
Abstract
Following infusion of the anti-CD28 superagonist monoclonal antibody TGN1412, three of six previously healthy, young male recipients developed gastrointestinal irritability associated with increased expression of ‘gut-homing’ integrin β7 on peripheral blood αβT cells. This subset of patients with intestinal symptoms also displayed a striking and persistent expansion of putative Vδ2+ γδT cells in the circulation which declined over a 2-year period following drug infusion, concordant with subsiding gut symptoms. These data demonstrate that TGN1412-induced gastrointestinal symptoms were associated with dysregulation of the ‘gut-homing’ pool of blood αβ and γδT cells, induced directly by the antibody and/or arising from the subsequent cytokine storm.
Similar content being viewed by others


Introduction
In higher primates, the blood T cell pool contains diverse αβT cells and semi-invariant ‘unconventional’ T cells that recognize either microbial peptides or metabolites, respectively [1]. In both cases, antigen activation can stimulate these cells to upregulate the gut-homing integrin α4β7 and traffic to the intestine [2,3,4]. Gut microbes, and the metabolic activities these perform, vary between host species. Consequently, the compounds generated and their conditioning effects on peripheral blood T cell responses are likely to differ between mice and humans [5, 6]. The influence of these microbial products may also diverge between individual recipients of agonist/antagonist immunotherapies; gut bacteria from patients with melanoma who respond to immune checkpoint blockade are enriched for anabolic functions proposed to stimulate host immunity [7]. However, the extent to which peripheral blood T cell responses contribute to these modulatory effects in vivo remains unclear.
Some of the most common antigen-specific lymphocytes in human blood are gut-tropic T cells, specialized to detect various bacterial metabolites [1, 8]. However, the frequencies and phenotypes adopted by these cells can differ between individuals and age groups [9, 10], and their impact on immunotherapeutic outcomes in treated patients is not well understood. It is now well recognized that checkpoint inhibitors can be associated with immune-related adverse events (irAEs) affecting the gut, most notably symptoms of diarrhea and colitis following blockade of cytotoxic T-lymphocyte antigen-4 (CTLA-4) or programmed cell death 1 (PD-1) in patients with melanoma [7, 11]. However, it is still unclear to what extent gastrointestinal irAEs are caused by disruption of local mucosal immunoregulation, versus systemic drug effects on gut-homing lymphocytes [12].
In March 2006, six healthy volunteers suffered from cytokine release syndrome (CRS) during a phase 1 first-in-man clinical trial of the monoclonal antibody TGN1412 [13]. In pre-clinical studies, this anti-CD28 superagonist induced preferential lymphocytosis of regulatory T cells in the absence of systemic inflammation [14, 15]; the immunological basis for antibody-induced CRS and resultant lymphopenia in the human trial has remained unclear. In addition to acute symptoms of CRS from which all six patients recovered [13], three patients suffered from prolonged gastrointestinal irritability of unknown etiology, suggesting unexpected TGN1412 antibody and/or CRS effects on gastrointestinal immunity. We, therefore, undertook a detailed investigation of peripheral blood distribution and expression levels of integrin β7 aiming to understand the immunological basis for these symptoms. These analyses revealed that blood αβT cells from patients who suffered from gut irritability displayed significantly enhanced levels of β7 expression that were not observed in either asymptomatic patients or healthy controls. In addition, TGN1412-induced gut symptoms were associated with a striking expansion of circulating γδT cells (putative phosphoantigen metabolite-responsive Vγ9Vδ2 + lineage) that was still evident 2 years after drug infusion. Together, these data suggest that in three of six recipients, the TGN1412 antibody or subsequent cytokine storm caused sustained dysregulation of the gut-homing T cell pool, which gradually normalized over the 2-year period following antibody infusion, concordant with subsiding gastrointestinal symptoms.
Methods
Clinical trial
Details of the first 30 days of clinical follow-up of the serious adverse event (SAE) have been reported previously. The patients presented herein correspond with those previously identified as follows [13]: 1-B, 2-A, 3-F, 4-E, 5-C, and 6-D. The TGN1412 antibody was produced by TeGenero AG (Wϋrzberg, Germany), manufactured by Boehringer Ingelheim (Germany), and the clinical trial was conducted by the contract research organization PAREXEL International (Waltham, MA, USA) on leased premises at Northwick Park Hospital, London, UK. The authors of this report were not involved in either pre-clinical or clinical testing of TGN1412.
Patients and data sources
Patients were clinically followed, off trial, and assessed as a cohort following the SAE [13]. Based on clinical need and requirements for SAE follow-up, the lead clinician (NP) requested immunological monitoring, including analysis of peripheral blood T cell subsets alongside intracellular and serum cytokine levels. Monitoring commenced 10 days after infusion of TGN1412 and the patients were evaluated at 21 time points over the subsequent 2 years. All patient blood samples were anonymized and the scientists performing the immunological tests were not aware of patient symptoms, signs, or clinical laboratory data. Patients were assessed by the lead clinician at the same intervals wherein blood was procured for monitoring. Control blood samples from healthy male volunteers (n = 24) were obtained in parallel with the patient samples after written informed consent. The six volunteers who received TGN1412 were male and had a median age of 29.5 years (range 19–34) at the time of recruitment into the first-in-man trial. Healthy control volunteers were male and had a median age of 30 years (range 19–42). All patients were well during the 2-week period preceding the clinical trial and were without significant medical history. Patients B and C were lost to immunological follow-up after 15 and 22 months, respectively. Following development of gastrointestinal symptoms in three of the six patients, additional assessment of β7 integrin expression on peripheral blood T cells was introduced for all patients at four separate time points over the 2-year follow-up. All clinical information was withheld from the scientists who performed these analyses (NEM, AJS, CLP, ERM, NLG, HOA, SCK) until laboratory investigations were complete. All six patients consented to immunological monitoring and have given written informed consent to the publication of data presented in this report.
Immune monitoring
Specific leukocyte subset monitoring began on Day + 10 following TGN1412 infusion and was repeated every 3 or 4 days for the first 2 weeks, then weekly for 4 weeks, then every 4 weeks for 3 months, then every 6 weeks for the remainder of 8 months (time points 1–17). In year two of monitoring, patients were evaluated every 3 months (time points 18–21). In the first 6 months, whole blood was assessed for T cell subsets, numbers, phenotypes, and intracellular cytokine expression. After 6 months, the tests were rationalized to those that were most informative. Additional correlates of immune function included assessment of T-cell receptor Vβ repertoire (kit kindly donated by Beckman Coulter), and T cell homing markers for the gastrointestinal tract and skin based on expression of β7 integrin and cutaneous leukocyte antigen (CLA), respectively. The bulk of these data are presented elsewhere (Cancer Immunol Immunotherapy, in press) —this report focuses on the gut-homing subsets. These studies were conducted in a laboratory that operates under Good Laboratory Practice (GLP) principles, undertakes exploratory research and were performed using established laboratory protocols that were Minimal Information About T cell Assays (MIATA) compliant (Supplementary MIATA information). The assays and reagents employed were previously validated and tested for assay performance during the course of standard general investigative research.
Flow cytometry
Peripheral whole blood was obtained by venipuncture into sodium-heparin Vacutainer™ tubes (Becton–Dickinson) and then directly labeled with monoclonal antibody (mAb; Supplementary Table 1) for 15 min at room temperature. After mAb labeling, Optilyse C reagent (Immunotech, Marseilles) was used to lyse erythrocytes for 15 min before washing the cells twice in cold FACS buffer (2% FCS, 0.02% sodium azide, and 1 mM EDTA in PBS) for 5 min at 300G. Cell pellets were fixed in 0.4 mL paraformaldehyde (1%) and stored at 4 °C in the dark until acquired on a FACSCalibur flow cytometer using CellQuest software (Becton–Dickinson). All analyses were performed using WinList software (Verity Software House, Maine, USA). Absolute cell counts were determined using Flow-Count™ Fluorospheres (Beckman Coulter) added to the cells immediately prior to acquisition.
Viable cells were gated according to their characteristic light-scatter properties, and individual leukocyte subsets identified based on expression of subset-specific surface antigens. Major CD3+ T cell subsets were identified based on differential expression of CD8 (CD8+ T cells were CD3+/CD8+ and putative CD4+ T cells were CD3+/CD8−), since CD8 is less susceptible than CD4 to down-regulation during T-cell stimulation used in the intracellular cytokine determination protocol. In each subset, naïve and memory populations were enumerated by further double staining; naïve cells were CD45RA+/CD45RO− and memory cells were CD45RA−/CD45RO+. Expression of CD69 in CD4+ and CD8+ T cell subsets was used to identify activated T cells.
Intracellular cytokine staining
Peripheral whole blood cells were cultured in complete medium (Dutch-modified RPMI-1640 medium, 10% FCS, 20 mM l-glutamine, 100u/mL penicillin, 100 μg/mL streptomycin) with or without monensin (3 μM), PMA (10 ng/mL), and ionomycin (2 μM) for 4 h at 37 °C, 5%CO2. Cells were then surface-labeled with anti-CD3 and anti-CD8 mAb for 15 min at room temperature. Optilyse C reagent (Immunotech, Marseilles) was used for lysis of erythrocytes (0.5 mL per 100 μL aliquot of blood) and the samples were incubated for 15 min at room temperature. The remaining cells were twice washed in cold FACS buffer for 5 min at 300G, re-suspended in 100μL Leucoperm A (Serotec, Oxford), and then incubated for 15 min at room temperature. The partially fixed cells were next washed twice in FACS buffer and re-suspended in 100 μL Leucoperm B (Serotec, Oxford). For intracellular staining, the cells were labeled with 5 μL anti-cytokine mAb for 30 min on ice then washed twice in FACS buffer and fixed in 0.4 mL paraformaldehyde (1%) prior to storage in the dark at 4 °C (acquisition by flow cytometry was performed within 24 h).
Statistics
Statistical analyses were conducted using SigmaStat™3.5 or SigmaPlot™11.0 software (Systat Software UK Ltd, London). The TCR-Vβ repertoire data were compared using Kruskal–Wallis one-way analysis of variance on ranks. One-way analysis of variance with all-pairwise multiple comparison procedures (Holm–Sidak method) was used to compare β7 integrin expression over time between patient subsets and ten healthy controls. Differences in expression of γδ-TCR between patients and controls were evaluated by Student’s t test.
Results
Three of six patients (A, B and E) who received TGN1412 suffered from gastrointestinal symptoms (Common Terminology Criteria for Adverse Events grade 1–2 irAE), manifesting as loose and frequent bowel motions or diarrhea (primarily after consuming spicy foods; hereafter described as’gut irritability’), not present prior to drug exposure. These symptoms started within one month of TGN1412 infusion and subsequently decreased in intensity over the 2-year follow-up period. Symptoms persisted in patients B and E at 2 years. Patient B displayed the most pronounced gut symptoms and in the 1st year of follow-up underwent a full gastrointestinal work-up including duodenal biopsies (which were normal), and removal of a colonic polyp which exhibited non-specific inflammation.
Gut irritability in TGN1412 recipients was associated with increased β7 expression by circulating αβT cells
Integrin α4β7 binding to MAdCAM-1 facilitates leukocyte recruitment into intestinal tissues [16]. Accordingly, T cell expression of α4β7 is significantly modulated during active gut inflammation, and inhibition of the α4β7:MAdCAM-1 axis has been an effective therapeutic strategy in patients with inflammatory bowel disease (IBD) [17, 18]. Development of gut symptoms in three of the six TGN1412 recipients prompted us to assess T-cell expression of β7 integrin at four separate time points over the 2-year follow-up period. CD45RA+ (predominately naïve) T cells in the blood of both patients and healthy controls uniformly expressed an intermediate level of β7, whereas CD45RA− (antigen-experienced effector/memory) T cells included both β7+ and β7− subsets, representing putative gut-homing and non-intestinal populations, respectively (Fig. 1a). In healthy volunteers, memory T cells were evenly distributed between β7+ and β7− subsets (median ratio 0.98, interquartile range 0.80–1.19; Fig. 1b). Patients C, D, and F, who did not exhibit gastrointestinal symptoms, were indistinguishable from control subjects at all time points analyzed. In contrast, patients with gut irritability (A, B, and E) displayed increased β7+ memory T cells at 8.6 months (p < 0.001) and 10.2 months (p = 0.003) post-TGN1412 infusion (Fig. 1b). At 8.6 months, both CD45RA+ and CD45RA− T cells from patients A, B, and E also exhibited higher levels of β7 integrin expression per cell (mean fluorescence) compared with T cells from healthy controls, although this had normalized by 1-year post-infusion (Supplementary Fig. 1a and b). Sustained changes in both CD8+ and CD8− (presumed CD4+) memory T cells contributed to the elevated β7 expression detected in patients with gut irritability (Supplementary Fig. 1c and d). Fewer than 8% of β7+ memory T cells from either patients or controls expressed CD103/αE integrin, the alternative binding partner for β7 (Fig. 1c), consistent with reports that β7 primarily forms complexes with the α4 subunit on blood T cells [16], and confirming that the data presented here reflected changes in the patients’ α4β7+ compartment. Together, these findings indicated that gut irritability in three of six patients infused with TGN1412 was associated with a sustained increase in gut-homing potential among both naïve and memory αβT cells.

available at time points 10.2 and 12.1 months, nor for patient A at 12.1 months
Blood T cell expression of β7 integrin and CD103 following TGN1412-induced cytokine storm. CD3+ T cells were identified in whole blood and β7 expression on memory (CD45RA−) and naïve (CD45RA+) subsets was assessed. a Representative data for a healthy control and two patients are shown, one in whom β7+ cells were prominent in the memory T cell population (patient B) and one in whom β7+ memory T cells appeared normal (patient C). Staining with isotype-matched control antibodies was contained within the boxed region in the lower left of the plots. b Summary data for memory T cells showing the ratio of β7+:β7− cells assessed at four separate time points over a period of 7 months. In patients with gut irritability (A, B, and E), the ratio of β7+:β7− cells was significantly higher at 8.6 and 10.2 months than was observed in healthy controls. c Proportion of β7+ memory T cells expressing CD103 in the patients, assessed over four separate time points. Patient B, the most symptomatic, displayed the highest ratio of β7+:β7− memory T cells and lowest percentage of CD103+ cells, suggesting a selective expansion of α4β7+ ‘gut-homing’ memory T cells. Data for Patient D were not
TGN1412-induced gut irritability correlated with peripheral blood expansion of putative Vδ2+ γδT cells
In addition to the features outlined above, the blood of patients who developed gut irritability after TGN1412 infusion contained a distinct subset of CD3hi T cells that was not present in the circulation of either asymptomatic patients or healthy controls (Fig. 2a). These cells displayed a CD4−CD8− ‘double negative’ phenotype characteristic of unconventional lymphocytes (data not shown) and expressed uniformly high levels of CD45RO (Fig. 2b) and β7 integrin (Fig. 2e), but lacked CD103 (Fig. 2f) and did not express any of the common Vβ-TCR repertoire variants assayed at 8.6 and 12.1 months following TGN1412 infusion (Supplementary Fig. 2). These features strongly implicated an expansion of ‘unconventional’ Vγ9Vδ2+T cells (hereafter Vδ2+T cells) which express high levels of α4β7 in human blood [3, 19] and are rapidly recruited to mucosal tissues in higher primates in vivo [20, 21]. Further support for this lineage identity was later provided by the absence of αβ-TCR (Fig. 2g) and lack of markers for natural killer cells (CD56) or invariant natural killer T cells (antibody 6B11; data not shown), but high expression levels of γδ-TCR [22] (Fig. 2h) as well as NKG2D, and CD161 [23, 24] (data not shown).

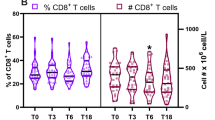
taken from 10 days post-TGN1412 infusion. CD3hiT cells in the patients with gut symptoms uniformly expressed β7 but not CD103/αE (e and f) indicating that these cells displayed the α4β7 heterodimer which mediates homing to the intestine (representative example from patient B). Staining with a specific mAb confirmed that the CD3hi cells were αβ-TCR− (g—R3) but strongly expressed γδ-TCR (h; black histogram—unfilled trace indicates isotype control) and mapped to the CD3hi population observed previously (a—R1; black histogram). While total T cell numbers in the patients were comparable at early time points (i) the CD3hi γδT cell subset was present only in patients A, B, and E, and persisted for up to 2 years post-TGN1412 infusion, decreasing slowly over time (j). In patient E, symptoms of gut irritability and diarrhea worsened at month 21, accompanied by an increase in CD3hi γδT cells at month 24 at which time symptoms had improved and he had more formed stool (although still not normal). Peak population size of CD3hi γδT cells (16% of the total T lymphocyte pool) was reached approximately 1.5 months post-TGN1412 exposure. Between 5 and 25% of this population displayed an activated (CD69+) phenotype that decreased steadily over 7 months (k). The median values and interquartile ranges of data obtained from healthy subjects (n = 24) are provided for reference (horizontal dashed lines)
Prolonged expansion of circulating γδT cells (putative Vδ2+) in patients with gut irritability after TGN1412-induced cytokine storm. Peripheral blood CD3+ T cells (a—R1) included a CD3hi subset (black histogram; identified by γδTCR-specific mAb used in h) which exhibited a CD4−CD8− ‘double negative’ phenotype characteristic of unconventional lymphocytes (data not shown). This discrete population uniformly expressed CD45RO and was clearly identifiable in the blood of patients A, B, and E (b—R2; representative example from patient A), but not in patient C, D, or F (c—R2; representative example from patient C), or in 24 healthy controls analysed in parallel (d—R2; representative example). Example analyses in (b), (c) and (d) are
Analysis at 15 months post-infusion confirmed that typical low numbers of γδT cells were present in peripheral blood from unaffected patients C, D, and F, as well as in six healthy controls analyzed in tandem, but these cells were not CD3hi (data not shown). In contrast, total γδT cells (including both CD3+ and CD3hi subsets) remained significantly increased in patients with gut irritability (A, B, E; 7.92–8.59%) compared with healthy controls (2.5–6.5%; p = 0.002) even at this late time point (more than 1 year post-infusion). In the blood of patients A, B, and E, the γδT cell pool reached peak numbers (16% of total T lymphocytes) approximately 1.5 months post-TGN1412 exposure, followed by a gradual decline coincident with improvement in gastrointestinal symptoms (Fig. 2i, j). Up to 25% of these γδT cells displayed an activated/CD69+ phenotype, and expression of this marker decreased steadily over the following 7 months (Fig. 2k). No other clinical or laboratory features correlated with γδT cell expansion as observed in the three symptomatic patients. Importantly, expansion of gut-homing lymphocytes in the patients with gut irritability was not restricted to γδT cells alone, because higher numbers of β7+T cells were still detected in these individuals (Fig. 1) when the γδT cell (CD3hi) population was excluded from this analysis (Supplementary Fig. 3).
The IFNγ-producing subset of blood Vδ2+T cells declines naturally with age and is lost more rapidly in men after the age of 30 [25], with both ethnic and environmental variables further impacting on the dynamics of this compartment [26]. To determine whether the expanded γδT cells detected in TGN1412 recipients remained functionally competent, and also to understand how these cells might be contributing to gastrointestinal irritability, we next assessed cytokine expression using a standard intracellular staining approach. The γδT cell population produced low levels of IL-10 in vitro in the absence of exogenous stimulation (Fig. 3), but did not appear to spontaneously produce either IFNγ or IL-4. However, γδT cell reactivation with phorbol myristate acetate and ionomycin resulted in substantial production of IFNγ across a wide range of time points analyzed, starting from 1 month post-infusion, when symptoms of gut irritability were first identified. Together, these data suggest that development of intestinal symptoms in patients infused with TGN1412 was associated with sustained expansion of circulating Vδ2+T cells with IFNγ-producing capacity, as well as increased gut-homing potential within the blood αβT cell pool.

The expanded γδT cell population spontaneously produced IL-10 and expressed IFNγ upon reactivation. To assess the functional potential of CD3hiβ7+γδT cells, whole blood cells were cultured with or without monensin and exogenous stimuli (PMA and ionomycin) for four hours prior to surface labelling and intracellular staining with anti-cytokine monoclonal antibodies for analysis by flow cytometry. CD3hiCD45RO+γδT cells spontaneously produced low levels of IL-10 in the absence of exogenous stimulation. By 1.5 months post-TGN1412 infusion, a substantial proportion of CD3hiγδT cells produced IFNγ upon reactivation with PMA and ionomycin. Dotted lines represent median and interquartile range of values obtained from conventional CD4+ αβT cells in all patients (no CD3hi cells were identifiable in healthy volunteers to serve as matched controls)
Discussion
This report provides unique evidence that TGN1412 antibody, or the subsequent CRS, dysregulated intestinal immunity in three of six drug recipients, outside of an identified infectious etiology; gut irritability was associated with sustained enhancement of mucosal trafficking in both conventional and unconventional T-cell subsets.
While the immunological response to TGN1412 infusion was surprisingly uniform in many respects [13], the long-term impact of the drug on mucosal immunity varied markedly between patients. Typical populations of β7+αβT cells [16] were present as expected in all trial patients, but enhanced β7 expression levels and the surprising expansion of γδT cells were unique to patients with symptoms of gut irritability. This variability of response may reflect patient-specific differences in homeostatic T-cell reconstitution after TGN1412-induced lymphopenia [13] and/or differential γδT cell responses to high levels of cytokines such as TNFα [27] following infusion of TGN1412. The peripheral blood location, kinetics, magnitude, and duration of these γδT-cell expansions, together with uniform expression of CD45RO [28] and high surface levels of β7, strongly implicate the Vγ9Vδ2+T cell lineage which responds to non-peptide ‘phosphoantigens’ (pAg) derived from microbes and stressed/transformed host cells [29, 30]. Vγ9Vδ2+ T cells undergo rapid polyclonal expansion in the first few weeks of human life, likely driven by pAg-producing bacteria within the gut microbiome; thereafter, the repertoire displays progressive selection of shared or ‘public’ pAg-reactive clones (defined by characteristic Vγ9JP and Vδ2 chains) [31]. With advancing age, the Vδ2+T cell compartment becomes increasingly oligoclonal, but different individuals may still display diverse or ‘private’ Vγ9Vδ2+ clonal expansions with distinct effector phenotypes [32], potentially including variable expression of gut-homing markers. It is, therefore, possible that the TGN1412 recipients with gut symptoms (A, B, and E) featured Vδ2+ clonotypes that were absent from the blood of those without (C, D and F). These cells may have exhibited different thresholds for pAg activation and intestinal recruitment in the context of cytokine storm. Rapid expansion of Vγ9JP+ γδT cells has also been observed during immune reconstitution of patients undergoing allogeneic hematopoietic stem cell transplantation, but the clonotypes generated following donor cell infusion were substantially different from those present either in the donor or the recipient pre-transplant [33]. Indeed, while Vδ2+T cells constitute only a minor fraction of total circulating lymphocytes in healthy individuals, their number and activation state in peripheral blood and body tissues has been correlated with therapeutic and clinical outcomes [34,35,36]. Work from our own laboratory has also demonstrated that activated blood Vδ2+T cells rapidly upregulate β7 and can populate human gut lamina propria where they induce substantial mucosal production of IFNγ [3]. In patients with Crohn’s disease, gut-homing potential and pro-inflammatory properties of Vδ2+T cells are enhanced [36], suggesting that they play a key role in human gut immunity and inflammation. In future, the microbiome composition of pAg-producing bacteria and blood γδT cell repertoire may also prove to be important determinants of clinical outcome in patients undergoing immunotherapy.
The Vδ2+ lineage is absent in rodents and does not recognize antigen in the context of MHC [37]. Instead, Vδ2+T cells respond to butyrophilin (BTN) proteins, considered as part of the B7 family of costimulatory receptors [37], with critical roles recently identified for both BTN3A1 and BTN2A1 [38]. Vδ2+T cells lack alloreactivity while displaying potent anti-tumor and anti-microbial functions, such that reconstitution of this lineage after chemotherapy-induced lymphopenia may reduce infection rates in patients receiving hematopoietic stem cell transplantation, without increased incidence of graft versus host-disease [33, 39]. Expansion of blood Vδ2+T cells has been observed in CRS, most notably in healthcare workers exposed to SARS-CoV-1 in the 2003 outbreak; these individuals displayed strikingly similar features including relatively stable αβT cell numbers and TCR-Vβ repertoire, whereas marked expansions of Vδ2+T cells with IFNγ-producing capacity were still evident 3 months after disease onset [40]. These findings resemble data from nonhuman primate models in which pAg injection stimulates blood Vδ2+T cell expansion in vivo [41], leading to accumulation of an IFNγ-producing subset both in lungs and intestinal mucosa [20], accompanied by robust Th1 immune protection against a range of different pathogens.
Vδ2+T cells undergo expansion in response to a variety of microbial infections and can dominate the blood lymphocyte pool for extended periods [10]. It is also now widely recognized that Vδ2+T cells display tissue-tropic phenotypes consistent with trafficking to barrier sites where pAg-producing microbes and tumors frequently originate [29, 30]. In particular, Vδ2+T cells are associated with effective host immunity to pAg-producing mycobacteria and robust responses to bacillus Calmette–Guérin (BCG) vaccination [42, 43], which induces population expansion and upregulation of CD69 and IFNγ expression in vitro. Notably, these responses are enhanced in BCG responders compared with non-sensitized controls [44, 45], and the pool of Vδ2+T cells generated lacks lymph node homing receptors while displaying homogenous expression of CD28 [45]. Therefore, TGN1412 may have directly stimulated Vδ2+T cells in the trial patients, and prior microbial exposures such as BCG may have influenced subsequent responses to mucosal pathogens and/or cytokine storm (as also postulated in the context of COVID-19 [46]). Indeed, while previous studies have primarily linked Vδ2+T cell expansion with host protection against bacterial pathogens, these lymphocytes can also lyse stressed host cells infected with viruses including influenza [47] and SARS-CoV-1 [40]. Together, these data suggest that monitoring of gut-homing αβ and γδT cell populations is likely to shed important new light on the initiation, propagation, monitoring, and resolution of mucosal symptoms in human subjects with irAEs or suffering CRS as a result of immunotherapy or severe infections such as COVID-19.
Code availability
Not applicable.
References
Godfrey DI, Uldrich AP, McCluskey J, Rossjohn J, Moody DB (2015) The burgeoning family of unconventional T cells. Nat Immunol 16:1114–1123. https://doi.org/10.1038/ni.3298
Berlin C, Berg EL, Briskin MJ, Andrew DP, Kilshaw PJ, Holzmann B, Weissman IL, Hamann A, Butcher EC (1993) Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell 74:185–195
McCarthy NE, Bashir Z, Vossenkamper A et al (2013) Proinflammatory Vdelta2+ T Cells populate the human intestinal mucosa and enhance IFN-gamma production by colonic alphabeta T Cells. J Immunol 191:2752–2763. https://doi.org/10.4049/jimmunol.1202959
Juno JA, Wragg KM, Amarasena T et al (2019) MAIT Cells Upregulate alpha4beta7 in response to acute simian immunodeficiency virus/Simian HIV infection but are resistant to peripheral depletion in pigtail macaques. J Immunol 202:2105–2120. https://doi.org/10.4049/jimmunol.1801405
Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, Siuzdak G (2009) Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U S A 106:3698–3703. https://doi.org/10.1073/pnas.0812874106
Schroeder BO, Backhed F (2016) Signals from the gut microbiota to distant organs in physiology and disease. Nat Med 22:1079–1089. https://doi.org/10.1038/nm.4185
Gopalakrishnan V, Spencer CN, Nezi L et al (2018) Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359:97–103. https://doi.org/10.1126/science.aan4236
McCarthy NE, Eberl M (2018) Human gammadelta T-Cell control of mucosal immunity and inflammation. Front Immunol 9:985. https://doi.org/10.3389/fimmu.2018.00985
Ryan PL, Sumaria N, Holland CJ, Bradford CM, Izotova N, Grandjean CL, Jawad AS, Bergmeier LA, Pennington DJ (2016) Heterogeneous yet stable Vdelta2(+) T-cell profiles define distinct cytotoxic effector potentials in healthy human individuals. Proc Natl Acad Sci U S A 113:14378–14383. https://doi.org/10.1073/pnas.1611098113
Morita CT, Jin C, Sarikonda G, Wang H (2007) Nonpeptide antigens, presentation mechanisms, and immunological memory of human Vgamma2Vdelta2 T cells: discriminating friend from foe through the recognition of prenyl pyrophosphate antigens. Immunol Rev 215:59–76. https://doi.org/10.1111/j.1600-065X.2006.00479.x
Berman D, Parker SM, Siegel J, Chasalow SD, Weber J, Galbraith S, Targan SR, Wang HL (2010) Blockade of cytotoxic T-lymphocyte antigen-4 by ipilimumab results in dysregulation of gastrointestinal immunity in patients with advanced melanoma. Cancer Immun 10:11
Abu-Sbeih H, Ali FS, Alsaadi D, Jennings J, Luo W, Gong Z, Richards DM, Charabaty A, Wang Y (2018) Outcomes of vedolizumab therapy in patients with immune checkpoint inhibitor-induced colitis: a multi-center study. J Immunother Cancer 6:142. https://doi.org/10.1186/s40425-018-0461-4
Suntharalingam G, Perry M, Ward S, Brett S, Castello-Cortes A, Brunner M, Panoskaltsis N (2006) Cytokine Storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. New En J Med 355:1018–1028
Beyersdorf N, Gaupp S, Balbach K et al (2005) Selective targeting of regulatory T cells with CD28 superagonists allows effective therapy of experimental autoimmune encephalomyelitis. J Exp Med 202:445–455
TGN1412 Investigator’s Brochure. TeGenero Immunotherapeutics. https://www.mhra.gov.uk/home/idcplg?IdcService=GET_FILE&dDocName=CON2023518&RevisionSelectionMethod=LatestReleased. (Accessed 5 May 2006)
Rott LS, Briskin MJ, Andrew DP, Berg EL, Butcher EC (1996) A fundamental subdivision of circulating lymphocytes defined by adhesion to mucosal addressin cell adhesion molecule-1. Comparison with vascular cell adhesion molecule-1 and correlation with b7 integrins and memory differentiation. J Immunol 156:3727–3736
Sandborn WJ, Feagan BG, Rutgeerts P et al (2013) Vedolizumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med 369:711–721. https://doi.org/10.1056/NEJMoa1215739
Feagan BG, Rutgeerts P, Sands BE et al (2013) Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med 369:699–710. https://doi.org/10.1056/NEJMoa1215734
Brandes M, Willimann K, Lang AB, Nam KH, Jin C, Brenner MB, Morita CT, Moser B (2003) Flexible migration program regulates gamma delta T-cell involvement in humoral immunity. Blood 102:3693–3701. https://doi.org/10.1182/blood-2003-04-1016
Ali Z, Shao L, Halliday L, Reichenberg A, Hintz M, Jomaa H, Chen ZW (2007) Prolonged (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate-driven antimicrobial and cytotoxic responses of pulmonary and systemic Vgamma2Vdelta2 T cells in macaques. J Immunol 179:8287–8296
Ryan-Payseur B, Frencher J, Shen L, Chen CY, Huang D, Chen ZW (2012) Multieffector-functional immune responses of HMBPP-specific Vgamma2Vdelta2 T cells in nonhuman primates inoculated with listeria monocytogenes {Delta}actA prfA*. J Immunol 189:1285–1293. https://doi.org/10.4049/jimmunol.1200641
Thibault G, Bardos P (1995) Compared TCR and CD3e expression of ab and gd T cells. Evidence for the association of two TCR heterodimers with three CD3e chains in the TCR/CD3 complex. J Immunol 154:3814–3820
Provine NM, Binder B, FitzPatrick MEB et al (2018) Unique and common features of innate-like human Vδ2+ γδT Cells and mucosal-associated invariant T Cells. Front Immunol. https://doi.org/10.3389/fimmu.2018.00756
Kong Y, Cao W, Xi X, Ma C, Cui L, He W (2009) The NKG2D ligand ULBP4 binds to TCRgamma9/delta2 and induces cytotoxicity to tumor cells through both TCRgammadelta and NKG2D. Blood 114:310–317. https://doi.org/10.1182/blood-2008-12-196287
Caccamo N, Dieli F, Wesch D, Jomaa H, Eberl M (2006) Sex-specific phenotypical and functional differences in peripheral human Vgamma9/Vdelta2 T cells. J Leukoc Biol 79:663–666. https://doi.org/10.1189/jlb.1105640
Esin S, Shigematsu M, Nagai S, Eklund A, Wigzell H, Grunewald J (1996) Different percentages of peripheral blood gamma delta + T cells in healthy individuals from different areas of the world. Scand J Immunol 43:593–596
Li H, Luo K, Pauza CD (2008) TNF-a is a positive reulatory factor for human Vg2Vd2 T cells. J Immunol 181:7131–7137
Miyawaki T, Kasahara Y, Taga K, Yachie A, Taniguchi N (1990) Differential expression of CD45RO (UCHL1) and its functional relevance in two subpopulations of circulating TCR-gamma/delta+ lymphocytes. J Exp Med 171:1833–1838
Eberl M, Hintz M, Reichenberg A, Kollas AK, Wiesner J, Jomaa H (2003) Microbial isoprenoid biosynthesis and human gammadelta T cell activation. FEBS Lett 544:4–10 (S0014579303004836 [pii])
Scheper W, Sebestyen Z, Kuball J (2014) Cancer immunotherapy using gammadeltaT cells: dealing with diversity. Front Immunol 5:601. https://doi.org/10.3389/fimmu.2014.00601
Papadopoulou M, Dimova T, Shey M et al (2020) Fetal public Vgamma9Vdelta2 T cells expand and gain potent cytotoxic functions early after birth. Proc Natl Acad Sci U S A 117:18638–18648. https://doi.org/10.1073/pnas.1922595117
Willcox CR, Davey MS, Willcox BE (2018) Development and selection of the human Vgamma9Vdelta2(+) T-cell repertoire. Front Immunol 9:1501. https://doi.org/10.3389/fimmu.2018.01501
Ravens S, Schultze-Florey C, Raha S et al (2017) Human gammadelta T cells are quickly reconstituted after stem-cell transplantation and show adaptive clonal expansion in response to viral infection. Nat Immunol 18:393–401. https://doi.org/10.1038/ni.3686
Davey MS, Lin CY, Roberts GW et al (2011) Human neutrophil clearance of bacterial pathogens triggers anti-microbial gammadelta T cell responses in early infection. PLoS Pathog 7:e1002040. https://doi.org/10.1371/journal.ppat.1002040
Laggner U, Di Meglio P, Perera GK et al (2011) Identification of a novel proinflammatory human skin-homing Vgamma9Vdelta2 T cell subset with a potential role in psoriasis. J Immunol 187:2783–2793. https://doi.org/10.4049/jimmunol.1100804
McCarthy NE, Hedin CR, Sanders TJ et al (2015) Azathioprine therapy selectively ablates human Vdelta2(+) T cells in Crohn’s disease. J Clin Invest 125:3215–3225. https://doi.org/10.1172/JCI80840
Rhodes DA, Reith W, Trowsdale J (2016) Regulation of immunity by butyrophilins. Annu Rev Immunol 34:151–172. https://doi.org/10.1146/annurev-immunol-041015-055435
Eberl M (2020) Antigen recognition by human gammadelta T cells: one step closer to knowing. Cell Biol Immunol. https://doi.org/10.1111/imcb.12334
Perko R, Kang G, Sunkara A, Leung W, Thomas PG, Dallas MH (2015) Gamma delta T cell reconstitution is associated with fewer infections and improved event-free survival after hematopoietic stem cell transplantation for pediatric leukemia. Biol Blood Marrow Trans 21:130–136. https://doi.org/10.1016/j.bbmt.2014.09.027
Poccia F, Agrati C, Castilletti C et al (2006) Anti-severe acute respiratory syndrome coronavirus immune responses: the role played by V gamma 9V delta 2 T cells. J Infect Dis 193:1244–1249. https://doi.org/10.1086/502975
Sicard H, Ingoure S, Luciani B, Serraz C, Fournie JJ, Bonneville M, Tiollier J, Romagne F (2005) In vivo immunomanipulation of V gamma 9V delta 2 T cells with a synthetic phosphoantigen in a preclinical nonhuman primate model. J Immunol 175:5471–5480
Shen L, Frencher J, Huang D et al (2019) Immunization of Vgamma2Vdelta2 T cells programs sustained effector memory responses that control tuberculosis in nonhuman primates. Proc Natl Acad Sci USA 116:6371–6378. https://doi.org/10.1073/pnas.1811380116
Dantzler KW, de la Parte L, Jagannathan P (2019) Emerging role of gammadelta T cells in vaccine-mediated protection from infectious diseases. Clin Trans Immunol 8:e1072. https://doi.org/10.1002/cti2.1072
Hoft DF, Brown RM, Roodman ST (1998) Bacille Calmette-Guerin vaccination enhances human gamma delta T cell responsiveness to mycobacteria suggestive of a memory-like phenotype. J Immunol 161:1045–1054
Martino A, Casetti R, Sacchi A, Poccia F (2007) Central memory Vgamma9Vdelta2 T lymphocytes primed and expanded by bacillus Calmette-Guerin-infected dendritic cells kill mycobacterial-infected monocytes. J Immunol 179:3057–3064
Netea MG, Giamarellos-Bourboulis EJ, Domínguez-Andrés J, Curtis N, van Crevel R, van de Veerdonk FL, Bonten M (2020) Trained immunity: a tool for reducing susceptibility and severity of SARS-CoV-2 infection. Cell. https://doi.org/10.1016/j.cell.2020.04.042
Li H, Xiang Z, Feng T et al (2013) Human Vgamma9Vdelta2-T cells efficiently kill influenza virus-infected lung alveolar epithelial cells. Cell Mol Immunol 10:159–164. https://doi.org/10.1038/cmi.2012.70
Acknowledgements
Financial support for some of the work was provided by The North West London Hospitals NHS Trust incorporating Northwick Park Hospital, Cancer Research UK, and The Northwick Park Hospital Leukaemia Research Trust Fund. We are also grateful to Sarah Clarson of Beckman Coulter for providing the TCR-Vβ repertoire kit used in these studies. Above all, we thank the six patients who have given consent for presentation of their personal data.
Funding
The North West London Hospitals NHS Trust; Cancer Research UK; The Northwick Park Hospital Leukemia Research Trust Fund.
Author information
Authors and Affiliations
Contributions
NEM and AJS were involved in the planning and execution of all experiments, interpretation of data, and in preparation of the manuscript. CLP, ERM, NLG, and HOA contributed to a number of experiments. NP had overall responsibility for the patients and clinical follow-up, and SCK and NP supervised the project, interpreted data, and prepared the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
None of the authors declare a financial conflict of interest. NP, SCK, CLP, HOA, ERM, and NG declare no conflicts of interest. NEM is supported by a Career Development Award from The Medical Research Council (Grant Ref: MR/R008302/1) and is in receipt of a project grant from Bart’s and The London Charity (MGU0465). He has also received consultancy fees and funding for research from ImCheck Therapeutics SAS. AJS research is supported by grants from Gilead Sciences, AbbVie, The Medical College of St Bartholomew’s Hospital Trust, Bowel and Cancer Research, and Bart’s Charity. SCK, NEM, and AJS have done contract work for Parexel pre-dating the work described in this report. At the time of this work and report, Parexel Clinical Trials Unit had a short-term contract with the Antigen Presentation Research Group (APRG) to use a Class II cabinet within the laboratory. The APRG has also been contracted to perform immunological studies by a pharmaceutical company, the tissue specimens for which were supplied on behalf of that company via Parexel which is located adjacent to the APRG department. There is no conflict of interest involved.
Ethics approval
Ethics approval had been obtained for the TGN1412 trial (by the investigators—none of the authors of this report were involved in the clinical trial). At the time of the trial-related serious adverse event, clinical and immune monitoring ensued as a matter of standard clinical care; no studies were done outside what was required for clinical care of the patients. Discussions between the Ethics Committee, MHRA and Expert Scientific Group set-up by the Minister of Health (UK) at the time to investigate the trial outcome unanimously concluded that the monitoring (as outlined in this report) should continue for standard of care, and that specific ethics approval was not required due to the extraordinary circumstances.
Consent to participate
Patients consented to clinical follow-up and immune monitoring. None of the authors of this work were involved with the conduct of the clinical trial or any of the pre-clinical testing of TGN1412. The patient cohort had consented to the TGN1412 first-in-man clinical trial that resulted in the cytokine storm serious adverse event. At the time of the start of sample collection for the current report, the patients had been removed from the trial and were being treated based on clinical need, rather than trial protocol.
Consent for publication
Patients have provided written informed consent to the publication of the clinical follow-up and immune monitoring data.
Availability of data and material
As this is a clinical cohort follow-up, and not data provided on a clinical trial, the data are unavailable due to personal privacy protections.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The work was done at the Antigen Presentation Research Group and the Department of Hematology, Imperial College London, Northwick Park and St. Mark’s Campus, London UK.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
McCarthy, N.E., Stagg, A.J., Price, C.L. et al. Patients with gastrointestinal irritability after TGN1412-induced cytokine storm displayed selective expansion of gut-homing αβ and γδT cells. Cancer Immunol Immunother 70, 1143–1153 (2021). https://doi.org/10.1007/s00262-020-02723-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-020-02723-4