Eltzschig HK, Eckle T (2011) Ischemia and reperfusion–from mechanism to translation. Nat Med 17:1391–1401
Farrar CA, Zhou W, Lin T, Sacks SH (2006) Local extravascular pool of C3 is a determinant of postischemic acute renal failure. FASEB J 20:217–226
Zhou W, Farrar CA, Abe K, Pratt JR, Marsh JE, Wang Y, Stahl GL, Sacks SH (2000) Predominant role for C5b–9 in renal ischemia/reperfusion injury. J Clin Invest 105:1363–1371
Howard MC, Nauser CL, Farrar CA, Wallis R, Sacks SH (2020) l-Fucose prevention of renal ischaemia/reperfusion injury in Mice. FASEB J 34:822–834
Farrar CA, Tran D, Li K, Wu W, Peng Q, Schwaeble W, Zhou W, Sacks SH (2016) Collectin-11 detects stress-induced L-fucose pattern to trigger renal epithelial injury. J Clin Invest 126:1911–1925
Thurman JM, Ljubanovic D, Edelstein CL, Gilkeson GS, Holers VM (2003) Lack of a functional alternative complement pathway ameliorates ischemic acute renal failure in mice. J Immunol 170:1517–1523
Panagiotou A, Trendelenburg M, Osthoff M (2018) The lectin pathway of complement in myocardial ischemia/reperfusion injury-review of its significance and the potential impact of therapeutic interference by C1 esterase inhibitor. Front Immunol 9:1151
Sproston NR, Ashworth JJ (2018) Role of C-reactive protein at sites of inflammation and infection. Front Immunol 9:754
Zhang M, Michael LH, Grosjean SA, Kelly RA, Carroll MC, Entman ML (2006) The role of natural IgM in myocardial ischemia-reperfusion injury. J Mol Cell Cardiol 41:62–67
Pepys MB, Hirschfield GM, Tennent GA, Gallimore JR, Kahan MC, Bellotti V, Hawkins PN, Myers RM, Smith MD, Polara A, Cobb AJ, Ley SV, Aquilina JA, Robinson CV, Sharif I, Gray GA, Sabin CA, Jenvey MC, Kolstoe SE, Thompson D, Wood SP (2006) Targeting C-reactive protein for the treatment of cardiovascular disease. Nature 440:1217–1221
Diepenhorst GM, van Gulik TM, Hack CE (2009) Complement-mediated ischemia-reperfusion injury: lessons learned from animal and clinical studies. Ann Surg 249:889–899
Collard CD, Vakeva A, Morrissey MA, Agah A, Rollins SA, Reenstra WR, Buras JA, Meri S, Stahl GL (2000) Complement activation after oxidative stress: role of the lectin complement pathway. Am J Pathol 156:1549–1556
Jordan JE, Montalto MC, Stahl GL (2001) Inhibition of mannose-binding lectin reduces postischemic myocardial reperfusion injury. Circulation 104:1413–1418
Schwaeble WJ, Lynch NJ, Clark JE, Marber M, Samani NJ, Ali YM, Dudler T, Parent B, Lhotta K, Wallis R, Farrar CA, Sacks S, Lee H, Zhang M, Iwaki D, Takahashi M, Fujita T, Tedford CE, Stover CM (2011) Targeting of mannan-binding lectin-associated serine protease-2 confers protection from myocardial and gastrointestinal ischemia/reperfusion injury. Proc Natl Acad Sci U S A 108:7523–7528
Lubbers R, van Essen MF, van Kooten C, Trouw LA (2017) Production of complement components by cells of the immune system. Clin Exp Immunol 188:183–194
Lehmann TG, Koeppel TA, Munch S, Heger M, Kirschfink M, Klar E, Post S (2001) Impact of inhibition of complement by sCR1 on hepatic microcirculation after warm ischemia. Microvasc Res 62:284–292
Arumugam TV, Woodruff TM, Stocks SZ, Proctor LM, Pollitt S, Shiels IA, Reid RC, Fairlie DP, Taylor SM (2004) Protective effect of a human C5a receptor antagonist against hepatic ischaemia-reperfusion injury in rats. J Hepatol 40:934–941
Fondevila C, Shen XD, Tsuchihashi S, Uchida Y, Freitas MC, Ke B, Busuttil RW, Kupiec-Weglinski JW (2008) The membrane attack complex (C5b–9) in liver cold ischemia and reperfusion injury. Liver Transpl 14:1133–1141
Thorgersen EB, Barratt-Due A, Haugaa H, Harboe M, Pischke SE, Nilsson PH, Mollnes TE (2019) The role of complement in liver injury, regeneration, and transplantation. Hepatology 70:725–736
Marshall KM, He S, Zhong Z, Atkinson C, Tomlinson S (2014) Dissecting the complement pathway in hepatic injury and regeneration with a novel protective strategy. J Exp Med 211:1793–1805
Diepenhorst GM, de Graaf W, Niessen HW, van Vliet AK, Hack CE, van Gulik TM (2014) Immunoglobulin M, C-reactive protein and complement activation in rat hepatic ischemia-reperfusion injury. Eur Surg Res 52:50–62
Killick J, Morisse G, Sieger D, Astier AL (2018) Complement as a regulator of adaptive immunity. Semin Immunopathol 40:37–48
Carroll MC, Isenman DE (2012) Regulation of humoral immunity by complement. Immunity 37:199–207
Toapanta FR, Ross TM (2006) Complement-mediated activation of the adaptive immune responses: role of C3d in linking the innate and adaptive immunity. Immunol Res 36:197–210
Liu J, Miwa T, Hilliard B, Chen Y, Lambris JD, Wells AD, Song WC (2005) The complement inhibitory protein DAF (CD55) suppresses T cell immunity in vivo. J Exp Med 201:567–577
Fang C, Miwa T, Shen H, Song WC (2007) Complement-dependent enhancement of CD8+ T cell immunity to lymphocytic choriomeningitis virus infection in decay-accelerating factor-deficient mice. J Immunol 179:3178–3186
Hopken UE, Lu B, Gerard NP, Gerard C (1996) The C5a chemoattractant receptor mediates mucosal defence to infection. Nature 383:86–89
Dunkelberger JR, Song WC (2010) Complement and its role in innate and adaptive immune responses. Cell Res 20:34–50
Grover SP, Mackman N (2019) Intrinsic pathway of coagulation and thrombosis. Arterioscler Thromb Vasc Biol 39:331–338
Degn SE, Jensenius JC, Bjerre M (2011) The lectin pathway and its implications in coagulation, infections and auto-immunity. Curr Opin Organ Transplant 16:21–27
Matsushita M, Endo Y, Fujita T (2013) Structural and functional overview of the lectin complement pathway: its molecular basis and physiological implication. Arch Immunol Ther Exp (Warsz) 61:273–283
Gulla KC, Gupta K, Krarup A, Gal P, Schwaeble WJ, Sim RB, O’Connor CD, Hajela K (2010) Activation of mannan-binding lectin-associated serine proteases leads to generation of a fibrin clot. Immunology 129:482–495
Krarup A, Wallis R, Presanis JS, Gal P, Sim RB (2007) Simultaneous activation of complement and coagulation by MBL-associated serine protease 2. PLoS One 2:e623
Pratt JR, Basheer SA, Sacks SH (2002) Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat Med 8:582–587
Farrar CA, Kupiec-Weglinski JW, Sacks SH. 2013. The innate immune system and transplantation. Cold Spring Harb Perspect Med 3: a015479
Tang Z, Lu B, Hatch E, Sacks SH, Sheerin NS (2009) C3a mediates epithelial-to-mesenchymal transition in proteinuric nephropathy. J Am Soc Nephrol 20:593–603
Peng Q, Li K, Smyth LA, Xing G, Wang N, Meader L, Lu B, Sacks SH, Zhou W (2012) C3a and C5a promote renal ischemia-reperfusion injury. J Am Soc Nephrol 23:1474–1485
Thurman JM, Lenderink AM, Royer PA, Coleman KE, Zhou J, Lambris JD, Nemenoff RA, Quigg RJ, Holers VM (2007) C3a is required for the production of CXC chemokines by tubular epithelial cells after renal ishemia/reperfusion. J Immunol 178:1819–1828
Arumugam TV, Shiels IA, Strachan AJ, Abbenante G, Fairlie DP, Taylor SM (2003) A small molecule C5a receptor antagonist protects kidneys from ischemia/reperfusion injury in rats. Kidney Int 63:134–142
De Vries B, Matthijsen RA, Wolfs TG, Van Bijnen AA, Heeringa P, Buurman WA (2003) Inhibition of complement factor C5 protects against renal ischemia-reperfusion injury: inhibition of late apoptosis and inflammation. Transplantation 75:375–382
Saadi S, Holzknecht RA, Patte CP, Platt JL (2000) Endothelial cell activation by pore-forming structures: pivotal role for interleukin-1alpha. Circulation 101:1867–1873
Heeger PS, Lalli PN, Lin F, Valujskikh A, Liu J, Muqim N, Xu Y, Medof ME (2005) Decay-accelerating factor modulates induction of T cell immunity. J Exp Med 201:1523–1530
Zhou W, Patel H, Li K, Peng Q, Villiers MB, Sacks SH (2006) Macrophages from C3-deficient mice have impaired potency to stimulate alloreactive T cells. Blood 107:2461–2469
Peng Q, Li K, Patel H, Sacks SH, Zhou W (2006) Dendritic cell synthesis of C3 is required for full T cell activation and development of a Th1 phenotype. J Immunol 176:3330–3341
Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, Shapiro VS, Dubyak GR, Heeger PS, Medof ME (2008) Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity 28:425–435
Peng Q, Li K, Anderson K, Farrar CA, Lu B, Smith RA, Sacks SH, Zhou W (2008) Local production and activation of complement up-regulates the allostimulatory function of dendritic cells through C3a–C3aR interaction. Blood 111:2452–2461
Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS (2008) Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood 112:1759–1766
Fang Y, Xu C, Fu YX, Holers VM, Molina H (1998) Expression of complement receptors 1 and 2 on follicular dendritic cells is necessary for the generation of a strong antigen-specific IgG response. J Immunol 160:5273–5279
Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT (1996) C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science 271:348–350
Marsh JE, Farmer CK, Jurcevic S, Wang Y, Carroll MC, Sacks SH (2001) The allogeneic T and B cell response is strongly dependent on complement components C3 and C4. Transplantation 72:1310–1318
Gosset C, Lefaucheur C, Glotz D (2014) New insights in antibody-mediated rejection. Curr Opin Nephrol Hypertens 23:597–604
Lachmann N, Terasaki PI, Budde K, Liefeldt L, Kahl A, Reinke P, Pratschke J, Rudolph B, Schmidt D, Salama A, Schonemann C (2009) Anti-human leukocyte antigen and donor-specific antibodies detected by luminex posttransplant serve as biomarkers for chronic rejection of renal allografts. Transplantation 87:1505–1513
Pepys MB (1974) Role of complement in induction of antibody production in vivo. Effect of cobra factor and other C3-reactive agents on thymus-dependent and thymus-independent antibody responses. J Exp Med 140:126–145
Loupy A, Lefaucheur C, Vernerey D, Prugger C, Duong van Huyen JP, Mooney N, Suberbielle C, Fremeaux-Bacchi V, Mejean A, Desgrandchamps F, Anglicheau D, Nochy D, Charron D, Empana JP, Delahousse M, Legendre C, Glotz D, Hill GS, Zeevi A, Jouven X (2013) Complement-binding anti-HLA antibodies and kidney-allograft survival. N Engl J Med 369:1215–1226
Li Q, Peng Q, Xing G, Li K, Wang N, Farrar CA, Meader L, Sacks SH, Zhou W (2010) Deficiency of C5aR prolongs renal allograft survival. J Am Soc Nephrol 21:1344–1353
Wang H, Arp J, Liu W, Faas SJ, Jiang J, Gies DR, Ramcharran S, Garcia B, Zhong R, Rother RP (2007) Inhibition of terminal complement components in presensitized transplant recipients prevents antibody-mediated rejection leading to long-term graft survival and accommodation. J Immunol 179:4451–4463
Dorling A (2012) Transplant accommodation–are the lessons learned from xenotransplantation pertinent for clinical allotransplantation? Am J Transplant 12:545–553
Locke JE, Magro CM, Singer AL, Segev DL, Haas M, Hillel AT, King KE, Kraus E, Lees LM, Melancon JK, Stewart ZA, Warren DS, Zachary AA, Montgomery RA (2009) The use of antibody to complement protein C5 for salvage treatment of severe antibody-mediated rejection. Am J Transplant 9:231–235
Dawson KL, Parulekar A, Seethamraju H (2012) Treatment of hyperacute antibody-mediated lung allograft rejection with eculizumab. J Heart Lung Transplant 31:1325–1326
Glotz D, Russ G, Rostaing L, Legendre C, Tufveson G, Chadban S, Grinyo J, Mamode N, Rigotti P, Couzi L, Buchler M, Sandrini S, Dain B, Garfield M, Ogawa M, Richard T, Marks WH, Group CS. 2019. Safety and efficacy of eculizumab for the prevention of antibody-mediated rejection after deceased-donor kidney transplantation in patients with preformed donor-specific antibodies. Am J Transplant 19: 2865-75
Stites E, Le Quintrec M, Thurman JM (2015) The complement system and antibody-mediated transplant rejection. J Immunol 195:5525–5531
Mastellos DC, Ricklin D, Lambris JD (2019) Clinical promise of next-generation complement therapeutics. Nat Rev Drug Discov 18:707–729
Hillmen P, Young NS, Schubert J, Brodsky RA, Socie G, Muus P, Roth A, Szer J, Elebute MO, Nakamura R, Browne P, Risitano AM, Hill A, Schrezenmeier H, Fu CL, Maciejewski J, Rollins SA, Mojcik CF, Rother RP, Luzzatto L (2006) The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med 355:1233–1243
Kaabak M, Babenko N, Shapiro R, Zokoyev A, Dymova O, Kim E. 2018. A prospective randomized, controlled trial of eculizumab to prevent ischemia-reperfusion injury in pediatric kidney transplantation. Pediatr Transplant 22
Barnett AN, Asgari E, Chowdhury P, Sacks SH, Dorling A, Mamode N (2013) The use of eculizumab in renal transplantation. Clin Transplant 27:E216–E229
Marks WH, Mamode N, Montgomery RA, Stegall MD, Ratner LE, Cornell LD, Rowshani AT, Colvin RB, Dain B, Boice JA, Glotz D, Group CS (2019) Safety and efficacy of eculizumab in the prevention of antibody-mediated rejection in living-donor kidney transplant recipients requiring desensitization therapy: a randomized trial. Am J Transplant 19:2876–2888
Patel H, Smith RA, Sacks SH, Zhou W (2006) Therapeutic strategy with a membrane-localizing complement regulator to increase the number of usable donor organs after prolonged cold storage. J Am Soc Nephrol 17:1102–1111
Kassimatis T, Qasem A, Douiri A, Ryan EG, Rebollo-Mesa I, Nichols LL, Greenlaw R, Olsburgh J, Smith RA, Sacks SH, Drage M (2017) A double-blind randomised controlled investigation into the efficacy of Mirococept (APT070) for preventing ischaemia reperfusion injury in the kidney allograft (EMPIRIKAL): study protocol for a randomised controlled trial. Trials 18:255
Kassimatis T, Greenlaw R, Hunter JP, Douiri A, Flach C, Rebollo-Mesa I, Nichols LL, Qasem A, Danzi G, Olsburgh J, Drage M, Friend PJ, Neri F, Karegli J, Horsfield C, Smith RA, Sacks SH (2021) Ex vivo delivery of Mirococept: a dose-finding study in pig kidney after showing a low dose is insufficient to reduce delayed graft function in human kidney. Am J Transplant 21:1012–1026
Tatapudi VS, Montgomery RA (2019) Therapeutic modulation of the complement system in kidney transplantation: Clinical indications and emerging drug leads. Front Immunol 10:2306
C1-Inhibitor (INH) for Refractory Antibody Mediated Renal Allograft Rejection. https://ClinicalTrials.gov/show/NCT02936479
Amyndas. accessed 20/09/21. https://www.amyndas.com/research-focus
Hillmen P, Szer J, Weitz I, Roth A, Hochsmann B, Panse J, Usuki K, Griffin M, Kiladjian JJ, de Castro C, Nishimori H, Tan L, Hamdani M, Deschatelets P, Francois C, Grossi F, Ajayi T, Risitano A, de la Tour RP (2021) Pegcetacoplan versus Eculizumab in Paroxysmal Nocturnal Hemoglobinuria. N Engl J Med 384:1028–37
Jaffe GJ, Sahni J, Fauser S, Geary RS, Schneider E, McCaleb M (2020) Development of IONIS-FB-LRx to treat geographic atrophy associated with AMD. Investig Ophthalmol Vis Sci 61:4305
Risitano AM, Kulasekararaj AG, Lee JW, Maciejewski JP, Notaro R, Brodsky R, Huang M, Geffner M, Browett P (2020) Danicopan: an oral complement factor D inhibitor for paroxysmal nocturnal hemoglobinuria. Haematologica Online ahead of print
Polycarpou A, Howard M, Farrar CA, Greenlaw R, Fanelli G, Wallis R, Klavinskis LS, Sacks S (2020) Rationale for targeting complement in COVID-19. EMBO Mol Med 12:e12642
Chan L, Chaudhary K, Saha A, Chauhan K, Vaid A, Zhao S, Paranjpe I, Somani S, Richter F, Miotto R, Lala A, Kia A, Timsina P, Li L, Freeman R, Chen R, Narula J, Just AC, Horowitz C, Fayad Z, Cordon-Cardo C, Schadt E, Levin MA, Reich DL, Fuster V, Murphy B, He JC, Charney AW, Bottinger EP, Glicksberg BS, Coca SG, Nadkarni GN, Mount Sinai CIC (2021) AKI in hospitalized patients with COVID-19. J Am Soc Nephrol 32:151–160
Author information
Authors and Affiliations
Contributions
S. Sacks conceived the idea for the article. M. Howard took the lead in writing the manuscript, alongside writing and literature searches from C. Nauser and C. Farrar. All authors provided critical feedback and helped shape the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
S. Sacks has previously consulted for Omeros and other companies with an interest in complement therapeutics. The other authors have no conflict of interest to disclose.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is a contribution to the Special issue on: Complement & Disease: Out of the Shadow into the Spotlight - Guest Editors: Daniel Ricklin & Richard B. Pouw
The original online version of this article was revised: In this article, the order of references 78 until 85 has been corrected.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Howard, M.C., Nauser, C.L., Farrar, C.A. et al. Complement in ischaemia–reperfusion injury and transplantation. Semin Immunopathol 43, 789–797 (2021). https://doi.org/10.1007/s00281-021-00896-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-021-00896-3