
Abstract
Bone marrow mesenchymal stromal cells (MSCs) have been described as potent regulators of T-cell function, though whether they could impede the effectiveness of immunotherapy against acute myeloid leukemia (AML) is still under investigation. We examine whether they could interfere with the activity of leukemia-specific clonal cytotoxic T-lymphocytes (CTLs) and chimeric antigen receptor (CAR) T cells, as well as whether the immunomodulatory properties of MSCs could be associated with the induction of T-cell senescence. Co-cultures of leukemia-associated Wilm’s tumor protein 1 (WT1) and tyrosine-protein kinase transmembrane receptor 1 (ROR1)-reactive CTLs and of CD123-redirected switchable CAR T cells were prepared in the presence of MSCs and assessed for cytotoxic potential, cytokine secretion, and expansion. T-cell senescence within functional memory sub-compartments was investigated for the senescence-associated phenotype CD28−CD57+ using unmodified peripheral blood mononuclear cells. We describe inhibition of expansion of AML-redirected switchable CAR T cells by MSCs via indoleamine 2,3-dioxygenase 1 (IDO-1) activity, as well as reduction of interferon gamma (IFNγ) and interleukin-2 (IL-2) release. In addition, MSCs interfered with the secretory potential of leukemia-associated WT1- and ROR1-targeting CTL clones, inhibiting the release of IFNγ, tumor necrosis factor alpha, and IL-2. Abrogated T cells were shown to retain their cytolytic activity. Moreover, we demonstrate induction of a CD28loCD27loCD57+KLRG1+ senescent T-cell phenotype by MSCs. In summary, we show that MSCs are potent modulators of anti-leukemic T cells, and targeting their modes of action would likely be beneficial in a combinatorial approach with AML-directed immunotherapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute myeloid leukemia (AML) is a form of hematological cancer derived from the abnormal expansion of myeloid precursor cells, resulting in invasion of the bone marrow and ultimately failure of normal hematopoiesis. As it stands, the 5-year survival rate is less than 30%, underlining a desperate need for alternative treatments. Chimeric antigen receptor (CAR) T cells are a promising avenue currently in early clinical trials [1]. Initial reports have noted however that while several refractory/relapsed AML patients achieve a response to CAR T-cell adoption, only a minority achieve complete remission and a relevant proportion fail to respond at all, highlighting the need for further research into mechanisms of resistance [2,3,4]. In addition to immunosuppressive pathways intrinsic to AML blasts, neighboring constituents of the bone marrow can provide additional protection. In particular, a lot of interest has been garnered for resident mesenchymal stromal cells (MSCs) which were found to possess an impressive immunomodulatory capacity in suppressing the proliferation and the inflammatory potential of T cells [5]. This along with other properties such as multipotency has highlighted the therapeutic potential of MSCs in regenerative medicine and the treatment of immune disorders [6].
It follows to ask if MSCs can interfere with the action of T-cell-based immunotherapies in the context of hematological malignancy. At the time of writing, few reports have come out addressing this topic. One early report investigated whether MSCs could ablate the cytotoxic potential of T cells specific for the AML antigen Wilm’s tumor protein 1 (WT1), of which they found no effect [7]. More recently, it was reported that infusion of MSCs did not affect CD19 CAR T-cell activity in acute lymphocytic leukemia xenografts despite being able to efficiently control inflammation in a acute colitis murine model [8]. In contrast, another study demonstrated that MSCs could in fact decrease cell lysis mediated by several CAR T-cell constructs against multiple myeloma [9]. To reconcile these contradictory reports, it is apparent that more research in the field is needed.
To gain deeper insight into this issue, firstly, we investigated the effect of MSCs on the proliferative, secretory, and cytotoxic potential of clonal CD8+ cytotoxic T-lymphocytes (CTLs) specific for the leukemia-associated antigens WT1 and tyrosine-protein kinase transmembrane receptor 1 (ROR1) [10, 11], as well a switchable CAR T-cell system redirected against the AML marker CD123 currently tested in a phase I clinical trial [12,13,14,15]. We demonstrate that MSCs induce a reduction in T-cell proliferation and release of inflammatory cytokines, but without ultimately influencing killing potential against AML targets. Secondly, aberrant senescent T cells have been identified in circulation and within the bone marrow of AML patients [16,17,18], and we demonstrate here that MSCs can induce the senescence-associated CD28loCD27loCD57+KLRG1+ phenotype in T cells.
Methods
Isolation of mesenchymal stromal cells and peripheral blood mononuclear cells
MSCs were isolated from bone marrow aspirates of healthy donors (EK307082018) and AML patients (EK98032010) after informed consent and approval by the local review board, as previously described [19]. MSCs were characterized based on the criteria set out by the International Society for Cellular Therapy [20]. Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Pancoll (PAN-Biotech, Germany) from blood donated by healthy volunteers (EK206082008).
PBMC co-culture
PBMCs and MSCs were co-cultured at a 5:1 or 100:1 ratio with CD3/CD28 Dynabeads (Gibco, USA) in RPMI: Roswell Park Memorial Institute 1640 (Gibco, USA) with 10% fetal bovine serum (FBS) (Merck, Germany). Proliferation assays were carried out as previously described [19]. The Transwell Permeable Support system with 0.4-μm polycarbonate membranes (Corning, USA) was used for non-contact cultures.
CTL clone co-culture
CTL clones were generated as described elsewhere [10, 11]. Matched HLA-A*02:01 T2 target cells were pulsed with 20 μg/mL WT1126 (RMFPNAPYL) or HIV Gag-Pol896 (ILKEPVHGL) nonamers and HLA-B*07:02 K562 with ROR1783 (NPRYPNYMF) or HIV Gag-Pol355 (GPGHKARVL) nonamers. For cytotoxicity assays, 100 μCi/mL 51Chromium (Hartmann Analytic, Germany) was added. CTLs and target cells were co-cultured with MSCs at an E:T:MSC ratio of 10:1:2 in RPMI. Supernatant was collected and mixed with Ultima Gold scintillation cocktail (PerkinElmer, USA), and 51Cr release was assessed with the MicroBeta 2 (PerkinElmer, USA). Supernatant was also collected for cytokine determination.
Switchable CAR T-cell co-culture
The switchable CAR T-cell system has been described elsewhere [12]. AML blasts were collected from a primary patient sample derived from a 74-year-old female diagnosed with NPM-1 and CEBPA-mutated AML. Phenotyping revealed > 90% positivity for CD123 within the marrow blast population. The patient had provided written consent to the banking of the material and to its use within the research project (EK98032010). Target MOLM-13 and AML blasts were labeled with eFluor 670 (Thermo Fisher, USA) and co-cultured with CAR T cells in the Transwell Permeable Support system with MSCs at a E:T:MSC ratio of 16:16:3 with 0.5-nM target module (TM) in RPMI/Complete (RPMI with 10% FBS, 100 μg/ml penicillin/streptomycin, 1% nonessential amino acids, 2-mM N-acetyl-l-alanyl-l-glutamine, and 1-mM sodium pyruvate (Biochrom, UK)). For indoleamine 2,3-dioxygenase 1 (IDO-1) blocking assays, 0.2-mM 1-methyl-L-tryptophan (1-MT) (Sigma-Aldrich, USA) diluted in 0.2-mM NaOH was added. For cytotoxicity determination, CAR T cells (PI−eGFP+) and target cells (PI−eFluor670+) were quantified with the MACSQuant X (Miltenyi Biotec, Germany). MSC viability was assessed with the PE Annexin V Apoptosis Detection Kit (BD, USA). Supernatant was collected for cytokine analysis. Fresh RPMI/Complete with 0.5-nM TM was added back to the cultures after each collection. Cultures were restimulated twice with target cells on day 2 and day 4 or 5.
Cytokine assessment
Cytokine concentrations were assessed with commercial kits. Human IFNγ ELISA Set and Human IL-2 ELISA Set (BD, USA) were measured on the Sunrise spectrophotometer and analyzed with Magellan software (Tecan, Switzerland). Interleukin (IL)-2, IL-17a, tumor necrosis factor alpha (TNFα), interferon gamma (IFNγ), soluble Fas ligand (sFasL), granzyme A, and perforin were measured with the LEGENDplex Human CD8/NK Panel (Biolegend, USA) on the MACSQuant X (Miltenyi Biotec, Germany) and analyzed with Legendplex software (Biolegend, USA).
Gene and cell surface protein expression
For cell surface protein expression, MSCs, PBMCs, and CAR T cells were resuspended with fluorochrome-labeled antibodies (Supplementary Table 1) and 40 ng/mL DAPI, acquired on the LSR II flow cytometers (BD, USA), and analyzed with FlowJo software (BD, USA).
For gene expression, RNA was isolated from MSCs with TRIzol reagent (Thermo Fisher, USA), and cDNA libraries were prepared with RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, USA). Quantitative polymerase chain reaction (qPCR) was performed with the Maxima SYBR Green/ROX qPCR Master Mix (Thermo Scientific, USA) on the QuantStudio 3 (Applied Biosystems, USA). Primers are listed in Supplementary Table 2.
Statistical analyses
Standard deviation (SD) and one-way or two-way ANOVA with Bonferroni’s correction for multiple comparisons were calculated with Graphpad Prism 6. p values ≤ 0.05 were considered statistically significant and were further stratified: p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), and p ≤ 0.0001 (****).
Results
MSC-mediated interference of anti-leukemic CD8+ T-cell activity
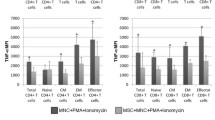
MSCs from healthy donors were selected for their ability to inhibit PBMC proliferation in co-culture (Supplementary Fig. 1). Next, we evaluated the immunomodulatory potential of these MSCs on the cytotoxic capabilities of WT1- and ROR1-specific CD8+ CTL clones, and Fig. 1a shows that the MSCs did not significantly affect cytotoxicity of the CTLs against their target cells. MSCs are also reported to decrease the release of inflammatory cytokines such as IFNγ, TNFα, and IL-2. Despite having no discernable effect on cytotoxicity, IFNγ release was significantly decreased after 24 h in the presence of MSCs for both CTL clones (Fig. 1b). The decrease in IFNγ secretion by ROR1-reactive CTLs could even be observed as early as 4 h of co-culture (Supplementary Fig. 2a), along with other cytokines (IL-2 and TNFα). In contrast, MSCs did not have a significant effect on the concentrations of the cytotoxic effector molecules sFasL, granzyme A, or perforin within these same cultures, corroborating the cytotoxicity data (Supplementary Fig. 2b).

MSCs modulate the inflammatory capabilities of WT1- and ROR1-reactive CD8+ T-cell clones without affecting cytotoxicity. a Antigen-specific WT1- and ROR1-reactive CTL clones were, respectively, incubated with 51Cr-loaded target T2 cells pulsed with WT1 peptide or K562 cells pulsed with ROR1 peptide in the presence or absence of 1 × 104 allogeneic MSCs from six healthy donors (MSC1-6) at a E:T:MSC ratio of 10:1:2. Target cells pulsed with irrelevant HIV Gag-Pol peptides served as a negative control. Efficacy of CTL-mediated lysis of target cells was determined by 51Cr release after 4 h as measured by β-counter and is expressed as a percentage of maximum lysis with correction for spontaneous release. b Cell cultures were prepared as in (a) without 51Cr loading. After 24 h, supernatants were collected and assessed for IFNγ concentration via ELISA. Data are representative of three independent experiments and are presented as the mean of technical triplicates ± SD. Asterisks represent statistically significant differences compared to the control (* p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; and **** p ≤ 0.0001; n.s. not significant)
MSC-mediated interference of AML-retargeted switchable CAR T-cell activity
The flow cytometry examination of the immunomodulatory potential of MSCs against switchable CAR T cells was conducted long term with repeated cytotoxic challenge in the Transwell cell culture system; MSCs and CAR T cells were thus limited to paracrine interactions. CAR T cells redirected against MOLM-13 cells with the anti-CD123 TM had significantly inhibited expansion kinetics in MSC co-cultures, approaching the minimal growth rate of the CAR T cells lacking TM-mediated stimulation (Fig. 2a). Intriguingly, the presence of MSCs abrogated the increased expansion of CD4+ CAR T cells compared to their CD8+ counterparts, maintaining a steady CD4+/CD8+ ratio over time similar to the unstimulated cultures (Fig. 2b). MSCs also decreased secretion of IFNγ and IL-2 by the CAR T cells, as observed previously with the CTL clones (Fig. 2c and d). Finally, the cytotoxic capabilities of the CAR T cells were studied by measuring the kinetics of MOLM-13 death after the third round of cytotoxic challenge. In line with the analysis of CTL clones, MSCs did not have an observable effect on the killing capacity of the CAR T cells, despite inducing overall lower proliferative and cytokine secretion capabilities (Fig. 2e). Similar trends could be observed when using patient-derived MSCs and AML blasts (Supplementary Fig. 3). In response to CAR T-cell activity, a large increase in gene expression was observed in MSCs for IDO-1 (Fig. 3a). To study the contribution of IDO-1 on suppression of CAR T-cell expansion, co-cultures were treated with the IDO-1 inhibitor 1-MT. In response, inhibition of expansion was almost completely reversed (Fig. 2f). To verify that this was not due to toxicity of the treatment, MSCs were subsequently assessed for apoptosis and necrosis and found to remain viable (Supplementary Fig. 4).

MSCs interfere with the proliferative and inflammatory capabilities of CD123-targeting switchable CAR T cells without affecting cytotoxicity. In the apical chamber of the Transwell cell culture system, eGFP+ CAR T cells were redirected with an anti-CD123 target module (+ TM, 0.5 nM) against eFluor670-labeled MOLM-13 cells in the presence or absence in the basolateral chamber of 7.5 × 103 MSCs from three allogeneic healthy donors (MSC1-3) at a E:T:MSC ratio of 16:16:3. Cultures without TM (-TM) served as a negative control. Additional eFluor670-labeled MOLM-13 cells were added on day 2 (1.2 × 105) and day 5 (variable at a E:T of 1:2). Surviving CAR T cells (PI−eGFP+) and MOLM-13 cells (PI−eFluor670+) were quantified via flow cytometry at 1, 2, 5, 6, and 7 days of culture. Shown are representative results of three independent assays. Data points are presented as the mean of technical triplicates ± SD. Asterisks represent statistically significant differences (**** p ≤ 0.0001; n.s. not significant). a CAR T-cell expansion was assessed as PI−eGFP+ cells/mL over time. b CD4+ and CD8+ CAR T-cell populations were assessed by flow cytometry on days 0 and 7. Data are presented as the ratio of CD4+/CD8+ fractions in the DAPI−eFluor670−CD45+eGFP+ cell population. c and d Supernatant was collected on days 1, 2, 5, 6, and 7 and assessed for IFNγ and IL-2 concentration via ELISA. e MOLM-13 killing kinetics after the third round of CAR T-cell stimulation on day 5 at a E:T of 1:2. Data points represent cumulative loss of target cells relative to the initial population on day 5. Negative values indicate MOLM-13 proliferation. f In a separate assay, co-cultures were treated with 0.2-mM NaOH (left) or 0.2-mM 1-MT/NaOH (right). CAR T-cell expansion was assessed as DAPI-eGFP + cells/mL over time

AML-activated CAR T-cell inflammatory stimuli induce the expression of immune checkpoint and lymphocyte adhesion molecules in MSCs. Co-cultures were prepared as in Fig. 2. a On day 7, mRNA was isolated from the MSCs from a single donor and reverse-transcribed into cDNA libraries. The expression of the indicated genes was assessed by qPCR, and the data are reported as expression levels relative to that of GAPDH. b and c On day 7, cells were collected and analyzed for cell surface expression of the indicated markers by flow cytometry. Shown are representative results of two independent experiments. b Cell surface expression of the indicated markers on DAPI− MSCs of the basolateral chamber. Data are presented as the mean % of positive MSCs from biological triplicates ± SD. Asterisks represent statistically significant differences (*** p ≤ 0.001 and **** p ≤ 0.0001). c Cell surface expression on day 0 and day 7 of PD-1 on CD4+ (left) and CD8+ (right) CAR T cells of the apical chamber (DAPI−eGFP+CD45+). Data are presented as the mean % of PD-1 positive CAR T cells
In addition to IDO-1, increase in gene expression in response to CAR T-cell activity was observed for the immunosuppressive factor cyclooxygenase 2 (COX-2), immune checkpoint ligands programmed cell death ligand 1 (PD-L1) and PD-L2, and intercellular adhesion molecule 1 (ICAM-1) (Fig. 3a). PD-L1, PD-L2, and ICAM-1, along with vascular cell adhesion protein 1 (VCAM-1) and lymphocyte function-associated antigen 3 (LFA-3), were further validated for cells surface expression (Fig. 3b). In parallel, programmed cell death 1 (PD-1) also increased significantly on the cell surface of CAR T cells over time (Fig. 3c).
Induction of the T-cell senescence-associated phenotype by MSCs
In addition to PD-1, investigation of co-receptor CD28 expression on the surface of activated CAR T cells was also conducted, revealing MSC-mediated decrease in CD28 median fluorescence intensity (MFI) (Supplementary Fig. 5). Using PBMCs, we investigated whether MSCs could induce an enrichment of senescence-associated CD28− and CD57+ T cells within the global CD4+ and CD8+ populations, as well as within further sub-compartments of T-cell differentiation and memory as defined by cell surface expression of CD45RA, CCR7, and CD45RO. Much like with the CAR T cells, MSC-mediated loss of CD28 could be observed on T cells within both the CD4+ and CD8+ fractions, as well as memory subfractions though CD4+ naïve and CD8+ terminal effector T cells remained largely unaffected (Fig. 4a). When referencing senescent T cells as CD8+CD28−CD57+ [16], we measured average fold changes ranging from 0.9 to 3.7 compared to the control within the differentiation subpopulations (Fig. 4b). Supplementary Fig. 6 shows example contour plots for CD28 and CD57 cell surface expression within the CD45RA−CCR7+CD45RO+ central memory compartment. We further investigated senescence-associated loss of CD27 and gain KLRG1, which we observed in conjunction with the above-mentioned senescence indicators (Fig. 4c and d), with a mean 8.1-fold increase in the CD8+CD28loCD27loCD57+KLRG1+ population in MSC co-cultures (Fig. 4d; 4 senescence indicators). Similar trends were observed in indirect co-cultures (Supplementary Fig. 7). Contour plots can be found in Supplementary Figs. 8 and 9. We further assessed the surface expression on MSCs of the CD28 ligands CD80 and CD86, which were negative, as well as HLA-DR which was positive (Supplementary Fig. 10).

MSCs induce senescence of unmodified T cells, as characterized by loss of CD28 and CD27, and gain of CD57 and KLRG1. Healthy donor PBMCs were cultured with or without 5 × 104 allogeneic MSCs from three healthy donors (MSC1-3) at a PBMC:MSC ratio of 5:1 and stimulated with anti-CD3/CD28 antibody-coated beads. After 6 days, cells were harvested and analyzed by flow cytometry. T-cell senescence (CD28lo, CD27lo, CD57+, KLRG1+) was assessed within the global CD4+ and CD8+ T-cell populations (global; DAPI−CD45+CD4+ and DAPI−CD45+CD8+), as well as within further T-cell memory subpopulations: naïve (N; CD45RA+CCR7+CD45RO−), stem cell memory (SCM; CD45RA+CCR7+CD45RO+), central memory (CM; CD45RA−CCR7+CD45RO+), effector memory (EM; CD45RA−CCR7−CD45RO+), and terminal effector (TE; CD45RA+CCR7−CD45RO−). a Above: Fraction size of CD28hi cells within the global and memory stages of CD4+ (blue) and CD8+ (red) T-cell population in direct MSC co-cultures relative to the control. Below: Median fluorescence intensity (MFI) of CD28 within the global and memory stages of CD4+ (above) and CD8+ (below) T-cell populations for control (black) MSC co-cultures (green). Data are presented as the mean of biological triplicates ± SD. φ indicates lack of sufficient number of events for assessment. b Fraction size of CD28loCD57+ cells within the global and memory stages of the CD8+ T-cell population in direct MSC co-cultures relative to the control. Results are representative of three independent experiments. c Fraction size of each senescence marker within the global CD4+ (above) and CD8+ (below) populations for control (black) and MSC co-cultures (red). d Fraction size based on the number of co-expressed senescence indicators within the global CD4+ (above) and CD8+ (below) populations for control (black) and MSC co-cultures (green). Data are presented as the mean of biological triplicates ± SD
Discussion
As light is shed onto mechanisms of AML-mediated immune escape, a new appreciation has developed regarding the reciprocal interactions within the local tumor microenvironment in suppressing the host immune response, and by extension targeted therapies with antibodies or effector immune cells. MSCs are emblematic of this activity with the ability to express a wide range of soluble and cell surface immune modulating molecules.
Herein, we demonstrated that MSCs significantly abrogate the T-cell-mediated release of several inflammatory molecules and the proliferative potency of AML-targeting T cells, activities which are likely detrimental to sustained, long-term immunotherapeutic response [21,22,23,24]. Curiously, even once activated, the paracrine mechanisms of MSCs were found insufficient to prevent directed cytolysis once the immunological synapse has formed, confirming previous findings [7, 25]. As certain MSC-associated molecules such as tumor growth factor β reduce both inflammatory and cytolytic molecules at the transcriptional level [26], it could be postulated that MSCs do not affect secretion events per se of granules already present in primed cytotoxic T cells. Alternatively, considering IFNγ promotes cytotoxicity [27], it is possible that reduced inflammation levels still surpass the threshold for efficient T-cell killing activity. As our assays preclude the involvement of direct contact mechanisms such as the PD-1/PD-L1/PD-L2 axis, their role cannot be discounted within the AML microenvironment.
Increase in both exhausted and senescent CD8+ lymphocytes has been identified within the peripheral blood and bone marrow of AML patients [16,17,18]. Exhaustion is denoted by inhibitory receptors such as PD-1, of which we found expression on CAR T cells independently of MSC activity, likely the result of priming and sustained stimulation. With regard to senescence, they can be identified by gain of KLRG1 and CD57, and loss of CD27 and CD28 [28]. We observed loss of CD28 and CD27 and gain of KLRG1 in MSC co-cultures in both CD4+ and CD8+ fractions. Enrichment of CD8+CD28loCD27loCD57+KLRG1+ cells could also be observed, though they remained at a low percentage relative to the total CD8+ population. Curiously, the CD28 ligands CD80 and CD86 are not involved due to lack of expression on MSCs [29], and rather our non-contact cultures imply activity from paracrine mechanisms. It should be noted that senescent T cells in AML have been found to possess increased IFNγ and TNFα potential, though lower IL-2 [18], which differs to what we and many others have demonstrated regarding MSC immunosuppression. It is clear that more study needs to be carried out to robustly conclude if CD28loCD27loCD57+KLRG1+ T cells under MSC activity are truly senescent. With regard to specific pathways by which MSCs and the AML microenvironment could induce the senescence-associated phenotype, tumor cells have been shown to induce senescence through mediation of cAMP activity within effector T cells [30]. This pathway is potentially shared by MSCs and regulatory T cells (Tregs) via T-cell adenylate cyclase activation by extracellular adenosine produced by the CD73-CD39 receptors [31], as well as through prostaglandin E2 release mediated by COX-2 [32] expressed by both MSCs and myeloid-derived suppressor cells. Indeed, Tregs have been demonstrated to induce CD27 and CD28 loss in both CD4+ and CD8+ T cells, along with inhibited proliferative capacity [33]. Blocking these pathways could potentially reverse the observed induction of the CD28loCD27loCD57+KLRG1+ phenotype.
Finally, we demonstrate that MSCs express a plethora of immune modulating factors in response to CAR T-cell activity. We show that IDO-1 inhibition almost completely reverses MSC-mediated suppression of CAR T-cell expansion, though the contribution of other mechanisms should be considered. Indeed, hierarchal clustering of gene expression data from AML bone marrow samples of patients receiving CD3/CD123 bispecific antibody flotetuzumab demonstrated higher immune scores for a number of MSC-associated modulatory signatures such as IDO1, TGFβ, and PDL2, as well a stromal biological signature, in non- and partial responders in comparison with complete responders [34]. Much of the paracrine regulation by MSCs is mediated through extracellular vesicles [35], which, in addition to facilitating the transfer of metabolic and peptidic factors, allow for epigenetic gene regulation via micoRNAs. A large number of extracellular microRNAs have recently been found to be differentially expressed in AML [36], some of which possess potent immunosuppressive activity. This includes bone marrow miR-21 [37], which is also abundantly present in MSC exosomes [38]. In addition to inducing T-cell apoptosis, AML-derived miR-21 can polarize T cells toward the regulatory phenotype very reminiscent of MSC activity [5]. These are just some examples that later could be investigated in relevant preclinical models to develop a combinatorial approach for optimized CAR T-cell therapy.
Data availability
Data are available upon reasonable request.
References
Koedam J, Wermke M, Ehninger A, Cartellieri M, Ehninger G (2022) Chimeric antigen receptor T-cell therapy in acute myeloid leukemia. Curr Opin Hematol 29:74–83. https://doi.org/10.1097/MOH.0000000000000703
Wang QS, Wang Y, Lv HY, Han QW, Fan H, Guo B, Wang LL, Han WD (2015) Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther 23:184–191. https://doi.org/10.1038/mt.2014.164
Budde L, Song JY, Kim Y, Blanchard S, Wagner J, Stein AS, Weng L, Del Real M, Hernandez R, Marcucci E, Shepphird JK, Wang X, Wood B, Marcucci G, Brown CE, Forman SJ (2017) Remissions of acute myeloid leukemia and blastic plasmacytoid dendritic cell neoplasm following treatment with CD123-specific CAR T cells: a first-in-human clinical trial. Blood 130:811–811. https://doi.org/10.1182/blood.V130.Suppl_1.811.811
Cummins KD, Frey N, Nelson AM, Schmidt A, Luger S, Isaacs RE, Lacey SF, Hexner E, Melenhorst JJ, June CH, Porter DL, Gill SI (2017) Treating relapsed/refractory (RR) AML with biodegradable anti-CD123 CAR modified T cells. Blood 130:1359–1359. https://doi.org/10.1182/blood.V130.Suppl_1.1359.1359
Müller L, Tunger A, Wobus M, von Bonin M, Towers R, Bornhäuser M, Dazzi F, Wehner R, Schmitz M (2021) Immunomodulatory properties of mesenchymal stromal cells: an update. Front Cell Dev Biol 9:1–9. https://doi.org/10.3389/fcell.2021.637725
Merimi M, Fahmi H, De Kock J, Beguin C, Burny A, Moll G, Poggi A, Najar M (2022) Mesenchymal stem/stromal cells as a therapeutic tool in cell-based therapy and regenerative medicine: an introduction expertise to the topical collection. Cells 11:11–13. https://doi.org/10.3390/cells11193158
Ramasamy R, Tong CK, Seow HF, Vidyadaran S, Dazzi F (2008) The immunosuppressive effects of human bone marrow-derived mesenchymal stem cells target T cell proliferation but not its effector function. Cell Immunol 251:131–136. https://doi.org/10.1016/j.cellimm.2008.04.009
Zanetti SR, Romecin PA, Vinyoles M, Juan M, Fuster JL, Cámos M, Querol S, Delgado M, Menendez P (2020) Bone marrow MSC from pediatric patients with B-ALL highly immunosuppress T-cell responses but do not compromise CD19-CAR T-cell activity. J Immunother cancer 8:1–13. https://doi.org/10.1136/jitc-2020-001419
Holthof LC, van der Schans JJ, Katsarou A, Poels R, Gelderloos AT, Drent E, van Hal-van Veen SE, Li F, Zweegman S, van de Donk NWCJ, Themeli M, Groen RWJ, Mutis T (2021) Bone marrow mesenchymal stromal cells can render multiple myeloma cells resistant to cytotoxic machinery of CAR T cells through inhibition of apoptosis. Clin Cancer Res. https://doi.org/10.1158/1078-0432.ccr-20-2188
Tunger A, Wehner R, von Bonin M, Kühn D, Heidenreich F, Matko S, Nauerth M, Rücker-Braun E, Dietz S, Link CS, Eugster A, Odendahl M, Busch DH, Tonn T, Bonifacio E, Germeroth L, Schetelig J, Bachmann MP, Bornhäuser M, Schmitz M (2017) Generation of high-avidity, WT1-reactive CD8+ cytotoxic T cell clones with anti-leukemic activity by streptamer technology. Leuk Lymphoma 58:1246–1249. https://doi.org/10.1080/10428194.2016.1233538
Heidenreich F, Rücker-Braun E, Walz JS, Eugster A, Kühn D, Dietz S, Nelde A, Tunger A, Wehner R, Link CS, Middeke JM, Stölzel F, Tonn T, Stevanovic S, Rammensee H-G, Bonifacio E, Bachmann M, Zeis M, Ehninger G, Bornhäuser M, Schetelig J, Schmitz M (2017) Mass spectrometry-based identification of a naturally presented receptor tyrosine kinase-like orphan receptor 1-derived epitope recognized by CD8 + cytotoxic T cells. Haematologica 102:e460–e464. https://doi.org/10.3324/haematol.2017.167312
Cartellieri M, Feldmann A, Koristka S, Arndt C, Loff S, Ehninger A, von Bonin M, Bejestani EP, Ehninger G, Bachmann MP (2016) Switching CAR T cells on and off: a novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J 6:e458. https://doi.org/10.1038/bcj.2016.61
Wermke M, Kraus S, Ehninger A, Bargou RC, Goebeler ME, Middeke JM, Kreissig C, von Bonin M, Koedam J, Pehl M, Bornhäuser M, Einsele H, Ehninger G, Cartellieri M (2021) Proof of concept for a rapidly switchable universal CAR-T platform with UniCAR-T-CD123 in relapsed/refractory AML. Blood 137:3145–3148. https://doi.org/10.1182/blood.2020009759
Koristka S, Cartellieri M, Feldmann A, Arndt C, Loff S, Michalk I, Aliperta R, von Bonin M, Bornhäuser M, Ehninger A, Ehninger G, Bachmann MP (2014) Flexible antigen-specific redirection of human regulatory T cells via a novel universal chimeric antigen receptor system. Blood 124:3494–3494. https://doi.org/10.1182/blood.v124.21.3494.3494
Feldmann A, Arndt C, Koristka S, Berndt N, Bergmann R, Bachmann MP (2019) Conventional CARs versus modular CARs. Cancer Immunol Immunother 68:1713–1719. https://doi.org/10.1007/s00262-019-02399-5
Beatty GL, Smith JS, Reshef R, Patel KP, Colligon TA, Vance BA, Frey NV, Johnson FB, Porter DL, Vonderheide RH (2009) Functional unresponsiveness and replicative senescence of myeloid leukemia antigen-specific CD8+ T cells after allogeneic stem cell transplantation. Clin Cancer Res 15:4944–4953. https://doi.org/10.1158/1078-0432.CCR-08-3332
Rutella S, Vadakekolathu J, Mazziotta F, Reeder S, Yau TO, Mukhopadhyay R, Altmann H, Kramer M, Knaus HA, Zeidner JF, Radojcic V, Arruda A, Minden MD, Tasian SK, Bornhäuser M, Gojo I, Luznik L (2021) Transcriptomic features of immune exhaustion and senescence predict outcomes and define checkpoint blockade-unresponsive microenvironments in acute myeloid leukemia. Blood 138:223–223. https://doi.org/10.1182/blood-2021-145854
Knaus HA, Berglund S, Hackl H, Blackford AL, Zeidner JF, Montiel-Esparza R, Mukhopadhyay R, Vanura K, Blazar BR, Karp JE, Luznik L, Gojo I (2018) Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. JCI insight 3:1–20. https://doi.org/10.1172/jci.insight.120974
Von Dalowski F, Kramer M, Wermke M, Wehner R, Röllig C, Alakel N, Stölzel F, Parmentier S, Sockel K, Krech M, Schmitz M, Platzbecker U, Schetelig J, Bornhäuser M, Von Bonin M (2016) Mesenchymal stromal cells for treatment of acute steroid-refractory graft versus host disease: clinical responses and long-term outcome. Stem Cells 34:357–366. https://doi.org/10.1002/stem.2224
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini FC, Krause DS, Deans RJ, Keating A, Prockop DJ, Horwitz EM (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy 8:315–317. https://doi.org/10.1080/14653240600855905
Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, Piha-Paul SA, Yearley J, Seiwert TY, Ribas A, McClanahan TK (2017) IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 127:2930–2940. https://doi.org/10.1172/JCI91190
Rutella S, Church SE, Vadakekolathu J, Viboch E, Sullivan AH, Hood T, Warren SE, Cesano A, La Motte-Mohs R, Muth J, Lelièvre H, Lowenberg B, DiPersio JF, Davidson-Moncada JK (2018) Adaptive immune gene signatures correlate with response to flotetuzumab, a CD123 × CD3 bispecific Dart® molecule, in patients with relapsed/refractory acute myeloid leukemia. Blood 132:444–444. https://doi.org/10.1182/blood-2018-99-111539
Vadakekolathu J, Minden MD, Hood T, Church SE, Reeder S, Altmann H, Sullivan AH, Viboch EJ, Patel T, Ibrahimova N, Warren SE, Arruda A, Liang Y, Smith TH, Foulds GA, Bailey MD, Gowen-MacDonald J, Muth J, Schmitz M, Cesano A, Pockley AG, Valk PJM, Löwenberg B, Bornhäuser M, Tasian SK, Rettig MP, Davidson-Moncada JK, DiPersio JF, Rutella S (2020) Immune landscapes predict chemotherapy resistance and immunotherapy response in acute myeloid leukemia. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aaz0463
Lamble AJ, Kosaka Y, Laderas T, Maffit A, Kaempf A, Brady LK, Wang W, Long N, Saultz JN, Mori M, Soong D, LeFave CV, Huang F, Adams H, Loriaux MM, Loriaux MM, Tognon CE, Lo P, Tyner JW, Tyner JW, Fan G, McWeeney SK, Druker BJ, Druker BJ, Druker BJ, Lind EF, Lind EF, Lind EF (2020) Reversible suppression of T cell function in the bone marrow microenvironment of acute myeloid leukemia. Proc Natl Acad Sci USA 117:14331–14341. https://doi.org/10.1073/pnas.1916206117
Karlsson H, Samarasinghe S, Ball LM, Sundberg B, Lankester AC, Dazzi F, Uzunel M, Rao K, Veys P, Le Blanc K, Ringdén O, Amrolia PJ (2008) Mesenchymal stem cells exert differential effects on alloantigen and virus-specific T-cell responses. Blood 112:532–541. https://doi.org/10.1182/blood-2007-10-119370
Thomas DA, Massagué J (2005) TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8:369–380. https://doi.org/10.1016/j.ccr.2005.10.012
Bhat P, Leggatt G, Waterhouse N, Frazer IH (2017) Interferon-γ derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis 8:e2836. https://doi.org/10.1038/cddis.2017.67
Liu X, Hoft DF, Peng G (2020) Senescent T cells within suppressive tumor microenvironments: emerging target for tumor immunotherapy. J Clin Invest 130:1073–1083. https://doi.org/10.1172/JCI133679
Tse WT, Pendleton JD, Beyer WM, Egalka MC, Guinan EC (2003) Suppression of allogeneic T-cell proliferation by human marrow stromal cells: implications in transplantation. Transplantation 75:389–397. https://doi.org/10.1097/01.TP.0000045055.63901.A9
Ye J, Ma C, Hsueh EC, Dou J, Mo W, Liu S, Han B, Huang Y, Zhang Y, Varvares MA, Hoft DF, Peng G (2014) TLR8 signaling enhances tumor immunity by preventing tumor-induced T-cell senescence. EMBO Mol Med 6:1294–1311. https://doi.org/10.15252/emmm.201403918
Saldanha-Araujo F, Ferreira FIS, Palma PV, Araujo AG, Queiroz RHC, Covas DT, Zago MA, Panepucci RA (2011) Mesenchymal stromal cells up-regulate CD39 and increase adenosine production to suppress activated T-lymphocytes. Stem Cell Res 7:66–74. https://doi.org/10.1016/j.scr.2011.04.001
Valitutti S, Dessing M, Lanzavecchia A (1993) Role of cAMP in regulating cytotoxic T lymphocyte adhesion and motility. Eur J Immunol 23:790–795
Ye J, Huang X, Hsueh EC, Zhang Q, Ma C, Zhang Y, Varvares MA, Hoft DF, Peng G (2012) Human regulatory T cells induce T-lymphocyte senescence. Blood 120:2021–2031. https://doi.org/10.1182/blood-2012-03-416040
Uy GL, Aldoss I, Foster MC, Sayre PH, Wieduwilt MJ, Advani AS, Godwin JE, Arellano ML, Sweet KL, Emadi A, Ravandi F, Erba HP, Byrne M, Michaelis L, Topp MS, Vey N, Ciceri F, Carrabba MG, Paolini S, Huls GA, Jongen-Lavrencic M, Wermke M, Chevallier P, Gyan E, Récher C, Stiff PJ, Pettit KM, Löwenberg B, Church SE, Anderson E, Vadakekolathu J, Santaguida M, Rettig MP, Muth J, Curtis T, Fehr E, Guo K, Zhao J, Bakkacha O, Jacobs K, Tran K, Kaminker P, Kostova M, Bonvini E, Walter RB, Davidson-Moncada JK, Rutella S, DiPersio JF (2021) Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 137:751–762. https://doi.org/10.1182/blood.2020007732
Harrell CR, Jovicic N, Djonov V, Arsenijevic N, Volarevic V (2019) Mesenchymal stem cell-derived exosomes and other extracellular vesicles as new remedies in the therapy of inflammatory diseases. Cells. https://doi.org/10.3390/cells8121605
Agha DM, Rouas R, Najar M, Bouhtit F, Naamane N, Fayyad-Kazan H, Bron D, Meuleman N, Lewalle P, Merimi M (2020) Identification of acute myeloid leukemia bone marrow circulating microRNAs. Int J Mol Sci 21:1–31. https://doi.org/10.3390/ijms21197065
Moussa Agha D, Rouas R, Najar M, Bouhtit F, Fayyad-Kazan H, Lagneaux L, Bron D, Meuleman N, Lewalle P, Merimi M (2020) Impact of bone marrow miR-21 expression on acute myeloid leukemia T lymphocyte fragility and dysfunction. Cells 9:1–18. https://doi.org/10.3390/cells9092053
Li G-Q, Fang Y-X, Liu Y, Meng F-R, Wu X, Zhang C-W, Zhang Y, Liu Y-Q, Liu D (2021) MicroRNA-21 from bone marrow mesenchymal stem cell-derived extracellular vesicles targets TET1 to suppress KLF4 and alleviate rheumatoid arthritis. Ther Adv Chronic Dis 12:20406223211007370. https://doi.org/10.1177/20406223211007369
Acknowledgements
We would like to thank Kristin Mӧbus, Katrin Müller, Claudia Richter, and Robert Kuhnert for excellent technical support.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the Mildred-Scheel Early Career Center (MSNZ) through the Mildred-Scheel Foundation and the Cluster4Future Consortium SaxoCell (funded by the Federal Ministry of Education and Research, BMBF). Grant number 70113306.
Author information
Authors and Affiliations
Contributions
RT designed the experiments, interpreted the data, and wrote the manuscript. RT, LT, MF, and A-LB performed the experiments. AT and MS provided the CTL clones, and AF and MB provided the CAR T cells. They, along with RW, FF, CA, RS, and FD, also provided critical analytical input. MVonB, MW, and MB conceptualized the study. All authors have provided their consent for publication of this manuscript.
Corresponding author
Ethics declarations
Conflict of interest
MB reports scientific advisory board memberships and speaker honoraria from Jazz Pharmaceuticals, MSD Sharp and Dome and Alexion.
Ethical approval and consent to participate
Primary cells were collected from donors and patients after informed consent and approval by the local review board (ethical approval EK307082018, EK98032010, and EK206082008).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Towers, R., Trombello, L., Fusenig, M. et al. Bone marrow-derived mesenchymal stromal cells obstruct AML-targeting CD8+ clonal effector and CAR T-cell function while promoting a senescence-associated phenotype. Cancer Immunol Immunother 73, 8 (2024). https://doi.org/10.1007/s00262-023-03594-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00262-023-03594-1