
Abstract
Staphylococcus aureus is one of the most relevant mastitis pathogens in dairy cattle, and the acquisition of antimicrobial resistance genes presents a significant health issue in both veterinary and human fields. Among the different strategies to tackle S. aureus infection in livestock, bacteriophages have been thoroughly investigated in the last decades; however, few specimens of the so-called jumbo phages capable of infecting S. aureus have been described. Herein, we report the biological, genomic, and structural proteomic features of the jumbo phage vB_SauM-UFV_DC4 (DC4). DC4 exhibited a remarkable killing activity against S. aureus isolated from the veterinary environment and stability at alkaline conditions (pH 4 to 12). The complete genome of DC4 is 263,185 bp (GC content: 25%), encodes 263 predicted CDSs (80% without an assigned function), 1 tRNA (Phe-tRNA), multisubunit RNA polymerase, and an RNA-dependent DNA polymerase. Moreover, comparative analysis revealed that DC4 can be considered a new viral species belonging to a new genus DC4 and showed a similar set of lytic proteins and depolymerase activity with closely related jumbo phages. The characterization of a new S. aureus jumbo phage increases our understanding of the diversity of this group and provides insights into the biotechnological potential of these viruses.
Key points
• vB_SauM-UFV_DC4 is a new viral species belonging to a new genus within the class Caudoviricetes.
• vB_SauM-UFV_DC4 carries a set of RNA polymerase subunits and an RNA-directed DNA polymerase.
• vB_SauM-UFV_DC4 and closely related jumbo phages showed a similar set of lytic proteins.
Similar content being viewed by others

Introduction
Staphylococcus aureus is a commensal microorganism inhabiting the skin, nares, and mucosae of livestock and humans. This bacterium can occasionally subvert the immune system and cause acute and chronic infections, ranging from skin wounds, pneumonia, and endocarditis to bloodstream invasion followed by sepsis (Kümmel et al. 2016; Sakr et al. 2018). Along with Enterococcus faecium, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp., S. aureus is part of the ESKAPE group, which includes opportunistic pathogens most commonly responsible for causing nosocomial infections (Maciejewska et al. 2018; Ma et al. 2020).
Besides its clinical relevance, S. aureus is also of major concern in agriculture, and it stands as one of the main causes of bovine mastitis in dairy cows, a multifactorial inflammatory disease where the animal, environment, udder microbiome, and pathogen-associated factors are key components of this complex disease (Haag et al. 2019; da Silva et al. 2020; Campos et al. 2022; Winther et al. 2022). S. aureus can encode a vast arsenal of virulence factors that are commonly correlated with the development of severe infections (Gill et al. 2011). Its virulence is based on the production of adhesion proteins and toxins, which avoid the capture by phagocytes and other immune system cells, as well as its common biofilm capability (Chung and Toh 2014; Kaźmierczak et al. 2014; Sakr et al. 2018).
The global burden of multidrug-resistant strains of S. aureus, such as methicillin-resistant S. aureus (MRSA), has been considered a strong public health concern and a priority pathogen by the World Health Organization (Sulis et al. 2022). Hence, the search for an effective and widely accessible new generation of antimicrobial compounds capable of controlling the growth of such pathogenic bacteria represents an urgent demand and deserves global attention (Bhandari and Suresh 2022).
Bacteriophages, also known as phages, are viruses that infect bacteria. Despite being considered the most abundant entities on Earth (about 1031 particles around the biosphere), there is still much to discover about phage biology (Hendrix 2002). “Jumbo phage” is a relatively recent classification of phages that possess exceptionally large genomes (greater than 200 kb in length) with unusual properties, such as a non-modular genome structure, presence of several genes that encode complex proteins (e.g., different types of DNA polymerases, multi-subunit RNA polymerases, RNA ligase and enzymes for NAD+ synthesis), and an even greater number of genes with unknown function (Yuan and Gao 2017). Currently, only six viruses classified as Staphylococcus jumbo bacteriophages have been meticulously characterized (Uchiyama et al. 2014; Korn et al. 2021; Lee et al. 2021; Zhang et al. 2022).
Jumbo phages and their unique genomic traits make them more independent of the host machine than the other phages and with still uncovered biological features of biotechnological interest (Iyer et al. 2021; Korn et al. 2021; Nazir et al. 2021). For instance, enzymes such as endolysins, virion-associated lysins, and polysaccharide depolymerases—phage-encoded proteins that act cleaving peptidoglycan bonds, or, in the case of depolymerases, the biofilm matrix—have been considered promising alternatives to tackle antimicrobial resistance, along with phage preparations (Fischetti 2018; Maciejewska et al. 2018; Melo et al. 2018; Akturk et al. 2019). Herein, the aim of this work is (i) to biologically characterize the jumbo phage vB_SauM-UFV_DC4 and (ii) to provide a genomic comparison with other jumbo phages that infect S. aureus to identify genomic traits of interest, such as lytic enzymes.
Materials and methods
Bacterial strains and culture conditions
In total, 26 isolates of Staphylococcus aureus obtained from different ecological niches (human hospital, n = 4; veterinary hospital, n = 14; meat products, n = 4; environmental, n = 4) were used in this study (Table S1). All strains were routinely cultivated on BHI broth (Kasvi, Curitiba, Brazil) with moderate shaking (100 rpm, Tecnal TE-420, Brazil) at 37 °C. In all experiments with phages, the temperature was set at 30 °C. The strain S. aureus 3059 was deposited in the Collection of Microorganisms, DNA and Cells of Universidade Federal de Minas Gerais (UFMG) belonging to the World Data Centre for Microorganisms under the accession number UFMG-CM-B275.
vB_SauM-UFV_DC4 isolation and physiological features
The virus DC4 was isolated as described by da Silva Duarte et al. (2020). In summary, samples taken from wastewater of a dairy farm located at the Departamento de Zootecnia, Universidade Federal de Viçosa (DZO/UFV) at Viçosa, Minas Gerais, Brazil, were used for viral isolation. Samples were centrifuged and diluted (1:4) in SM buffer. The viral suspension was double-filtered through pore-size PES membranes (0.45 and 0.22 µm) and added to an early log-phase culture of S. aureus 3059 grown in BHI broth. The mixture was incubated at 30 °C for phage attachment and then plated using the standard soft agar overlay technique. The bacteriophage phage vB_SauM-UFV_DC4 is available upon request and can be obtained by contacting the corresponding author.
One-step growth curve
The one-step growth curve protocol was performed as described by Kropinski (2018), with some adaptations. Briefly, the virus DC4 was added to a bacterial culture (S. aureus 3059) at the beginning of the exponential growth phase (OD600nm 0.2; 1.6 × 108 CFU/mL), at an MOI of 0.00001. After 10 min of incubation at 30 °C with moderate agitation (100 rpm) aiming for phage adsorption, the moisture was centrifuged at 10,000 g for 10 min at room temperature (Eppendorf 5804 Benchtop Centrifuge), the supernatant discarded, and the pellet was resuspended in 30 mL of fresh and pre-warmed BHI medium. Afterward, the mixture was incubated at 30 °C, setting-up an agitation of 100 rpm, for 5 h. Viral titration was carried out through the double-layer agar assay, where 100 µL of the sample was collected at different time points with an interval of 20 min and immediately plated. The latent period (delay between phage adsorption and phage particle release) was calculated by measuring the time interval between the phage infection and the beginning of the phage titer increase. The burst size (number of phages released after each infection round) was calculated by dividing the number of phages at the first peak of the one-step growth curve graphic, by the titer of phages at time zero of the experiment. Data obtained over three independent biological replicates, with three technical replicates for each, were used for graph construction and latent period/burst size determination.
Host range determination and efficiency of plating (EOP)
In total, 26 isolates of S. aureus were used to evaluate the host range of the DC4 phage. Thus, 100 µL of 1 × 108 PFU/mL phage suspension was used to perform ten-fold serial dilutions that were then plated with the different hosts, using the double-agar layer assay. The BHI top agar was made using 0.35% of agar, and the plates were incubated at 30 °C, overnight. After determining the phage titer in the different hosts, the EOP was calculated using the titer obtained in the strain S. aureus 3059 (host of isolation) as reference. The average EOP value for the combination DC4-bacterium was classified according to the criteria established by Mirzaei and Nilsson (2015). Data obtained over three independent biological replicates, with three technical replicates for each, were used for EOP calculation.
Phage stability at different temperatures and pH
The stability protocols were performed as described by Jurczak-Kurek et al. (2016). In brief, to evaluate the stability of viral particles at different thermal conditions, 100 µL of 1 × 108 PFU/mL DC4 phage suspension was diluted in 900 µL of BHI medium. The microcentrifuge tubes were kept, in triplicate, at − 20 °C for 12 h, 40 °C and 62 °C for 2 h, and 95 °C for 5 min. Tubes kept at room temperature for 2 h were used as control. To evaluate the stability of viral particles in different pH values, 100 µL of the DC4 phage suspension was diluted in 900 µL of BHI medium with different pH values (2, 4, 10, and 12). Phages diluted in BHI medium with pH 7 were used as control. The tubes were kept at room temperature for 2 h, in triplicate. After the respective treatments, the quantification of viable viral particles was performed using the double-agar layer assay. The phage titer after the treatments was compared to the initial titer.
Statistical analysis
To evaluate the significance of the DC4 titers after the stability treatments, a one-way ANOVA and Dunnet’s post-hoc tests were performed (p-value < 0.05).
vB_SauM-UFV_DC4 DNA extraction and sequencing
For viral DNA extraction, the protocol described by Amend et al. (2023) was followed and adapted with the necessary modifications. Briefly, a high viral titer (~ 1010 PFU/mL) was obtained after phage propagation via plate lysate and viral concentration from the bacterial lysate by using Vivaspin® 20 centrifugal concentrators with a molecular size cut-off of 100 kDa (GE Healthcare Life Sciences, UK). To the viral suspension (1 mL), 0.0125 M MgCl2 was added and mixed gently. Afterward, the lysate-MgCl2 was treated with 0.8 µL of DNase I (2000 U/mL) (Sigma-Aldrich, USA), and incubated at 37 °C for 30 min. To this mixture, 40 µL of 0.5 M EDTA, 5 µL of Proteinase K (10 mg/mL), and 50 µL of 10% SDS were added, vigorously vortexed, and incubated at 55 °C for 60 min. After this period, 500 µL of the mixture was transferred to 1.5-mL microcentrifuge tubes previously filled with the same volume of phenol:chloroform:isoamyl-alcohol (PCI) (25:24:1) and centrifuged (5 min at room temperature at 13,000 rpm, Eppendorf 5415C Centrifuge). The top aqueous phase layer was collected and transferred to a new 1.5-mL microcentrifuge tube with 500 µL of chloroform and a new centrifugation was conducted (5 min at room temperature at 13,000 rpm). To the top aqueous phase obtained from the previous step, DNA was precipitated by adding 50 µL of 3 M sodium acetate solution and 1 mL of 95% (v/v) ethanol. The sample was incubated for 5 min on ice and centrifuged at room temperature for 10 min at 13,000 rpm. The DNA pellet was washed with 500 µL of 70% (v/v) ethanol and finally in SpeedVac prior to resuspension in 50 µl of DNase/RNase-free distilled water. The purity and quality of the DNA were checked by electrophoresis on 0.8% (p/v) agarose gel, and DNA concentration was estimated by measuring the absorbance at 260 nm by using NanoDrop 2000 (Thermo Fisher, USA).
Viral DNA was sent to the Molecular Research DNA (Shallowater, TX, USA; mrdna.com). Sequencing was conducted with the Illumina MiSeq platform using PE reads (2 X 150 bp) and Nextera library preparation.
Genomic analysis
The raw sequence data was evaluated and assembled de novo using the software SeqMan NGen 15 (DNAStar, Madison, WI, USA). Afterward, the contigs were inspected for internal errors, end trimmed, and reassembled using SeqManPro15. The obtained consensus sequence was annotated using Prokka (Seemann 2014). The sequence was further manually inspected for potential alternative start codons or the presence of non-annotated CDSs. The genome was then manually curated using Geneious 2022.0.2 (Biomatters Ltd.). BLASTX (Altschul 1997) was used to check for missing genes in intergenic regions. Encoded proteins were queried against protein sequences in BLASTP (Altschul 1997), PFAM (Finn et al. 2016), InterPro (Paysan-Lafosse et al. 2023), and HHpred (Söding et al. 2005) for homology search, protein families, signatures, and structure prediction. The potential coding sequences (CDSs) were also searched against Hidden Markov model profiles downloaded from the prokaryotic Virus Orthologous Groups (pVOGs) database (Grazziotin et al. 2017) using hmmscan (Eddy 2011). Protein sequences were further analyzed with TMHMM (Krogh et al. 2001), Phobius (Käll et al. 2007), and OCTOPUS (Viklund and Elofsson 2008) to predict transmembrane domains and SignalP (Petersen et al. 2011) and SPOCTOPUS (Viklund et al. 2008) to predict signal peptide cleavage sites. Intron sequences were predicted with Rfam (Kalvari et al. 2021).
The DC4 lifecycle (i.e., virulent, temperate, or chronic) was predicted by adopting a combination of Phage AI (machine learning models trained on 4694 manually selected bacteriophages from different species and families) (Tynecki et al. 2020) and BACPHLIP (searching for a particular set of temperate-specific protein domains) (Hockenberry and Wilke 2021). The proportion of non-coding ORFs present in DC4 was estimated using the Coding Potential Calculator 2 (CPC2) web server (Kang et al. 2017). Genes encoding tRNA were identified by using tRNAscan-SE (Lowe and Chan 2016) and ARAGORN (Laslett and Canback 2004). ResFinder 4.1 (Bortolaia et al. 2020) and HostPhinder 1.1 (Villarroel et al. 2016) were used to identify, respectively, the presence of acquired resistance genes (threshold for identity: 90%; minimum length: 60%; selected species: Staphylococcus aureus) and the in silico spectrum of phage hosts. Codon usage of DC4 and its hosts was calculated with SMS (sequence manipulation suite) taking into consideration the bacterial genetic code (Stothard 2000).
For phylogenomic purposes, phage genomes were downloaded and filtered from the National Center for Biotechnology Information (NCBI: txid10239) using inphared (INfrastructure for a PHAge REference Database, release 3 Oct 2021) (Cook et al. 2021). After a manual inspection, 83 whole-genome sequences (74 Staphylococcus phages and 9 Bacillus phages) were chosen based on their families (Myoviridae, Rountreeviridae, and Siphoviridae) and genome sizes (16.8 to 497.5 kb) (Table S2). At the time of selecting and filtering phages, tailed bacteriophages were still classified inside the former order Caudovirales (Turner et al. 2021). Therefore, the classical families Myoviridae and Siphoviridae were considered and mentioned throughout the text except when stated otherwise. Their relevant features were retrieved, and accession numbers were forwarded to VICTOR (Virus Classification and Tree Building Online Resource) (Meier-Kolthoff and Göker 2017) setting up amino acid as data type. VICTOR’s phylogenetic tree was annotated using IToL v6 (Interactive Tree of Life, version 6) (Letunic and Bork 2021). For taxonomic purposes, VIRIDIC (Moraru et al. 2020) and ViPTree (Nishimura et al. 2017) were adopted. The DC4 linear genomic map was constructed by using the ViPTree tool “genomic alignment view” and ORFs manually colored according to their function with INKSCAPE (version 1.2.2). Lastly, the R package corrplot was used for correlation analysis using the GC content and genome size as factors.
For comparative genomics, a pairwise comparison of all the Staphylococcus viruses’ genomes previously selected was carried out with the fastANI v1.3 algorithm (Jain et al. 2018) and based on the average nucleotide identity (ANI) method. Euclidean distances were computed among ANI results for clustering. progressiveMauve (Darling et al. 2010) within Geneious Prime 2022.0.2 was used to align and compute locally collinear blocks (LCBs) between PALS2 and DC4 genomes using default settings (match seed weight: 15; minimum LCB score: 30,000). Lastly, comparison and annotation of orthologous gene clusters among the six S. aureus jumbo phages (PALS2, SA1, Machias, Madawaska, MarsHill, and DC4) was conducted with OrthoVenn2 (E-value: 1e-2; Inflation value: 1.5) (Xu et al. 2019). BLAST Ring Image Generator (BRIG, version 0.95) was used to display circular comparisons among S. aureus jumbo genomes (Alikhan et al. 2011). The prediction of lytic and biofilm-degrading enzymes was based on the identification of specific and conserved domains such as CHAP (cysteine, histidine-dependent amidohydrolases/peptidases), SH3 (Src homology 3), and peptidoglycan binding domain (PGBD) using the InterPro protein families and domain database (Blum et al. 2021). Selected proteins were concatenated and aligned using Geneious Prime version 2022.0.2 with the BLOSUM62 cost matrix.
The complete genome sequence of vB_SauM-UFV_DC4 was deposited in GenBank under the accession number MZ779063.
Phage purification and proteomic analysis
After virus propagation and concentration (item 2.3), the viral suspension was purified by a three-step protocol using ion exchange and desalting columns in a chromatography system (ÄKTAprime plus, GE Healthcare Life Sciences, Uppsala, Sweden) as adopted previously for virus vB_EcoM-UFV13 (da Silva Duarte et al. 2018b; da Silva Duarte et al. 2018a). Salts present in the sample were removed (step 1) using the HiTrap Desalting prepacked column (GE Healthcare Life Sciences, Uppsala, Sweden). The first two peaks were collected and purified using an anion exchange chromatography column (step 2), with specific fractions (7 to 12) carried out to the third and final step, a new desalting procedure. For the anion exchange column, start (20 mM Tris-HC, pH 8.0) and elution buffers (20 mM Tris-HC, 1 M NaCl, pH 8.0) were used, whereas in desalting steps, a phosphate buffer (20 mM sodium phosphate, 0.15 M NaCl, pH 7.0) was prepared. A flow rate of 5 mL.min−1 was adopted for both columns. The final viral titer was carried out by the double-agar overlay method using S. aureus 3059 as the host strain. Plates were incubated at 30 °C overnight.
After phage purification, viral particles were lyophilized and sent to GenOne (Rio de Janeiro, RJ, Brazil) for DC4 structural proteins extraction, quantification, and enzymatic digestion using trypsin. Peptides were injected in LCMS-NanoUltra-HPLC ultimate 3000 and Quadropole-Orbitrap QExactive Plus Gradient. Finally, bioinformatic analysis was carried out with Proteome Discoverer (Thermo Fisher Scientific, Waltham, MA, USA) software for protein identification using DC4-annotated proteins as a reference.
Results
vB_SauM-UFV_DC4 physiological features and host range analysis
One-step growth curve for the phage DC4 with the host strain S. aureus 3059 showed a latent period of 40 min followed by a small burst of 3.4 PFUs/infected cell (Fig. 1A, B). The virus DC4 displayed multiple small bursts after two additional lag periods of 40 min (t80–t120 = 1.5 PFU/infected cell; t120–t160 = 5.7 PFUs/infected cell). The physical and chemical stability of phage DC4 was determined to provide more information regarding phage storage and potential applications at different conditions (Fig. 1C, D). More than 94% of DC4 particles survived at pH values ranging from 7 to 12, and 71.4% survived pH 4; however, DC4 could not survive pH 2. DC4 was stable at temperatures ranging from − 20 to 40 °C but not at 62 and 95 °C. The lytic activity of phage DC4 against 26 strains of Staphylococcus aureus is shown in Table S1. Phage DC4 could infect 11 out of 26 S. aureus strains (10 from a veterinary hospital and 1 from a hospital for humans). The relative EOP of phage DC4 was also measured for 11 different strains, and it was found that DC4 can efficiently propagate in nine isolates (Table 1).

One-step growth curve and physical/chemical stability assays. A The data shown for DC4 one-step growth curve are the mean values from three independent measurements performed in triplicate. For a better plotting, only data collected until the first 200 min of experiment are shown and expressed in PFU per infected cell (B). The virus DC4 demonstrated a latent period of 40 min followed by a small burst of progeny (3 PFUs per infected cell). The stability of vB_SauM-UFV_DC4 under different temperatures (C) and pH (D) was evaluated. *p < 0.05 (n = 9). The error bars represent the standard deviations
Genomic features of vB_SauM-UFV_DC4
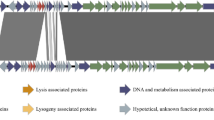
The phage DC4 genome is a 263,185 bp contiguous sequence of linear, double-stranded DNA with an overall G + C content of 25% (Fig. 2; Fig. S1) and encodes 263 predicted CDSs. The correlation analysis between large genomes and low GC content relationship carried out with Staphylococcus viruses revealed a significant and moderate negative correlation between genome size and GC content (Pearson R value = − 0.59, p < 0.001; Table S3). The gene-coding potential of the global genome is 91.9% with only eight overlapping genes and displays. Among all the CDSs, 207 initiated translation with an ATG start codon, and most genes (79.8%) are transcribed from the positive strand. The DC4 genome also encodes 1 tRNA (Phe-tRNA: genome coordinates − 52,143–52,236 kb) and has a predicted pseudo-tRNA (Leu-tRNA: 72,405–72,475 kb). Following gene inspection and annotation with different approaches, 54 genes received a final functional assignment (Table 2), 185 were assigned as “hypothetical protein,” and 24 were predicted as “hypothetical membrane protein” (proteins with predicted transmembrane domains). No antimicrobial resistance genes were identified, as well as genes with predicted lysogeny functions (e.g., integrase, transposase, and excisionase). Indeed, BACPHLIP predicted that DC4 has a lower probability of being a temperate phage (23.75%). On the contrary, the tool PhageAI, which infers bacteriophage lifestyle with approximately 98% of accuracy, has predicted DC4 as a temperate virus. Considering the prediction of mobile self-splicing elements, Rfam did not identify any intronic sequences in the DC4 genome. However, after protein screening using InterPro, five sequences stand out and the C-terminal RSV-like homing endonuclease domain (Gene3D: G3DSA:3.40.960.10). The UFVDC4_00129 gene encodes the putative HNH homing endonuclease (Pfam: PF13392). Only one intein was observed, and it is present within the protein-coding gene gyrA (DNA gyrase subunit A). An endonuclease domain LAGLIDADG_3 (Pfam: PF14528, IPR004860) was also identified.

The linear genome map of phage DC4 showing position of CDSs. The CDSs are colored according to their functional category
Functional features
At least nine multi-subunit DNA-dependent RNA polymerases (RNAPs) and a Sigma70 factor (UFVDC4_00065) were identified on phage DC4 genome. One of these subunits (UFVDC4_00067) was also identified packaged into the virion (vRNAP) along with a transcriptional factor (UFVDC4_00011) belonging to the AAA family ATPase. The virus also displays the four DNA-directed RNAP subunits with high similarities to those enzymes described in the PALS2 genome (Lee et al. 2021) (β: UFVDC4_00159, PALS2_067 – 99.72%; β′: UFVDC4_00179, PALS2_089 – 100%; ω: DC4_00013, PALS2_188 – 99.27%; δ: UFVDC4_00049, PALS2_228 – 98.12%). The annotation of the other five genes showed that ORFs 23 and 69 encode for β subunits and show high homology with those found in the Staphylococcus phage Madawaska (98.41 and 98.04%), while the genes UFVDC4_00067 and UFVDC4_00178 encode for β subunits and display high identity with coding sequences found in the Staphylococcus phage Madawaska (98.45%) and Staphylococcus phage MarsHill (98.52%), respectively. Lastly, UFVDC4_00087 was annotated as a non-viral RNA polymerase subunit (Staphylococcus phage MarsHill, 98.44%). Another noteworthy feature on DC4 genome is the presence of a predicted RNA-dependent DNA polymerase (reverse transcriptase) (Pfam: PF00078). This is a conserved protein among the S. aureus jumbo phages encompassed in this study sharing 84% of identity. In the case of DC4, this corresponds to locus tag UFVDC4_00113 (UAJ17040.1), a basic 58.9 kDa protein.
In silico bacterial host prediction using HostPhinder revealed that DC4 might have several bacterial genera as hosts, including Staphylococcus and Bacillus (Table S2). The initial functional annotation at the protein level of the phage DC4 revealed a homology of 22% with the phages AR9 (KU878088) and PBS1 (MF360957) and 21% with the phage vB_BpuM-BpSp (KT895374), both infecting Bacillus sp. However, genomic comparison including 74 Staphylococcus phages and nine Bacillus jumbo phages showed that the phage DC4 is phylogenetically related to five recently reported Staphylococcus jumbo phages (Machias, MW349128; Madawaska, MW349129; MarsHill, MW248466; PALS2, MN091626; and vB_StaM_SA1, MW218148) (Fig. 3).

Interactive Tree Of Life (iTOL) was constructed based on VICTOR’s phylogenetic tree using 83 whole-genome sequences (74 Staphylococcus phages and nine Bacillus phages). Outer and inner rings show viral families and genera, respectively. The color saturation indicates the degree of difference in terms of genome size
Phylogenomic
The whole-genome similarity among the DC4 and other Staphylococcus phages was estimated based on phylogenomic analysis. As depicted in Fig. 4, it is possible to identify at least four relevant clusters showing high ANI scores based on gene content. Interestingly, each cluster is characterized by a different range of GC content combined with genome size (cluster 1: 30–35% GC and 25–50 Kb; cluster 2: 25–30% GC and < 25 Kkb; cluster 3: 30–35% GC and 25–50 kb; cluster 4: 25–30% GC and > 200 kb). Overall, viruses belonging to the genera Triavirus, Rosenblumvirus, and Rockefellervirus were grouped in the cluster 1, cluster 2, and cluster 3, respectively. Specifically, regarding cluster 4, which includes only Staphylococcus jumbo phages, VIRIDIC and VIPtree analyses (Fig. 5) revealed that the DC4 genome is one of four new species in a new genus and shows high similarity with the phage PALS2 (Fig. S2). Based on the frequency of codon usage for the amino acid phenylalanine, DC4 and its hosts (S. aureus 2030RH1 and 3059) show very similar fraction values for the codons TTT (~ 0.69) and TTC (~ 0.31) (Fig. S3).

Heatmap of the average nucleotide identity (ANI) between the whole genome sequences of bacteriophages displaying S. aureus as host (n = 74)

VIRIDIC (A) generated tree (BLASTN homology) of S. aureus jumbo phages (red stars) and unrelated viruses infecting the different hosts (phylum Firmicutes and class Gammaproteobacteria) shows that DC4 is one of four new species in a new genus. ViPTree (B) based on the proteome of S. aureus jumbo phages (TBLASTX homology) reveals that DC4 is probably part of a new family composed of two subfamilies and one genus
Orthologous and vB_SauM-UFV_DC4 lytic proteins
The comparative analysis of orthologous genes among the six Staphylococcus jumbo phages revealed 303 clusters (133 orthologous clusters present in at least two viruses) and 170 single-copy gene clusters (Fig. S4). Investigating the biological processes of the biggest cluster of genes (172 genes), the most enriched gene ontology (GO) terms were related to DNA replication (GO:0006260) and metabolic process (GO:0008152), both crucial for the phage life cycle (Weigel and Seitz 2006). Lastly, three genes were not included in any cluster (singletons: DC4_00155, DC4_00190, and DC4_00218) for the phage DC4; all of them were annotated as hypothetical proteins.
Regarding the set of putative lytic enzymes encoded in the DC4 genome, nine proteins stand out and were further aligned and compared with their homolog proteins identified in the other four S. aureus jumbo phages (cluster 4, Figs. 4 and 6). Due to their low overall identity with DC4 proteins, phages vB_StaM_SA1 and Machias were not included in this analysis. Three out of nine proteins were classified as cell wall hydrolases and displayed CHAP + SH3 (DC4_0060, endolysin), amidase_2 + SH3 (DC4_0061, N-acetylmuramoyl-L-alanine amidase), and amidase_2 + PG_binding (DC4_00110, N-acetylmuramoyl-L-alanine amidase) domains. In two hypothetical proteins (DC4_00236 and DC4_00241), a PGBD domain was identified. The DC4_00248 encodes a putative transglycosylase IsaA. Lastly, three tail-associated enzymes were identified: DC4_0062 encodes a tail fiber with a pectin lyase–like domain, whereas a tail lysozyme (DC4_00259) and a tail tape measure protein with peptidase_M23 domain (DC4_00260) were also predicted. After concatenation and alignment, the identity matrix revealed that together the set of lytic enzymes predicted for the virus DC4 shares 97%, 88%, and 84% of identity with the phages PALS2, Madawaska, and MarsHill, respectively. As depicted in Fig. 5A, several amino acid substitutions can be observed comparing the four S. aureus jumbo isolates, and most of them are in domain regions, which might impact substrate recognition and binding. Between DC4 and PALS2, 169 residues were not identical, whereas, between DC4 and Madawaska/MarsHill, 774 and 844 differences were noticed, correspondingly. Noteworthy, a tail fiber protein containing a pectin lyase–like domain (DC4_0062) showed a very low conserved region when compared to the other viruses. A manual inspection revealed that in the phages DC4, PALS2, and MarsHill, this protein displays, on average, 773 aa, while the virus Madawaska encodes a protein with 1139 aa with an exclusive glycoside hydrolase/deacetylase domain (Fig. 5B).

The nine putative lytic proteins (A) identified in the DC4 proteome were concatenated, aligned, and compared with their homologs found in the phages PALS2, Madawaska, and MarsHill. B Detailed analysis of the tail fiber protein with a pectin lyase domain among the four closely related S. aureus jumbo phages. Mean pairwise identity over all pairs in the column: green: 100% identity; greeny-brown: 30–100% identity; red: below 30% identity
With regard to the proteins identified as endolysins in this study, a high degree of conservation in terms of length and the primary sequence were observed among the 4 isolates. The viruses DC4 and PALS2 differ in 9 residues, whereas between DC4 and Madawaska/MarsHill, 11 and 45 differences were noticed, respectively.
DC4 structural proteome
Phage structural proteome analysis revealed that 20 putative genes in the DC4 genome potentially code for virion proteins, such as portal vertex protein (ORF_0049), base plate wedge subunit (ORF_0083), tail fiber protein (ORF_0216), prohead core scaffold protein and protease (ORF_235), contractile tail sheath (ORF_251), precursor of major head subunit (ORF_0238), and tail tape measure protein (ORF_256).
To verify the structural and packaged proteins in the mature phage virion of DC4, purified phage particles were enzymatically treated and analyzed using LCMS. In total, 26 peptides were mapped to 15 proteins (Table 3), including the precursor of the major head subunit (UFVDC4_00242), tail fiber protein (UFVDC4_00062; UFVDC4_00220), and prohead core scaffold protein and protease (UFVDC4_00239). Among the other phage-coding proteins, six of them were identified as hypothetical proteins (UFVDC4_00056, UFVDC4_00234, UFVDC4_00107, UFVDC4_00019, UFVDC4_00134, and UFVDC4_00153), one as Ig-like domain-containing protein (UFVDC4_00059), and the other four proteins are GroEL (UFVDC4_00086), UvsX-like recombinase (UFVDC4_00158), a transcription factor belonging to AAA family ATPase (UFVDC4_00011) and RNAP β subunit (UFVDC4_00067). In addition, we also checked the peptides obtained for the protein GroEL against the genome of the host strains S. aureus 2030RH1 and 3059 to verify whether this protein is of bacterial origin; however, no similar pattern was observed in both strains.
Discussion
Jumbo phages have attracted great attention over the last years due to their wide lytic activity (broad host range) and partial independence from the host enzymes in terms of gene expression (encoding their own RNA polymerases) (Yuan and Gao 2017; Sokolova et al. 2020). Furthermore, some isolates of jumbo viruses such as phiKZ-like phages infecting Pseudomonas can assemble a nucleoid structure during viral infection as an adaptive mechanism to evade bacterial defense systems like CRISPR-Cas restriction enzymes (Malone et al. 2019; Guan and Bondy-Denomy 2020). Overall, jumbo phages have been characterized as tailed bacteriophages (myovirus and siphovirus) with a genome size greater than 200 kb, have large particles, and show low similarity to those already described (Hendrix 2009). In terms of jumbo phages targeting Staphylococcus, genomic information of only six viruses can be found in public databases. The Machias (MW349128), Madawaska (MW349129), and MarsHill (MW248466) were isolated in the USA from swine barn; SA1 (accession no: MW218148) and bacteriophages S6 (accession no: LC680885) were obtained from sewage in China and Japan, respectively. The virus PALS2 (accession no: MN091626.1) was isolated from bird feces in South Korea. Among them, only PALS2 was completely characterized (Lee et al. 2021), which reflects the current scarcity of biological information about jumbo phages infecting S. aureus, one of the main important pathogens in human and veterinary medicine (Park and Ronholm 2021).
The physiological analysis showed that DC4 phage has similarities to other jumbo phages. When compared to the latest available data for Staphylococcus jumbo bacteriophages, DC4 shows the smallest burst size among PALS2 (12 PFUs/infected cell) and SA1 (140 PFU/infected cell), with an intermediate latency period (PALS2: 30 min; SA1: 55 min) The phage also presents some additional small burst events, which is a commonly observed feature for jumbo phages with small burst sizes (Shkoporov et al. 2018; Sharma et al. 2019; Lee et al. 2021). Despite data scarcity specifically related to jumbo phages stability, different studies have reported an apparent higher sensitivity of this group of phages to higher temperatures but not at basic pH values. The jumbo phage MIJ3 is stable at temperatures ranging from 4 to 60 °C, while more than 90% of MIJ3 phage survived at pH values ranging from pH 3 to ~ 10 (pH values above 10 were not tested) (Imam et al. 2019). The Xanthomonas jumbo phage LucasX remained viable up to 45 °C with a pH optimum of 7 but after 30 min at 50 °C, just a few particles were still viable. This phage also was completely inactivated on pH 3 and 11 (Marquioni et al. 2022). For the Salmonella jumbo phage pSal-SNUABM-04, viral particles were stable at 4–27 °C, whereas stability dropped at 37 °C within 2 h (Kwon et al. 2020). Our data shown that besides the DC4 phage present (as others jumbo phages) a relatively low resistance to high temperatures, it is still stable on pH 12, appearing to show greater resistance in this field than the phages discussed above. Also, the wide host range against staphylococci from the veterinary environment indicates that phage DC4 has the potential to be explored as an anti-staphylococcal agent for veterinary usage.
The GC content of the phage DC4 is within the range of other Staphylococcus jumbo phages (24.8–26.8%), which is significantly lower than that of their hosts (S. aureus 3059, 32.8%; S. aureus 2030RH1, 32.7%) and other phages with dsDNA genomes larger than 200 kb (Lavysh et al. 2016; Wojtus et al. 2019). The GC content of 25% is also only slightly higher than the Staphylococcus phage Machias (24.8%). As reported by Almpanis et al. (2018), bacteriophages with large genome sizes are more likely to present lower content of guanine and cytosine, whereas larger bacterial genomes tend to have higher GC content. Our results reinforce this idea, once a significant and moderate negative correlation between genome size and GC content was found among the Staphylococcus viruses evaluated on the correlation analysis. Although the evolutionary explanation for this phenomenon is still a matter of debate, environmental factors and physiological capabilities have a major influence in shaping the GC content of the phage genome, which can impact codon usage for protein synthesis (Almpanis et al. 2018). Phage DC4 also presents a considerably low gene density (genes per kilobase pair of nucleotide sequence) when compared to other jumbo phages (gene density of 1.5 ± 0.25) (Iyer et al. 2021), once its 263,185 bp genome encodes 263 predict CDSs.
Although the large size, the DC4 genome only encodes 1 tRNA and one predicted pseudo-tRNA. The low number of tRNAs encoded in DC4 genome is not exclusive for this virus but was also noticed or even not predicted for others Staphylococcus jumbo phages such as PALS2, S6 and Madawaska (1 tRNA-Asn), MarsHill (0 tRNA), Machias (2 pseudo tRNA-Asn), and vB_StaM_SA1 (tRNA-Ser). This result shows that these phages have underwent different selective pressures and retained different tRNAs based on host and virus codon usage. Moreover, it is quite intriguing that such “autonomous” phages, a common characteristic stated for jumbo phages, have a such low number of tRNA even when compared to other jumbo phages. In a comparative genomic study, Iyer et al. (2021) showed that phage reliance on self-encoded tRNAs usually differs between phages, and their number can range between 4 and 22 per genome.
The divergence between BACPHLIP and PhageAI on the DC4 lifestyle raised the question of whether DC4 shows a prolonged propagation or pseudolysogeny/carrier state event as observed for Pseudomonas aeruginosa PA5oct jumbo phage (Olszak et al. 2019). Another interesting point that must be considered and could explain the discrepancy between both approaches is that these tools employ machine learning algorithms trained on a specific dataset of virulent and temperate phage genomes. Therefore, the inclusion of genomes from jumbo phages must be contemplated and the higher number of uncharacterized proteins carefully considered. The high identity (96.4%) between the DC4 gyrA intein and those identified in gyrA phages PALS2, Madawaska, and MarsHill suggest that both phages have acquired it by common ancestor as hypothesized for the phages Madawaska and MarsHill (Korn et al. 2021).
One of the interesting aspects of DC4 biology is the annotation of at least nine multi-subunit DNA-dependent RNA polymerases (RNAPs) and a Sigma70 factor, which suggests that DC4 does not rely exclusively on the host transcription machinery to express its genes. Although, the presence of multiple RNAPs is considered a common feature across the genomes of jumbo phages, and a higher diversity of virion, nonvirion RNAPs (nvRNAPs), and transcriptional factors have been reported across different jumbo viruses (Iyer et al. 2021). The presence of more than one paralogous for RNAP might reflect the use of different subunits according to the lifecycle and type of genes (early, middle, or late) that have been expressed in a certain moment (Miller et al. 2003). Interestingly, it is assumed that the presence of these multi-subunits might broaden the jumbo phages host spectrum due to their overall lower dependence on the host machinery in terms of metabolism (Yuan and Gao 2017).
Another remarkable feature encoded in the DC4 genome that can broaden the viral spectrum of hosts is a predicted RNA-dependent DNA polymerase (reverse transcriptase). This phenomenon was observed for phages that infect the genus Bordetella, where the selective mutagenesis of the phage tail fiber (VR1 region of the mtd gene) was dependent of a retron-like reverse transcriptase to extend the phage host range (Liu et al. 2004). As argued by Korn et al. (2021), its role and association with host switching in jumbo phages infecting S. aureus have not been confirmed and need further investigation mainly due to the absence of characteristic regions that would include it in the diversity-generating retroelements (DGRs) group. Interestingly, based upon nucleoside analysis conducted by Uchiyama et al. (2014) which demonstrated the presence of deoxyuridine rather than thymidine in the nucleic acid of Staphylococcus phage S6 and its sequence, similarity to DC4 indicates that the latter phage possesses a similar modification. Korn et al. (2021) commented that the genomes of S. aureus phages MarsHill, Madawaska, and Machias have “presumably hypermodified DNA which inhibits sequencing by several different common platforms,” though the hypermodification is not supported by the above mentioned nucleoside analysis. Analysis of the staphylococcal jumbo phage proteomes reveals that they all encode a reverse transcriptase (RNA-dependent DNA polymerase).
The in silico prediction of several bacterial hosts (obtained using HostPhinder) is a quite uncommon feature for a phage. According to Villarroel et al. (2016), this tool assumes that bacteriophages with similar genomic features are prone to share bacterial hosts. Since jumbo phages generally lack the typical modular organization found in viruses with smaller-genome sizes, a high number of uncharacterized and presumed to have uracil-substituted DNA which interferes with DNA sequencing genes (Uchiyama et al. 2014; Naknaen et al. 2021), the use of in silico tools to predict phage hosts must be considered with caution. According to the host range analysis, DC4 showed antimicrobial activity mainly against S. aureus isolated from the veterinary ecosystem, which is indeed where this virus was first isolated (da Silva Duarte et al. 2020) and may reflect its predilection to S. aureus infecting animal husbandry. It is important to stress out that due to the lack of Bacillus strains from veterinary sources, the capability of DC4 to infect Bacillus species remains to be investigated.
Besides phage DC4 presents homology both with Bacillus and Staphylococcus infecting phages, it is possible to observe that jumbo phages infecting Staphylococcus are closely related to jumbo viruses infecting Bacillus. This suggests that a set of core genes may be shared between viruses infecting both bacterial genera. Additionally, according to previous studies (Iyer et al. 2021; Weinheimer and Aylward 2022), genes related to replication machinery and infection apparatus are specifically shared between jumbo phages and small genome phages, which supports the creation of a distinct clade for jumbo phages within the class Caudovirecetes (tailed viruses of bacteria and archaea).
Based on the phylogenomic analysis, we further estimated the whole-genome similarity among the DC4 and other Staphylococcus phages (NCBI: taxid10239), by calculating the mean nucleotide identity of orthologous gene pairs shared between two viral genomes. At least four relevant clusters were grouped based on high ANI scores. Phages from different clusters exhibited extremely low or no similarity among them, which indicates a divergent origin (Yuan and Gao 2017). According to the International Committee on Taxonomy of Viruses (ICTV) (Adriaenssens and Brister, 2017), an average nucleotide identity (ANI) higher than 95% is used to classify bacteriophages at the species level. Because of this, and based on VIRIDIC and VIPtree results, DC4 phage was considered one of four new species in a new genus and shows high similarity with the phage PALS2. Notwithstanding that both viruses share high homology, it is noteworthy to mention some differences between them. The virus PALS2 was isolated from bird feces, whereas DC4 was obtained from dairy cattle farm waste (da Silva Duarte et al. 2020). Furthermore, DC4 encodes a Phe-tRNA and has an apparent narrower host spectrum among different isolates of S. aureus, while PALS2 encodes an Asn-tRNA and can infect different Staphylococcus species (Lee et al. 2021). As discussed by Delesalle et al. (2016), the presence of tRNA genes in bacteriophages has been justified based on codon or amino acid usage. One of the hypotheses suggests that phages with codon usages like their hosts would not retain such tRNA genes, which is not observed for the virus DC4.
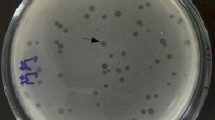
The comparative analysis of orthologous genes among the six Staphylococcus jumbo phages revealed 303 clusters and 170 single-copy gene clusters. Regarding lytic enzymes, nine proteins on phage DC4 genomes could be aligned and present homology with other four S. aureus jumbo phages. Three of these proteins were classified as cell wall hydrolases, two have PGBD domains, one encodes a putative transglycosylase IsaA, and the last tree possessed tail-associated enzymes. Different domains were identified among the S. aureus jumbo phages lytic enzymes. Deacetylases and pectate lyase are classified as phage-encoded polysaccharide depolymerases (PSDs) that allow phages to degrade and overcome bacterial barriers such as capsular polysaccharides, exopolysaccharides, and lipopolysaccharides (Danis-Wlodarczyk et al. 2021). Pectate/pectin lyases are characterized by cleavage of the α-1,4 bonds of polygalacturonic acid and have been identified in tail proteins of bacteriophages infecting different bacterial genera such as Salmonella, Acinetobacter, and Campylobacter and might function in the structure of receptor-binding proteins (RBPs) (Latka et al. 2017; Oliveira et al. 2017; Thanki et al. 2019; Sørensen et al. 2021). The presence of PSD in jumbo phages is a quite common feature reported during functional annotation of viral genes. Depolymerase activity is typically verified by the presence of phage plaque-surrounding halo zones due to its production and diffusion in a soluble form. It is noteworthy mentioning that after plating and incubation for 24 h, the presence of halo zones surrounding phage plaques have been recorded for DC4, and its diameter size changed according to the host used for propagation (Fig. S5).
Bacterial peptidoglycan cleavage by hydrolases (e.g., glycosidase, amidase, and endopeptidase) is essential along the bacteriophage lifecycle and relies on the activity of specific enzymes classified as virion-associated peptidoglycan hydrolases (VAPGHs) or endolysins (Danis-Wlodarczyk et al. 2021). VAPGHs are structural components of the virion particle involved in the initial steps of phage infection and are characterized by a modular structure composed of one or two N-terminal catalytic domains (EAD, enzymatically active domain) but commonly lacking one C-terminal cell wall-binding domain (CBD); on the contrary, most staphylococcal phage endolysins display both EAD and CBD domains (Gutiérrez et al. 2018). Undoubtedly, the use of phage lytic enzymes has been one of the most studied classes of new antimicrobials against S. aureus over the last few years, mainly aimed at the control of methicillin-resistant S. aureus (MRSA) and vancomycin-resistant S. aureus (VRSA) (Duarte et al. 2021; Gutiérrez et al. 2021).
For the control of bovine mastitis caused by S. aureus, Donovan et al. (2006) demonstrated that the endolysin phi11 is active at the physiological pH and Ca2+ concentration of milk and is efficient in controlling bacterial growth in turbidity assays. When evaluated in animal models of mastitis, the phage lytic protein LysRODI outperformed CHAPSH3b displaying a pronounced activity to prevent mammary infections by S. aureus and Staphylococcus epidermidis. For the enzymes λSA2-E-Lyso-SH3b/λSA2-E-LysK-SH3b, a pronounced effect in terms of reducing bacterial load was also observed (Schmelcher et al. 2012). Although little information is available regarding field trials, Fan et al. (2016) report that Trx-SA1 could effectively control mild clinical mastitis caused by S. aureus in dairy cows by reducing somatic cell and pathogen levels after treatment. Overall, the identification and characterization of catalytic domains, mainly from newly isolated jumbo phages, can help in the design of new chimeric enzymes and their evaluation against S. aureus and biofilms formed by this species.
Phage structural proteome analysis has been used to better comprehend and annotate the set of proteins found on phage particles after a complete life cycle (Lavigne et al. 2009). The DC4 genome possesses 20 putative genes related to virion proteins; purified phage particles were analyzed using LCMS to verify the mature phage virion of DC4 and identified 26 peptides, including a vRNAP. The presence of vRNAP is a common feature of jumbo phages and is associated with the transcription of early genes (Ceyssens et al. 2014; Sokolova et al. 2017); however, the presence of the other three co-packaged proteins must be further confirmed since the presence of viral chaperons following viral purification using anion exchange chromatography has been reported (da Silva Duarte et al. 2018a). UvsX-like proteins are involved in homologous recombination and error-free repair of DNA double-strand breaks (Maher and Morrical 2013), whereas MoxR-like ATPase and GroEL act as chaperone-like proteins helping the maturation of metabolic protein complexes and the assembly of phage capsid proteins, respectively (Ang et al. 2000; Snider and Houry 2006).
In conclusion, bacteriophages are the most abundant organisms on Earth which makes them an incredible source of biodiversity with still many uncovered biological features to be explored. Thanks to the next-generation tools and bioinformatic pipelines, there has been a recent increase, although with still relatively very few specimens, in the number of phages classified as S. aureus jumbo phages. The virus vB_SauM-UFV_DC4 was isolated from the dairy environment in Vicosa, Minas Gerais, Brazil. This virus has a small burst size of progeny of 3 PFUs per infected cell, shows a great stability in basic pHs, and shares a high identity with the virus PALS2 (93.5%). This study reports that the viruses DC4, PALS2, MarsHill, and Madawaska can be considered viral species belonging to a new genus within the class Caudoviricetes. DC4 carries a set of virion and non-virion RNA polymerase subunits, an intein disrupted gene (gyrA), an endonuclease (VSR and LAGLIDADG), and an RNA-directed DNA polymerase. Lastly, DC4 and closely related jumbo phages showed a similar set of lytic proteins (tailed-associated or not), except for the presence of a deacetylase domain present in the tail of the phage Madawaska. The identification and characterization of catalytic domains from newly isolated jumbo phages can help in the design of new chimeric enzymes against S. aureus and biofilms formed by this species.
Data availability
The authors declare that sequence data that support the findings of this study have been deposited in GenBank with the accession number MZ779063. All other data supporting the findings of this study are available within the article and its supplementary information files.
References
Akturk E, Oliveira H, Santos SB, Costa S, Kuyumcu S, Melo LDR, Azeredo J (2019) Synergistic action of phage and antibiotics: parameters to enhance the killing efficacy against mono and dual-species biofilms. Antibiotics 8. https://doi.org/10.3390/antibiotics8030103
Alikhan NF, Petty NK, ben Zakour NL, Beatson SA (2011) BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genom 12. https://doi.org/10.1186/1471-2164-12-402
Almpanis A, Swain M, Gatherer D, McEwan N (2018) Correlation between bacterial G+C content, genome size and the G+C content of associated plasmids and bacteriophages. Microb Genom 4. https://doi.org/10.1099/mgen.0.000168
Altschul S (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. https://doi.org/10.1093/nar/25.17.3389
Amend AM, Bifone JP, Brewer JC, Denton MN, Gilbert EB, Grimm AC, Hogan JM, Kelley RM, Kelly-Brooner LJ, Mukerji JA, Osterhoudt M, Senn CR, Smith BR, Stillwell OG, Vo J, Watt DK, Connerly PL, Rueschhoff EE (2023) Genome eequence of Gordonia rubripertincta phage survivors, a cluster ct siphovirus. Microbiol Resour Announc. https://doi.org/10.1128/mra.01086-22
Ang D, Keppel F, Klein G, Richardson A, Georgopoulos C (2000) Genetic analysis of bacteriophage-encoded cochaperonins. Annu Rev Genet 34:439–456. https://doi.org/10.1146/annurev.genet.34.1.439
Bhandari V, Suresh A (2022) Next-generation approaches needed to tackle antimicrobial resistance for the development of novel therapies against the deadly pathogens. Front Pharmacol 13. https://doi.org/10.3389/fphar.2022.838092
Blum M, Chang HY, Chuguransky S, Grego T, Kandasaamy S, Mitchell A, Nuka G, Paysan-Lafosse T, Qureshi M, Raj S, Richardson L, Salazar GA, Williams L, Bork P, Bridge A, Gough J, Haft DH, Letunic I, Marchler-Bauer A, Mi H, Natale DA, Necci M, Orengo CA, Pandurangan AP, Rivoire C, Sigrist CJA, Sillitoe I, Thanki N, Thomas PD, Tosatto SCE, Wu CH, Bateman A, Finn RD (2021) The InterPro protein families and domains database: 20 years on. Nucleic Acids Res 49. https://doi.org/10.1093/nar/gkaa977
Bortolaia V, Kaas RS, Ruppe E, Roberts MC, Schwarz S, Cattoir V, Philippon A, Allesoe RL, Rebelo AR, Florensa AF, Fagelhauer L, Chakraborty T, Neumann B, Werner G, Bender JK, Stingl K, Nguyen M, Coppens J, Xavier BB, Malhotra-Kumar S, Westh H, Pinholt M, Anjum MF, Duggett NA, Kempf I, Nykäsenoja S, Olkkola S, Wieczorek K, Amaro A, Clemente L, Mossong J, Losch S, Ragimbeau C, Lund O, Aarestrup FM (2020) ResFinder 4.0 for predictions of phenotypes from genotypes. J Antimicrob Chemother 75. https://doi.org/10.1093/jac/dkaa345
Campos B, Pickering AC, Rocha LS, Aguilar AP, Fabres-Klein MH, de Oliveira Mendes TA, Fitzgerald JR, de Oliveira Barros Ribon A (2022) Diversity and pathogenesis of Staphylococcus aureus from bovine mastitis: current understanding and future perspectives. BMC Vet Res 18:115. https://doi.org/10.1186/s12917-022-03197-5
Ceyssens P-J, Minakhin L, van den Bossche A, Yakunina M, Klimuk E, Blasdel B, de Smet J, Noben J-P, Blasi U, Severinov K, Lavigne R (2014) Development of giant bacteriophage KZ is independent of the host transcription apparatus. J Virol 88. https://doi.org/10.1128/jvi.01347-14
Chung PY, Toh YS (2014) Anti-biofilm agents: recent breakthrough against multi-drug resistant Staphylococcus aureus. Pathog Dis 70:231–239. https://doi.org/10.1111/2049-632X.12141
Cook R, Brown N, Redgwell T, Rihtman B, Barnes M, Clokie M, Stekel DJ, Hobman J, Jones MA, Millard A (2021) INfrastructure for a PHAge REference Database: identification of large-scale biases in the current collection of cultured phage genomes. PHAGE. https://doi.org/10.1089/phage.2021.0007
da Silva Duarte V, Dias RS, Kropinski AM, Campanaro S, Treu L, Siqueira C, Vieira MS, da Silva PI, Santana GR, Martins F, Crispim JS, da Silva XA, Ferro CG, Vidigal PMP, da Silva CC, de Paula SO (2018a) Genomic analysis and immune response in a murine mastitis model of vB_EcoM-UFV13, a potential biocontrol agent for use in dairy cows. Sci Rep 8:6845. https://doi.org/10.1038/s41598-018-24896-w
da Silva Duarte V, Dias RS, Kropinski AM, da Silva Xavier A, Ferro CG, Vidigal PMP, da Silva CC, de Paula SO (2018b) AT4virus prevents biofilm formation by Trueperella pyogenes. Vet Microbiol 218. https://doi.org/10.1016/j.vetmic.2018.03.025
da Silva Duarte V, Treu L, Sartori C, Dias RS, da Silva PI, Vieira MS, Santana GR, Marcondes MI, Giacomini A, Corich V, Campanaro S, da Silva CC, de Paula SO (2020) Milk microbial composition of Brazilian dairy cows entering the dry period and genomic comparison between Staphylococcus aureus strains susceptible to the bacteriophage vB_SauM-UFV_DC4. Sci Rep 10:5520. https://doi.org/10.1038/s41598-020-62499-6
Danis-Wlodarczyk KM, Wozniak DJ, Abedon ST (2021) treating bacterial infections with bacteriophage-based enzybiotics: in vitro, in vivo and clinical application. Antibiotics 10:1497. https://doi.org/10.3390/antibiotics10121497
Darling AE, Mau B, Perna NT (2010) progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. https://doi.org/10.1371/journal.pone.0011147
Delesalle VA, Tanke NT, Vill AC, Krukonis GP (2016) Testing hypotheses for the presence of tRNA genes in mycobacteriophage genomes. Bacteriophage 6. https://doi.org/10.1080/21597081.2016.1219441
Donovan DM, Lardeo M, Foster-Frey J (2006) Lysis of staphylococcal mastitis pathogens by bacteriophage phi11 endolysin. FEMS Microbiol Lett 265. https://doi.org/10.1111/j.1574-6968.2006.00483.x
Duarte AC, Fernández L, de Maesschalck V, Gutiérrez D, Campelo AB, Briers Y, Lavigne R, Rodríguez A, García P (2021) Synergistic action of phage phiIPLA-RODI and lytic protein CHAPSH3b: a combination strategy to targetStaphylococcus aureus biofilms. NPJ Biofilms Microbiomes 7. https://doi.org/10.1038/s41522-021-00208-5
Eddy SR (2011) Accelerated profile HMM searches. PLoS Comput Biol 7. https://doi.org/10.1371/journal.pcbi.1002195
Fan J, Zeng Z, Mai K, Yang Y, Feng J, Bai Y, Sun B, Xie Q, Tong Y, Ma J (2016) Preliminary treatment of bovine mastitis caused by Staphylococcus aureus, with trx-SA1, recombinant endolysin of S. aureus bacteriophage IME-SA1. Vet Microbiol 191. https://doi.org/10.1016/j.vetmic.2016.06.001
Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, Salazar GA, Tate J, Bateman A (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44:D279–D285. https://doi.org/10.1093/nar/gkv1344
Fischetti VA (2018) Development of phage lysins as novel therapeutics: a historical perspective. Viruses 10. https://doi.org/10.3390/v10060310
Gill SR, McIntyre LM, Nelson CL, Remortel B, Rude T, Reller LB, Fowler VG (2011) Potential associations between severity of infection and the presence of virulence-associated genes in clinical strains ofStaphylococcus aureus. PLoS One 6. https://doi.org/10.1371/journal.pone.0018673
Grazziotin AL, Koonin Ev, Kristensen DM (2017) Prokaryotic virus orthologous groups (pVOGs): aresource for comparative genomics and protein family annotation. Nucleic Acids Res 45. https://doi.org/10.1093/nar/gkw975
Guan J, Bondy-Denomy J (2020) Intracellular organization by jumbo bacteriophages. J Bacteriol 203. https://doi.org/10.1128/JB.00362-20
Gutiérrez D, Fernández L, Rodríguez A, García P (2018) Are Phage Lytic Proteins the Secret Weapon To Kill Staphylococcus aureus? mBio 9. https://doi.org/10.1128/mBio.01923-17
Gutiérrez D, Rodríguez-Rubio L, Ruas-Madiedo P, Fernández L, Campelo AB, Briers Y, Nielsen MW, Pedersen K, Lavigne R, García P, Rodríguez A (2021) Designand selection of engineered lytic proteins with Staphylococcus aureus decolonizing activity. Front Microbiol 12. https://doi.org/10.3389/fmicb.2021.723834
Haag AF, Fitzgerald JR, Penadés JR (2019) Staphylococcus aureus in animals. Microbiol Spectr 7. https://doi.org/10.1128/microbiolspec.gpp3-0060-2019
Hendrix RW (2009) Jumbo bacteriophages. In: van Etten JL (ed) Lesser known large dsDNA viruses. Springer Berlin Heidelberg, Berlin, Heidelberg, pp 229–240. https://doi.org/10.1007/978-3-540-68618-7_7
Hendrix RW (2002) Bacteriophages: evolution of the majority. Theor Popul Biol 61. https://doi.org/10.1006/tpbi.2002.1590
Hockenberry AJ, Wilke CO (2021) BACPHLIP: Predicting bacteriophage lifestyle from conserved protein domains. Peer J 9. https://doi.org/10.7717/peerj.11396
Imam M, Alrashid B, Patel F, Dowah ASA, Brown N, Millard A, Clokie MRJ, Galyov EE (2019) vB_PaeM_MIJ3, anovel jumbo phage infecting Pseudomonas aeruginosa, possesses unusual genomic features. Front Microbiol 10. https://doi.org/10.3389/fmicb.2019.02772
Iyer LM, Anantharaman V, Krishnan A, Maxwell Burroughs A, Aravind L (2021) Jumbo phages: acomparative genomic overview of core functions and adaptions for biological conflicts. Viruses 13. https://doi.org/10.3390/v13010063
Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S (2018) High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun 9. https://doi.org/10.1038/s41467-018-07641-9
Jurczak-Kurek A, Gasior T, Nejman-Faleńczyk B, Bloch S, Dydecka A, Topka G, Necel A, Jakubowska-Deredas M, Narajczyk M, Richert M, Mieszkowska A, Wróbel B, Wȩgrzyn G, Wȩgrzyn A (2016) Biodiversity of bacteriophages: morphological and biological properties of a large group of phages isolated from urban sewage. Sci Rep 6. https://doi.org/10.1038/srep34338
Käll L, Krogh A, Sonnhammer ELL (2007) Advantages of combined transmembrane topology and signal peptide prediction–the Phobius web server. Nucleic Acids Res 35:W429–W432. https://doi.org/10.1093/nar/gkm256
Kalvari I, Nawrocki EP, Ontiveros-Palacios N, Argasinska J, Lamkiewicz K, Marz M, Griffiths-Jones S, Toffano-Nioche C, Gautheret D, Weinberg Z, Rivas E, Eddy SR, Finn RD, Bateman A, Petrov AI (2021) Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res 49. https://doi.org/10.1093/nar/gkaa1047
Kang YJ, Yang DC, Kong L, Hou M, Meng YQ, Wei L, Gao G (2017) CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. https://doi.org/10.1093/nar/gkx428
Kaźmierczak Z, Górski A, Dabrowska K (2014) Facing antibiotic resistance:Staphylococcus aureus phages as a medical tool. Viruses 6. https://doi.org/10.3390/v6072551
Korn AM, Hillhouse AE, Sun L, Gill JJ (2021) Comparative genomics of three novel jumbo bacteriophages infectingStaphylococcus aureus. J Virol 95. https://doi.org/10.1128/jvi.02391-20
Krogh A, Larsson B, von Heijne G, Sonnhammer ELL (2001) Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J Mol Biol 305:567–580. https://doi.org/10.1006/jmbi.2000.4315
Kropinski AM (2018) Practical advice on the one-step growth curve. Methods Mol Biol. https://doi.org/10.1007/978-1-4939-7343-9_3
Kümmel J, Stessl B, Gonano M, Walcher G, Bereuter O, Fricker M, Grunert T, Wagner M, Ehling-Schulz M (2016) Staphylococcus aureus entrance into the Dairy Chain: tracking S. aureus from dairy cow to cheese. Front Microbiol 7. https://doi.org/10.3389/fmicb.2016.01603
Kwon J, Kim SG, Kim HJ, Giri SS, Kim SW, Lee S, bin, Park SC, (2020) Isolation and characterization of Salmonella jumbo-phage pSal-SNUABM-04. Viruses 13:27. https://doi.org/10.3390/v13010027
Laslett D, Canback B (2004) ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. https://doi.org/10.1093/nar/gkh152
Latka A, Maciejewska B, Majkowska-Skrobek G, Briers Y, Drulis-Kawa Z (2017) Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl Microbiol Biotechnol 101:3103–3119. https://doi.org/10.1007/s00253-017-8224-6
Lavigne R, Ceyssens PJ, Robben J (2009) Phage proteomics: applications of mass spectrometry. Methods Mol Biol 502. https://doi.org/10.1007/978-1-60327-565-1_14
Lavysh D, Sokolova M, Minakhin L, Yakunina M, Artamonova T, Kozyavkin S, Makarova KS, Koonin Ev, Severinov K (2016) The genome of AR9, a giant transducing Bacillus phage encoding two multisubunit RNA polymerases. Virology. https://doi.org/10.1016/j.virol.2016.04.030
Lee Y, Son B, Cha Y, Ryu S (2021) Characterization andgenomic analysis of PALS2, a novel Staphylococcus jumbo bacteriophage. Front Microbiol 12. https://doi.org/10.3389/fmicb.2021.622755
Letunic I, Bork P (2021) Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49. https://doi.org/10.1093/nar/gkab301
Liu M, Gingery M, Doulatov SR, Liu Y, Hodes A, Baker S, Davis P, Simmonds M, Churcher C, Mungall K, Quail MA, Preston A, Harvill ET, Maskell DJ, Eiserling FA, Parkhill J, Miller JF (2004) Genomic andgenetic analysis of Bordetella bacteriophages encoding reverse transcriptase-mediated tropism-switching cassettes. J Bacteriol 186. https://doi.org/10.1128/JB.186.5.1503-1517.2004
Lowe TM, Chan PP (2016) tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res 44:W54–W57. https://doi.org/10.1093/nar/gkw413
Ma Y, Wang C, Li Y, Li J, Wan Q, Chen J, Tay FR, Niu L (2020) Considerations and caveats in combating ESKAPE pathogens against nosocomial infections. Advanced Science 7. https://doi.org/10.1002/advs.201901872
Maciejewska B, Olszak T, Drulis-Kawa Z (2018) Applications of bacteriophages versus phage enzymes to combat and cure bacterial infections: an ambitious and also a realistic application? Appl Microbiol Biotechnol 102:2563–2581. https://doi.org/10.1007/s00253-018-8811-1
Maher RL, Morrical SW (2013) Coordinated binding of single-stranded and double-stranded DNA by UvsX recombinase. PLoS One 8. https://doi.org/10.1371/journal.pone.0066654
Malone LM, Warring SL, Jackson SA, Warnecke C, Gardner PP, Gumy LF, Fineran PC (2019) A jumbo phage that forms a nucleus-like structure evades CRISPR–Cas DNA targeting but is vulnerable to type III RNA-based immunity. Nat Microbiol 5:48–55. https://doi.org/10.1038/s41564-019-0612-5
Marquioni V, Rossi FPN, Mendonça DC, Martins LF, Behlau F, Setubal JC, da Silva AM, Novo-Mansur MTM (2022) Isolation and characterization of vB_XciM_LucasX, a new jumbo phage that infects Xanthomonas citri and Xanthomonas fuscans. PLoS One 17:e0266891. https://doi.org/10.1371/journal.pone.0266891
Meier-Kolthoff JP, Göker M (2017) VICTOR: genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 33. https://doi.org/10.1093/bioinformatics/btx440
Melo LDR, Brandão A, Akturk E, Santos SB, Azeredo J (2018) Characterization of a newStaphylococcus aureus Kayvirus harboring a lysin active against biofilms. Viruses 10. https://doi.org/10.3390/v10040182
Miller ES, Kutter E, Mosig G, Arisaka F, Kunisawa T, Rüger W (2003) Bacteriophage T4 genome. Microbiology and Molecular Biology Reviews 67. https://doi.org/10.1128/mmbr.67.1.86-156.2003
Mirzaei MK, Nilsson AS (2015) Isolation of phages for phage therapy: acomparison of spot tests and efficiency of plating analyses for determination of host range and efficacy. PLoS One 10. https://doi.org/10.1371/journal.pone.0118557
Moraru C, Varsani A, Kropinski AM (2020) VIRIDIC—A novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses 12. https://doi.org/10.3390/v12111268
Naknaen A, Suttinun O, Surachat K, Khan E, Pomwised R (2021) A novel jumbo phage PhiMa05 inhibits harmful Microcystis sp. front microbiol 12. https://doi.org/10.3389/fmicb.2021.660351
Nazir A, Ali A, Qing H, Tong Y (2021) Emerging aspects of jumbo bacteriophages. Infect Drug Resist 14. https://doi.org/10.2147/IDR.S330560
Nishimura Y, Yoshida T, Kuronishi M, Uehara H, Ogata H, Goto S (2017) ViPTree: the viral proteomic tree server. Bioinformatics 33. https://doi.org/10.1093/bioinformatics/btx157
Oliveira H, Costa AR, Konstantinides N, Ferreira A, Akturk E, Sillankorva S, Nemec A, Shneider M, Dötsch A, Azeredo J (2017) Ability of phages to infectAcinetobacter calcoaceticus-Acinetobacter baumannii complex species through acquisition of different pectate lyase depolymerase domains. Environ Microbiol 19. https://doi.org/10.1111/1462-2920.13970
Olszak T, Danis-Wlodarczyk K, Arabski M, Gula G, MacIejewska B, Wasik S, Lood C, Higgins G, Harvey BJ, Lavigne R, Drulis-Kawa Z (2019)Pseudomonas aeruginosa PA5oct jumbo phage impacts planktonic and biofilm population and reduces its host virulence. Viruses 11. https://doi.org/10.3390/v11121089
Park S, Ronholm J (2021) Staphylococcus aureus in agriculture: Lessons in evolution from a multispecies pathogen. Clin Microbiol Rev 34. https://doi.org/10.1128/CMR.00182-20
Paysan-Lafosse T, Blum M, Chuguransky S, Grego T, Pinto BL, Salazar GA, Bileschi ML, Bork P, Bridge A, Colwell L, Gough J, Haft DH, Letunić I, Marchler-Bauer A, Mi H, Natale DA, Orengo CA, Pandurangan AP, Rivoire C, Sigrist CJA, Sillitoe I, Thanki N, Thomas PD, Tosatto SCE, Wu CH, Bateman A (2023) InterPro in 2022. Nucleic Acids Res 51:D418–D427. https://doi.org/10.1093/nar/gkac993
Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. https://doi.org/10.1038/nmeth.1701
Sakr A, Brégeon F, Mège J-L, Rolain J-M, Blin O (2018) Staphylococcus aureus nasal colonization: An update on mechanisms, epidemiology, risk factors, and subsequent infections. Front Microbiol 9. https://doi.org/10.3389/fmicb.2018.02419
Schmelcher M, Powell AM, Becker SC, Camp MJ, Donovan DM (2012) Chimeric phage lysins act synergistically with lysostaphin to kill mastitis-causingStaphylococcus aureus in murine mammary glands. Appl Environ Microbiol 78. https://doi.org/10.1128/AEM.07050-11
Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30. https://doi.org/10.1093/bioinformatics/btu153
Sharma R, Pielstick BA, Bell KA, Nieman TB, Stubbs OA, Yeates EL, Baltrus DA, Grose JH (2019) A novel, highly related jumbo family of bacteriophages that were isolated againstErwinia. Front Microbiol 10. https://doi.org/10.3389/fmicb.2019.01533
Shkoporov AN, Khokhlova E v., Fitzgerald CB, Stockdale SR, Draper LA, Ross RP, Hill C (2018) ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat Commun 9. https://doi.org/10.1038/s41467-018-07225-7
Snider J, Houry WA (2006) MoxR AAA+ ATPases: anovel family of molecular chaperones? J Struct Biol 156. https://doi.org/10.1016/j.jsb.2006.02.009
Söding J, Biegert A, Lupas AN (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33:W244–W248. https://doi.org/10.1093/nar/gki408
Sokolova M, Borukhov S, Lavysh D, Artamonova T, Khodorkovskii M, Severinov K (2017) A non-canonical multisubunit RNA polymerase encoded by the AR9 phage recognizes the template strand of its uracil-containing promoters. Nucleic Acids Res 45. https://doi.org/10.1093/nar/gkx264
Sokolova ML, Misovetc I, Severinov KV (2020) Multisubunit RNA Polymerases of Jumbo Bacteriophages. Viruses 12:1064. https://doi.org/10.3390/v12101064
Sørensen MCH, Vitt A, Neve H, Soverini M, Ahern SJ, Klumpp J, Brøndsted L (2021)Campylobacter phages use hypermutable polyG tracts to create phenotypic diversity and evade bacterial resistance. Cell Rep 35. https://doi.org/10.1016/j.celrep.2021.109214
Stothard P (2000) The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28. https://doi.org/10.2144/00286ir01
Sulis G, Sayood S, Katukoori S, Bollam N, George I, Yaeger LH, Chavez MA, Tetteh E, Yarrabelli S, Pulcini C, Harbarth S, Mertz D, Sharland M, Moja L, Huttner B, Gandra S (2022) Exposure to World Health Organization’s AWaRe antibiotics and isolation of multidrug resistant bacteria: a systematic review and meta-analysis. Clin Microbiol Infect. https://doi.org/10.1016/j.cmi.2022.03.014
Thanki AM, Brown N, Millard AD, Clokie MRJ (2019) Genomic characterization of jumbo Salmonella phages that effectively target United Kingdom pig-associated Salmonella serotypes. Front Microbiol 10. https://doi.org/10.3389/fmicb.2019.01491
Turner D, Kropinski AM, Adriaenssens EM (2021) A roadmap for genome-based phage taxonomy. Viruses 13. https://doi.org/10.3390/v13030506
Tynecki P, Guziński A, Kazimierczak J, Jadczuk M, Dastych J, Onisko A (2020) PhageAI - bacteriophage life cycle recognition with machine learning and natural language processing. bioRxiv. https://doi.org/10.1101/2020.07.11.198606
Uchiyama J, Takemura-Uchiyama I, Sakaguchi Y, Gamoh K, Kato SI, Daibata M, Ujihara T, Misawa N, Matsuzaki S (2014) Intragenus generalized transduction in Staphylococcus spp. by a novel giant phage. ISME J 8. https://doi.org/10.1038/ismej.2014.29
Viklund H, Bernsel A, Skwark M, Elofsson A (2008) SPOCTOPUS: A combined predictor of signal peptides and membrane protein topology. Bioinformatics 24. https://doi.org/10.1093/bioinformatics/btn550
Viklund H, Elofsson A (2008) OCTOPUS: Improving topology prediction by two-track ANN-based preference scores and an extended topological grammar. Bioinformatics 24. https://doi.org/10.1093/bioinformatics/btn221
Villarroel J, Kleinheinz KA, Jurtz VI, Zschach H, Lund O, Nielsen M, Larsen MV (2016) HostPhinder: aphage host prediction tool. Viruses 8. https://doi.org/10.3390/v8050116
Weigel C, Seitz H (2006) Bacteriophage replication modules. FEMS Microbiol Rev 30:321–381. https://doi.org/10.1111/j.1574-6976.2006.00015.x
Weinheimer AR, Aylward FO (2022) Infection strategy and biogeography distinguish cosmopolitan groups of marine jumbo bacteriophages. ISME J. https://doi.org/10.1038/s41396-022-01214-x
Winther AR, Narvhus JA, Smistad M, da Silva Duarte V, Bombelli A, Porcellato D (2022) Longitudinal dynamics of the bovine udder microbiota. Anim Microbiome 4:26. https://doi.org/10.1186/s42523-022-00177-w
Wojtus JK, Frampton RA, Warring S, Hendrickson H, Fineran PC (2019) Genome sequence of a jumbo bacteriophage that infects the kiwifruit phytopathogen Pseudomonas syringae pv. actinidiae. Microbiol Resour Announc. https://doi.org/10.1128/mra.00224-19
Xu L, Dong Z, Fang L, Luo Y, Wei Z, Guo H, Zhang G, Gu YQ, Coleman-Derr D, Xia Q, Wang Y (2019) OrthoVenn2: aweb server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res 47. https://doi.org/10.1093/nar/gkz333
Yuan Y, Gao M (2017) Jumbo bacteriophages: an overview. Front Microbiol 8. https://doi.org/10.3389/fmicb.2017.00403
Zhang B, Sun H, Zhao F, Wang Q, Pan Q, Tong Y, Ren H (2022) Characterization andgenomic analysis of a novel jumbo bacteriophage vB_StaM_SA1 infecting Staphylococcus aureus with two lysins. Front Microbiol 13. https://doi.org/10.3389/fmicb.2022.856473
Acknowledgements
We are grateful to the financial support of the following Brazilian agencies: Fundação de Amparo à Pesquisa do Estado de Minas Gerais (Fapemig), Coordenacão de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Financiadora de Estudos e Projetos (Finep), and Sistema Nacional de Laboratórios em Nanotecnologias (SisNANO)/Ministério da ciência, tecnologia e Informação (MCTI).
Funding
Open access funding provided by Norwegian University of Life Sciences This study was supported by the Portuguese Foundation for Science and Technology (FCT) under the scope of the strategic funding of UIDB/04469/2020 unit, and Project PTDC/BIA-MIC/2312/2020. Luís D. R. Melo acknowledges funding from the FCT through the Scientific Employment Stimulus Program (2021.00221.CEECIND).
Author information
Authors and Affiliations
Contributions
VSD, LDRM, SBS, and AMK conceived and designed the research. VSD, JDS, MFX, ISP, MSV, and JJFS conducted the experiments. RSD, DP, SOP, JDS, VSD, LDRM, SBS, and AMK analyzed data. JDS and VSD wrote the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval
This article does not contain any studies with animals performed by any of the authors.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
da Silva, J.D., Melo, L.D.R., Santos, S.B. et al. Genomic and proteomic characterization of vB_SauM-UFV_DC4, a novel Staphylococcus jumbo phage. Appl Microbiol Biotechnol 107, 7231–7250 (2023). https://doi.org/10.1007/s00253-023-12743-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-023-12743-6