
Abstract
Single-cell genomics and transcriptomics can provide reliable context for assembled genome fragments and gene expression activity on the level of individual prokaryotic genomes. These methods are rapidly emerging as an essential complement to cultivation-based, metagenomics, metatranscriptomics, and microbial community-focused research approaches by allowing direct access to information from individual microorganisms, even from deep-branching phylogenetic groups that currently lack cultured representatives. Their integration and binning with environmental ‘omics data already provides unprecedented insights into microbial diversity and metabolic potential, enabling us to provide information on individual organisms and the structure and dynamics of natural microbial populations in complex environments. This review highlights the pitfalls and recent advances in the field of single-cell omics and its importance in microbiological and biotechnological studies.
Key points
• Single-cell omics expands the tree of life through the discovery of novel organisms, genes, and metabolic pathways.
• Disadvantages of metagenome-assembled genomes are overcome by single-cell omics.
• Functional analysis of single cells explores the heterogeneity of gene expression.
• Technical challenges still limit this field, thus prompting new method developments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microbial dark matter (MDM)
Prokaryotic microorganisms harbor an enormous potential for biotechnological applications, such as novel natural product discovery, bioenergy production, and bioremediation of harmful anthropogenic-introduced substances (Singh et al. 2014; Katz and Baltz 2016; Kumar and Kumar 2017; Mullis et al. 2019; Stincone and Brandelli 2020). However, despite their global quantity and importance, it is estimated that over 99% of all microbial species remain uncultured or even completely uncharacterized (Wu et al. 2009; McDonald et al. 2012; Hug et al. 2016; Lloyd et al. 2018). These microorganisms are referred to as microbial dark matter (MDM). Very often, attempts to grow microbes under laboratory conditions fail, or they grow too slowly to obtain sufficient biomass for analysis. Although new cultivation methods have been recently developed (Nichols et al. 2010; Sherpa et al. 2015; Wiegand et al. 2020), genome sequences for the vast majority of prokaryotes have been inaccessible, obscuring our knowledge of microbial diversity, metabolism, (eco)physiology, inter-organism interactions, and adaptive evolution.
Cultivation-independent approaches for exploring MDM
The development of shotgun sequencing of DNA extracted from environmental samples—so-called metagenomics—has provided extensive gene content information from natural microbial communities (Temperton and Giovannoni 2012; Hedlund et al. 2014; Anantharaman et al. 2016; Vollmers et al. 2017a; Quince et al. 2017). Unfortunately, assembling individual discrete genomes from metagenomics data (so-called MAGs, metagenome-assembled genomes) is often difficult and rather costly, especially for high diversity samples or low abundant organisms (Gilbert and Dupont 2011; Narasingarao et al. 2012; Iverson et al. 2012). Binning algorithms, which group contigs and assign them to operational taxonomic units (OTUs), have massively improved over the past years, but MAGs are still likely consensus genomes derived from multiple cells that are equally abundant and share high-homologous regions (Venter et al. 2004; Rusch et al. 2010; Iverson et al. 2012). Large structural variants (SVs) such as genome rearrangements, gene insertions, duplications, or losses which can vary in highly homologous cells due to mutations, horizontal gene transfer (HGT), and recombination cannot be analyzed and mobile genomic elements such as plasmids or transposable elements cannot be accurately binned (Fraser et al. 2007; Vergin et al. 2007; Martinez-Garcia et al. 2012b; Shapiro et al. 2012; Dam et al. 2020). In recent years, long-read sequencing technologies such as Pacific Biosciences’ (PacBio) single-molecule real-time (SMRT) sequencing and Oxford Nanopore Technologies’ (ONT) nanopore sequencing have improved the detection and study of large SVs that lead to heterozygosity on the strain level (Amarasinghe et al. 2020; Ho et al. 2020). However, these technologies are sometimes still limited in use due to the lack of efficient extraction of high-molecular-weight nucleic acids and required library input amounts, as well as higher error rates (Frank et al. 2016; Amarasinghe et al. 2020).
Analyzing the RNA of a microbial community—so-called metatranscriptomics—can provide information of the actual gene expression, function, and metabolic activity given enough sequencing depth and repetition of sequenced samples. It even allows the determination of relative expression levels between active genes, making it a useful complement to metagenomics for studying ecological and metabolic interactions within microbial communities (Shi et al. 2011; Mason et al. 2012). However, even isogenic microbial populations show substantial cell-to-cell heterogeneities in transcriptional activity (Roberfroid et al. 2016; González-Cabaleiro et al. 2017). Since genomic heterogeneity is a common characteristic of microorganisms to adapt to environments with constant and rapid changes, standard metagenomic and metatranscriptomic analyses alone are not always well suited to obtain genomes of different species and deliver unequivocal information about the organization and activity of discovered genes within genomes, making it difficult to predict functional genes, metabolic pathways, and potential benefits of microbial species. This holds especially true for microorganisms with low abundance in a habitat since the quality of genome reconstruction is largely dependent on sequence coverage for assembly as well as coverage covariance-based binning (Albertsen et al. 2013; Vollmers et al. 2017b; Dam et al. 2020).
Omics on the single-cell level
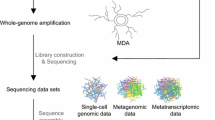
Single-cell genomics (SCG) has emerged as a powerful technique to overcome disadvantages of genome reconstruction from metagenomes, by enabling genome analysis of an individual prokaryotic cell, referred to as single amplified genomes (SAGs) (Woyke et al. 2010; Stepanauskas 2012; Blainey 2013; Kaster et al. 2014; Woyke et al. 2017; Sewell et al. 2017; Piel and Cahn 2019). It allows for the physical separation of single cells directly from environmental samples, followed by sequencing and assembly of their individual genomes and consists of a series of integrated processes (Fig. 1). Since its first application on microorganisms in 2005 (Raghunathan et al. 2005), SCG has become a powerful tool for studying uncultivable organisms and delineating complex populations. An increasing number of SAGs are available from public databases such as the National Center for Biotechnology Information (NCBI) GenBank (Sayers et al. 2020), and/or the Joint Genome Institute Genomes OnLine Database (GOLD) (Mukherjee et al. 2019), which includes all data from Integrated Microbial Genomes (IMG). As of July 2020, over 9000 SAG sequencing projects have been deposited in GOLD (Mukherjee et al. 2019), of which many are classified as uncultured and potentially novel taxonomic groups (Swan et al. 2011; McLean et al. 2013; Hedlund et al. 2014; Becraft et al. 2016; León-Zayas et al. 2017; Landry et al. 2017) (Fig. 2). Recently, a new reference database containing over 12,000 SAGs from the euphotic ocean was published (Pachiadaki et al. 2019), greatly expanding our knowledge on the diversity and complexity of marine microorganisms. SAGs of some species are however still difficult to obtain, for example, minority members in a microbial community or anaerobic organisms. This could be due to the difficulty in labeling them (Müller and Nebe-Von-Caron 2010) and/or the increased chance that they will lyse when exposed to oxygen, risking the DNA to be damaged during lysis steps prior to the downstream applications of SCG. In addition, the success of genome recovery from single cells, in general, varies widely from 0% to a finished genome.

Overview of a single-cell omics pipeline including respective challenges and advancements. a Unless analyzed immediately, environmental samples require deep-freezing in the presence of a cryoprotectant that preserves the integrity of the cell and its nucleic acids. b Cells are stained with a fluorescent dye, such as DAPI or SYBR® Green; however, they can also be specifically labeled. c Physical isolation of a single cell is typically performed by fluorescence-activated cell sorting (FACS) into multi-well plates. d After separation, the single cells are lysed to release their DNA and RNA. Today, most cell lysis in SC omics relies on an alkaline solution. e For single-cell transcriptomics (SCT), RNA must first be converted to double-stranded cDNA via reverse transcription prior to amplification. f Since a typical prokaryotic cell only contains a few fg grams of DNA and RNA, whole genome or transcriptome amplification (WGA/WTA) is needed by a factor of about 106. Multiple displacement amplification (MDA) is the most widely used reaction. g PCR is used to screen for specific loci after amplification, usually with broad eubacterial and archaeal 16S rRNA primers, followed by Sanger sequencing. h After DNA or RNA library preparation, next-generation sequencing technologies like Illumina, Oxford Nanopore, or PacBio are available for genome/transcriptome sequencing. i After quality assessment, trimming, and/or normalization of the sequencing reads, bioinformatics tools can conduct the assembly, ORF calling, and annotation of the genes, as well as pathway reconstruction and gene comparisons. Created with BioRender

Cladogram of prokaryotes (Bacteria and Archaea) showing the relative proportions of isolate genomes, single-amplified genomes (SAGs), and metagenome-assembled genomes (MAGs) that make up the total number of genomes in each phylum. The taxonomy is based on National Center for Biotechnology Information (NCBI) (Sayers et al. 2020). Total genome numbers for each phylum are shown at the top of each bar. Data extracted from the Genomes OnLine Database (GOLD) in July 2020 (Mukherjee et al. 2019). Cladogram created with Interactive Tree of Life (iTOL) version 5 (Letunic and Bork 2019). -proteo -proteobacteria. Asgard Lokiarchaeota-Thorarchaeota-Odinarchaeota-Heimdallarchaeota. DPANN Diapherotrites-Parvarchaeota-Aenigmarchaeota-Nanoarchaeota-Nanohaloarchaeota. TACK Thaumarchaeota-Aigarchaeota-Crenarchaeota-Korarchaeota. FCB Fibrobacteres-Chlorobi-Bacteroidetes. PVC Planctomycetes-Verrucomicrobia-Chlamydiae. CPR Candidate Phyla Radiation
Technical problems, recent advances, and future challenges
In general, the workflow involves (a) sampling and preservation, (b) non-specific staining of microbial populations, (c) cell sorting, (d) cell lysis, (e, f) whole genome amplification (WGA), (g) screening for SAGs of interest, and (h, i) sequencing and analysis (Fig. 1). For cell isolation, several methods can be used such as microfluidics, micromanipulation, and—conventionally—fluorescence-activated cell sorting (FACS). Microfluidic- and optofluidic-based systems (Marcy et al. 2007b; Marcy et al. 2007a; Blainey et al. 2011; Landry et al. 2013; Gole et al. 2013; Xu et al. 2016; Riba et al. 2016; Lan et al. 2017), as well as micromanipulation (Woyke et al. 2010; Grindberg et al. 2011), have the advantage of sorting cells based on their morphology and less physical stress applied to the cell. Most setups even allow for cell separation, lysis, and amplification performed in one closed system at nanoliter or even picoliter volumes (Marcy et al. 2007b; Marcy et al. 2007a; Blainey et al. 2011; Landry et al. 2013; Xu et al. 2016); however, some limitations remain in cell throughput and the successful recovery of non-contaminated amplified products. FACS has become the most commonly used method for separation due to its high speed and flexibility with the use of different fluorescence signals (Stepanauskas and Sieracki 2007; Rinke et al. 2014; Woyke et al. 2017). The main limitations of this technology include potential contamination due to the open-plate workflow, the inability to microscopically examine cells, the strong physical stress applied to cells, and further miniaturization of downstream reaction volumes (Blainey 2013; Woyke et al. 2017).
Cell lysis efficiency plays an important role in the success rate of SCG owing to the challenging natural diversity of microbial cell walls. Environmental sample preservation has a significant impact on lysis, and conditions have to be optimized for each organism of interest (Stepanauskas 2012; Rinke et al. 2014). Cell lysis methods need to accomplish releasing DNA and RNA from the cell while maintaining its integrity (Clingenpeel et al. 2014a) and must not interfere with downstream reactions. Alkaline lysis is the most widely used method; however, more efficient lysis of cells from complex communities can be accomplished by using a combination of different lysis methods (i.e., physical-like freeze-thaw cycles, chemical, or enzymatic) (Hall et al. 2013; He et al. 2016; Stepanauskas et al. 2017).
In general, WGA via multiple displacement amplification (MDA) to obtain a sufficient quantity of genomic DNA for sequencing remains the major limitation of the SCG pipeline. In addition to the high costs, this method often results in incomplete and uneven genome amplification and is biased against high GC regions of the genome (Lasken and Stockwell 2007; Lasken 2009; Sabina and Leamon 2015). Therefore, the average completeness of genomes obtained by SCG is often lower than that of MAGs from the same sample (Blainey et al. 2011; Bowers et al. 2017; Landry et al. 2017; Alneberg et al. 2018; Dam et al. 2020). One of the main sequencing limitations is due to MDA artifacts such as chimeras, uncontrolled bias, and non-specific products (Lasken and Stockwell 2007; Sabina and Leamon 2015). Amplification bias is thought to occur predominantly by random mechanisms, as sequences that are over-represented in one MDA reaction can be under-represented in another. However, there are some reproducible differences in the average level of representation of different sequences, and certain sequences are not amplified at all for unknown reasons (Dean et al. 2001; Lasken and Stockwell 2007; Lasken 2009). Several approaches can be taken to lessen the amplification bias of the MDA reaction such as reducing the reaction volume to improve the specificity of the reaction (Marcy et al. 2007a; Landry et al. 2013; Leung et al. 2016; Yu et al. 2017; Lan et al. 2017), post-amplification endonuclease treatment to reduce chimeric sequences (Zhang et al. 2006), and post-amplification normalization by nuclease degradation of dsDNA to reduce most abundant sequences (Rodrigue et al. 2009). To overcome the problem of amplification bias against high GC content, a more thermotolerant phi29 DNA polymerase was recently described, resulting in higher quality draft genomes of single sorted cells (Stepanauskas et al. 2017). The hybrid amplification method called multiple annealing and looping-based amplification cycles (MALBAC) can also amplify high GC regions and showed amplification of more than 90% of the genome of single human cells; however, the method is very costly and not that suitable for high-throughput applications for prokaryotes. Many amplification methods have been compared in the past, but the general consensus is that the amplification method of choice is dependent on the questions being asked as the methods each have their distinct advantages and disadvantages (De Bourcy et al. 2014; Huang et al. 2015; Estévez-Gómez et al. 2018).
Low throughput for low abundant cells: phylogeny-driven targeted labeling
Due to the amplification bias, statistically less than 30% of the 16S rRNA gene gets amplified (Swan et al. 2011). This makes screening challenging, especially for rare organisms of the population or cells that are hard to lyse. Like metagenomics, SCG is therefore often not a cost-efficient approach to unravel microbial community members which are not abundant, especially in complex environments. Given the limitations of cultivation-dependent and cultivation-independent techniques, minority members of microbial communities are therefore often overlooked and understudied. Nevertheless, they may still play important roles in many biogeochemical processes (e.g., due to high enzyme affinities to certain substrates) or might have biotechnological relevance (Frias-Lopez et al. 2008; Shi et al. 2011; Pratscher et al. 2018). Generating samples enriched for the specific microorganism of interest will increase the likelihood of actually sorting those cells and obtaining positive amplifications, which will ultimately reduce experimental costs.
Recently, Dam et al. (2020) implemented a fluorescent in situ hybridization (FISH) labeling, coupled with targeted cell sorting to obtain genomes of low abundant community members of the phylum Chloroflexi using a modified protocol (Podar et al. 2007; Yilmaz et al. 2010; Haroon et al. 2013). Usually, fluorescent labeling of bacterial cells requires fixatives such as paraformaldehyde to increase the fluorescent signal by allowing stronger permeabilization of the cell membrane and penetration of the probe. However, since the process weakens the cell wall, it can lead to the lysis of the labeled cells during the sorting process. Furthermore, paraformaldehyde compromises the downstream applications for SCG, namely, the amplification of the genomic DNA via MDA (Clingenpeel et al. 2014b; Doud and Woyke 2017). Dam et al. (2020) demonstrated that an in-solution, fixation-free FISH protocol allowed phylogenetic labeling of targeted cells to remain intact during the sorting process and that their fluorescent signals were sufficiently high for multiple sorts. This was achieved by longer hybridization times and higher probe concentrations to overcome the problem of low cell membrane permeability when no additional fixatives and lysing agents could be used.
Furthermore, catalyzed reporter deposition (CARD)-FISH (Pernthaler et al. 2002) could overcome weak FISH signals, which occur if the cells are in a low activity state, as is the case for most samples from oligotrophic environments. In some cases, the enrichment of low abundant bacterial cells after CARD-FISH and FACS doubled in comparison to the original sample (Wallner et al. 1997; Sekar et al. 2004). Through the use of CARD-FISH and laser microdissection, researchers were able to confirm the previously unknown identity of a bacterium that produces the cytotoxin calyculin, by using the biosynthetic gene cluster as the probe (Wakimoto et al. 2014). However, like with traditional FISH, the paraformaldehyde used is not compatible with whole genome amplification, which is why multiple cells were needed to avoid the amplification step. To combat this issue but still maintain comparable signal intensities, a modified hybridization chain reaction (HCR)-FISH protocol which does not require paraformaldehyde as a fixative was recently used and combined with FACS and MDA to successfully enrich bacteria from an environmental sample (Yamaguchi et al. 2015b; Yamaguchi et al. 2015a; Grieb et al. 2020).
Another labeling tool for gene-specific targeting is recognition of individual genes (RING)-FISH which can be combined with targeted cell sorting (Pratscher et al. 2009). This technique was used to target a previously unculturable methane oxidizer in soils—USCα—(Kolb et al. 2005), where the specific methane monooxygenase enzyme (pMMO) was known, but the 16S rRNA gene sequence was not. Application of a fluorescently labeled suicide substrate for pMMO was used to specifically sort the labeled cells via FACS and obtain the 16S rRNA sequence. For the first time, 16S rRNA FISH allowed then a direct link to the high-affinity activity of methane oxidation by USCα cells in situ. Analysis of the global biogeography of this group by 16S rRNA then further revealed its presence in previously unrecognized habitats, such as subterranean and volcanic biofilm environments, indicating a potential role of these environments in the biological sink for atmospheric methane.
Cherry-picking: function-driven single-cell genomics
Recently, different techniques for functional-driven SCG have been used to select and characterize single cells based on a specific functional trait prior to and in conjunction with WGS (Woyke and Jarett 2015; Lee et al. 2015; Doud and Woyke 2017; Couradeau et al. 2019; Hatzenpichler et al. 2020). Some of these methods have provided important ecological and biotechnological findings, which help to expand our knowledge on previously unknown gene functions (Woyke et al. 2018).
Raman micro-spectroscopy in combination with cell sorting, called Raman activated cell ejection (RACE), has been used to provide phenotypical and biochemical information of single cells without damaging the cells (Huang et al. 2004; Huang et al. 2010; Li et al. 2012). Song et al. (2017b) reported the first use of RACE coupled with SCG to detect novel carotenoid and isoprenoid synthesizing bacteria from the Red Sea as a label-free approach (Song et al. 2017b). They later showed that stable isotope labeling with D2O in combination with RACE and WGA could detect antimicrobial-resistant bacteria in the Thames River (Song et al. 2017a).
Substrate analog probing (SAP) uses synthetic molecules that are similar to naturally occurring molecules to study substrate uptake in organisms. This method has been combined with FACS and SCG by the use of fluorescently labeled substrates to uncover the significance of polysaccharide degradation by Verrucomicrobia (Martinez-Garcia et al. 2012a) and a novel cellulose-degrading bacterium from the candidate phylum Goldbacteria with less than 1% abundance in the sample (Doud et al. 2019). However, the greater use of this method is mainly limited by the uncertainty in how/if the fluorescent tag interferes with enzyme-substrate binding (Hatzenpichler et al. 2020). Bio-orthogonal non-canonical amino acid tagging (BONCAT) is one approach that has been used to overcome this limitation because detecting the uptake of these molecules is independent of the labeling approach. In general, this method uses synthetic amino acids that are taken up by bacterial cells so that they are incorporated into newly synthesized proteins which then become fluorescent due to azide-alkyne click chemistry (Mahdavi et al. 2014; Hatzenpichler et al. 2014). BONCAT has been previously used to identify pathogenic bacteria and their secreted proteins (Mahdavi et al. 2014; Sherratt et al. 2017). Recently, this targeting method was also shown to be compatible with FACS and MDA which enabled the authors to sort methane-oxidizing archaea-bacterial consortia from deep sea sediments and identify in situ activity and novel interactions between these organisms (Hatzenpichler et al. 2016). However, the current limitations, such as signal strength, substrate analog specificity, and quantification of activity rates, restrict the widespread use of these methods and need to be further improved for high-throughput applications (Doud and Woyke 2017; Hatzenpichler et al. 2020).
Viral-host interactions in single-cells
Studying viral-host interactions via metagenomics has proven to be difficult due to the naturally high diversity in most environments. Studies comparing MAGs and SAGs from the same samples find that the MAGs do not contain a significant portion of genes, indicative of prophage infection found within the SAGs, even within largely incomplete SAGSs (Labonté et al. 2015; Dam et al. 2020). SCG has therefore revolutionized the study of viruses in many environments, as well as their interactions within individual prokaryotic cells. In the marine subsurface, where cell abundance and cell activity is low, a particular Firmicute, Desulforudis, interestingly undergoes frequent HGT and viral infections (Labonté et al. 2015). Also, up to 32% of genes that were recovered from SAGs, were not present in the metagenome of the same sample. Munson-Mcgee et al. (2018) studied viral-host interactions within a hot spring environment and found that 60% of SAGs contained at least one virus, showcasing the broad host range of viruses (Munson-Mcgee et al. 2018). The authors also estimated that for low-level completed SAGs, only a 300 bp viral sequence minimum is needed to detect a virus-host interaction. By directly sequencing single viral genomes (vSAGs) from the Mediterranean Sea and the Atlantic Ocean, highly abundant and ecologically significant viruses could be discovered, which had been previously missed through metagenomics (Martinez-Hernandez et al. 2017). When the vSAGs were compared to public metagenome datasets, they found that their vSAGs best represent the diversity of viruses in global oceans. Notably, a large portion of viral diversity has still gone undiscovered in the human virome. De La Cruz Peña et al. (2018) found that most publicly available viral isolates did not match vSAGs in the oral virome. The authors also found that the viral community was highly variable and interpersonal between patients because none of the vSAGs obtained in this study were found in all viromes.
The next step: single-cell transcriptomics (SCT)
Although SCG has been successfully applied in microbial ecology studies, it only provides information related to genetic architecture and metabolic potential of a cell, but does not reveal the actual gene expression and activity of a cell under different environmental conditions. This, and the fact that the transcriptomic profile of individual cells can vary even if they are genetically homogenous underlines the necessity of studying the transcriptomic profile at single-cell resolution (de Jager and Siezen 2011; Kanter and Kalisky 2015). However, compared to genomic analysis, transcriptomic analysis for microorganisms at the single-cell level is much more challenging. Single-cell transcriptomics (SCT) in eukaryotic organisms has already been successfully applied (Kanter and Kalisky 2015; Tsang et al. 2015; Fan et al. 2015). Cells are first individually sorted and lysed; then, the RNA is converted to cDNA with oligo (dT) primers and amplified prior to library preparation and sequencing. Whilst a single eukaryotic cell contains up to 50 picograms (pg) of total RNA (Kang et al. 2011), the RNA concentration in prokaryotes is typically in the low femtogram range (1–5 fg) (Kang et al. 2011; Wang et al. 2015). Unfortunately, only a small fraction of total RNA is mRNA, as rRNA and tRNA molecules usually represent over 90% of the total RNA. Since most SCT methods target polyadenylated transcripts, the lack of a polyA tail on most mRNAs in prokaryotes means that the large fraction of rRNA and tRNA cannot be depleted using the methods established for eukaryotic transcriptomics. Furthermore, this structural difference of the mRNA molecule and the fact that prokaryotic mRNA can be polycistronic explains why not all methods for eukaryotic SCT can be applied to microorganisms. In addition, mRNA molecules have low stability over time. Nevertheless, in the recent past, several new approaches have been developed for SCT studies in prokaryotic cells (Kang et al. 2011; Kang et al. 2015; Wang et al. 2015; Liu et al. 2019). One method developed a rolling circle amplification (RCA) protocol that used the phi29 polymerase and random primers to amplify transcripts from single Burkholderia thailandensis cells (Kang et al. 2011). Even though this method was only used with microarray analysis, the authors report that including an rRNA depletion step would make this protocol compatible with next-generation sequencing (Kang et al. 2011; Kang et al. 2015). Another method combined SCT with RNA-seq, where RNA-based, single primer isothermal amplification (Ribo-SPIA) was used to amplify transcripts from Synechocystis sp. PCC 6803 (Wang et al. 2015). The authors used this method to show cell heterogeneity under environmental stress due to nitrogen starvation and successfully detected up to 98% of all putative genes within the genome. The most recently published method used RNA-seq in combination with whole transcriptome amplification (WTA) with the REPLI-g WTA kit (Qiagen) to amplify total RNA from Porphyromonas somerae (Liu et al. 2019). However, only 0.3% of rRNA transcripts were detected, which might have been due to the slow growth rate of P. somerae.
In general, these studies already provide the basic framework for SCT; however, they have so far only been carried out on model organisms, and many technical challenges still remain such as the inefficient transcriptome coverage, biased amplification, and throughput (Picelli 2017; Chen et al. 2017; Zhang et al. 2018). Direct-RNA methods that do not require pre-amplification, e.g., Oxford Nanopore sequencing systems, would be ideal to cut out amplification bias (Garalde et al. 2018). This method however still requires at least 100 nanograms (ng) of RNA and is based on polyadenylated mRNA transcripts. Nevertheless, it is possible that future improvements will decrease the input amount of RNA needed, making the technique more applicable for single-cell analysis.
Back to multiple cells?
Until improved technologies become available, completing the assembly of the microbial genomes and transcriptomes for most species likely requires a combination of approaches such as reconstruction from multiple homologous cells if available or additional metagenomic data. Metagenomic reads and contigs can be used to recover missing genomic regions and greatly improve the SAG assemblies (Dodsworth et al. 2013; Nurk et al. 2013; Becraft et al. 2016; Xu and Zhao 2018). Likewise, metagenomic bins can also be improved by SAG datasets (Pachiadaki et al. 2019). Another approach is the so-called mini-metagenomic approach, where a few cells (50–1000) are sorted into one well (Yu et al. 2017; Schulz et al. 2018; Alteio et al. 2020; Geesink et al. 2020; Grieb et al. 2020). Sorting multiple cells of the same type by targeted labeling might circumvent the necessity of additional amplification of DNA/RNA, therefore allowing single genomes/transcriptomes to be bioinformatically binned from those mini-metagenomes. This not only reduces the cost but also lessens the chance for contamination and increases genome coverage (Podar et al. 2007; Yu et al. 2017; Grieb et al. 2020) and might be a sufficient solution for some biological questions.
Outlook
The integration and binning of SCG with environmental omics data can already provide unprecedented insights into microbial diversity, metabolic features, and processes. SCG and SCT have tremendous potential to bring more clarity to the contested discussion about the nature of prokaryotic species and their metabolic potentials which will enable us to provide information on individual organisms and the structure and dynamics of natural microbial populations in all kinds of environments. SC omics holds great promise in microbial microevolution studies, industrial bioprospecting, and selection of suitable heterologous expression systems, with potential for novel and environmentally responsible energy solutions, bioremediation of toxins, and natural products. It can also help to identify evolutionary histories, inter-organismal interactions, and quantitative information on genomic variability in natural microbial populations. High-quality results currently require state-of-the-art instrumentation such as cell sorters, robotic liquid handlers, DNA sequencers, and a cleanroom. The amount of data to obtain new biological insights requires high-performance information technology with computer clusters for conducting assemblies and analyses of the sequences. Undoubtedly, the future for SC omics is exceptionally bright, but significant technical and conceptual challenges still have to be resolved. In order to expand the range of microorganisms amenable to SC omics, improvements of the methods are necessary. The rapidly growing number of applications that use limited quantities of DNA in genomic detection and analysis highlights the continued need for innovation in the field of DNA amplification and library preparation, e.g., nanoscale library preparation via microfluidics (Kim et al. 2017). This may also result in the omission of the amplification step, or at least its minimization, resulting in less biased sequencing results. In addition, further automation and miniaturization of sequencing processes along with the development of new labeling methods are required for emerging research needs. This will help create a more phylogenetically balanced representation of genomes in databases, which will ultimately help to improve models for computational gene annotation and taxonomic assignment (Woyke et al. 2009; de Jager and Siezen 2011; Wang and Navin 2015). It will also help with the cultivation of microorganisms of interest through facilitating a more informed development of culturing methods by revealing the nutritional needs and metabolic capabilities of the organism (Pratscher et al. 2018).
References
Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH (2013) Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31:533–538. https://doi.org/10.1038/nbt.2579
Alneberg J, Karlsson CMG, Divne AM, Bergin C, Homa F, Lindh MV, Hugerth LW, Ettema TJG, Bertilsson S, Andersson AF, Pinhassi J (2018) Genomes from uncultivated prokaryotes: a comparison of metagenome-assembled and single-amplified genomes 06 biological sciences 0604 genetics. Microbiome 6. https://doi.org/10.1186/s40168-018-0550-0
Alteio LV, Schulz F, Seshadri R, Varghese N, Rodriguez-Reillo W, Ryan E, Goudeau D, Eichorst SA, Malmstrom RR, Bowers RM, Katz LA, Blanchard JL, Woyke T (2020) Complementary metagenomic approaches improve reconstruction of microbial diversity in a forest soil. mSystems 5:768–787. https://doi.org/10.1128/msystems.00768-19
Amarasinghe SL, Su S, Dong X, Zappia L, Ritchie ME, Gouil Q (2020) Opportunities and challenges in long-read sequencing data analysis. Genome Biol 21:1–16
Anantharaman K, Brown CT, Hug LA, Sharon I, Castelle CJ, Probst AJ, Thomas BC, Singh A, Wilkins MJ, Karaoz U, Brodie EL, Williams KH, Hubbard SS, Banfield JF (2016) Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Commun 7:1–11. https://doi.org/10.1038/ncomms13219
Becraft ED, Dodsworth JA, Murugapiran SK, Ohlsson JI, Briggs BR, Kanbar J, De Vlaminck I, Quake SR, Dong H, Hedlund BP, Swingley WD (2016) Single-cell-genomics-facilitated read binning of candidate phylum EM19 genomes from geothermal spring metagenomes. Am Soc Microbiol 82:992–1003. https://doi.org/10.1128/AEM.03140-15
Blainey PC (2013) The future is now: single-cell genomics of bacteria and archaea. FEMS Microbiol Rev 37:407–427
Blainey PC, Mosier AC, Potanina A, Francis CA, Quake SR (2011) Genome of a low-salinity ammonia-oxidizing archaeon determined by single-cell and metagenomic analysis. PLoS One 6:6. https://doi.org/10.1371/journal.pone.0016626
Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, Schulz F, Jarett J, Rivers AR, Eloe-Fadrosh EA, Tringe SG, Ivanova NN, Copeland A, Clum A, Becraft ED, Malmstrom RR, Birren B, Podar M, Bork P, Weinstock GM, Garrity GM, Dodsworth JA, Yooseph S, Sutton G, Glöckner FO, Gilbert JA, Nelson WC, Hallam SJ, Jungbluth SP, Ettema TJG, Tighe S, Konstantinidis KT, Liu WT, Baker BJ, Rattei T, Eisen JA, Hedlund B, McMahon KD, Fierer N, Knight R, Finn R, Cochrane G, Karsch-Mizrachi I, Tyson GW, Rinke C, Lapidus A, Meyer F, Yilmaz P, Parks DH, Eren AM, Schriml L, Banfield JF, Hugenholtz P, Woyke T (2017) Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol 35:725–731
Chen Z, Chen L, Zhang W (2017) Tools for genomic and transcriptomic analysis of microbes at single-cell level. Front Microbiol 8
Clingenpeel S, Clum A, Schwientek P, Rinke C, Woyke T (2014a) Reconstructing each cell’s genome within complex microbial communities-dream or reality? Front Microbiol 5. https://doi.org/10.3389/fmicb.2014.00771
Clingenpeel S, Schwientek P, Hugenholtz P, Woyke T (2014b) Effects of sample treatments on genome recovery via single-cell genomics. ISME J 8:2546–2549. https://doi.org/10.1038/ismej.2014.92
Couradeau E, Sasse J, Goudeau D, Nath N, Hazen TC, Bowen BP, Chakraborty R, Malmstrom RR, Northen TR (2019) Probing the active fraction of soil microbiomes using BONCAT-FACS. Nat Commun 10:1–10. https://doi.org/10.1038/s41467-019-10542-0
Dam HT, Vollmers J, Sobol MS, Cabezas A, Kaster A-K (2020) Targeted cell sorting combined with single cell genomics captures low abundant microbial dark matter with higher sensitivity than metagenomics. Front Microbiol 11:1377. https://doi.org/10.3389/fmicb.2020.01377
De Bourcy CFA, De Vlaminck I, Kanbar JN, Wang J, Gawad C, Quake SR (2014) A quantitative comparison of single-cell whole genome amplification methods. PLoS One 9:e105585. https://doi.org/10.1371/journal.pone.0105585
de Jager V, Siezen RJ (2011) Single-cell genomics: unravelling the genomes of unculturable microorganisms. Microb Biotechnol 4:431–437. https://doi.org/10.1111/j.1751-7915.2011.00271.x
De La Cruz Peña MJ, Martinez-Hernandez F, Garcia-Heredia I, Gomez ML, Fornas Ò, Martinez-Garcia M (2018) Deciphering the human virome with single-virus genomics and metagenomics. Viruses 10. https://doi.org/10.3390/v10030113
Dean FB, Nelson JR, Giesler TL, Lasken RS (2001) Rapid amplification of plasmid and phage DNA using Phi29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res 11:1095–1099. https://doi.org/10.1101/gr.180501
Dodsworth JA, Blainey PC, Murugapiran SK, Swingley WD, Ross CA, Tringe SG, Chain PSG, Scholz MB, Lo CC, Raymond J, Quake SR, Hedlund BP (2013) Single-cell and metagenomic analyses indicate a fermentative and saccharolytic lifestyle for members of the OP9 lineage. Nat Commun 4:1854. https://doi.org/10.1038/ncomms2884
Doud DFR, Woyke T (2017) Novel approaches in function-driven single-cell genomics. FEMS Microbiol Rev 41:538–548. https://doi.org/10.1093/femsre/fux009
Doud DFR, Bowers RM, Schulz F, De Raad M, Deng K, Tarver A, Glasgow E, Vander Meulen K, Fox B, Deutsch S, Yoshikuni Y, Northen T, Hedlund BP, Singer SW, Ivanova N, Woyke T (2019) Function-driven single-cell genomics uncovers cellulose-degrading bacteria from the rare biosphere. ISME J 14:659–675. https://doi.org/10.1038/s41396-019-0557-y
Estévez-Gómez N, Prieto T, Guillaumet-Adkins A, Heyn H, Prado-López S, Posada D (2018) Comparison of single-cell whole-genome amplification strategies. bioRxiv:443754. https://doi.org/10.1101/443754
Fan X, Zhang X, Wu X, Guo H, Hu Y, Tang F, Huang Y (2015) Single-cell RNA-seq transcriptome analysis of linear and circular RNAs in mouse preimplantation embryos. Genome Biol 16:148. https://doi.org/10.1186/s13059-015-0706-1
Frank JA, Pan Y, Tooming-Klunderud A, Eijsink VGH, McHardy AC, Nederbragt AJ, Pope PB (2016) Improved metagenome assemblies and taxonomic binning using long-read circular consensus sequence data. Sci Rep 6:1–10. https://doi.org/10.1038/srep25373
Fraser C, Hanage WP, Spratt BG (2007) Recombination and the nature of bacterial speciation. Science (80-. ) 315:476–480
Frias-Lopez J, Shi Y, Tyson GW, Coleman ML, Schuster SC, Chisholm SW, Delong EF (2008) Microbial community gene expression in ocean surface waters. Proc Natl Acad Sci U S A 105:3805–3810. https://doi.org/10.1073/pnas.0708897105
Garalde DR, Snell EA, Jachimowicz D, Sipos B, Lloyd JH, Bruce M, Pantic N, Admassu T, James P, Warland A, Jordan M, Ciccone J, Serra S, Keenan J, Martin S, McNeill L, Wallace EJ, Jayasinghe L, Wright C, Blasco J, Young S, Brocklebank D, Juul S, Clarke J, Heron AJ, Turner DJ (2018) Highly parallel direct RNA sequencing on an array of nanopores. Nat Methods 15:201–206. https://doi.org/10.1038/nmeth.4577
Geesink P, Wegner CE, Probst AJ, Herrmann M, Dam HT, Kaster AK, Küsel K (2020) Genome-inferred spatio-temporal resolution of an uncultivated Roizmanbacterium reveals its ecological preferences in groundwater. Environ Microbiol 22:726–737. https://doi.org/10.1111/1462-2920.14865
Gilbert JA, Dupont CL (2011) Microbial metagenomics: beyond the genome. Annu Rev Mar Sci 3:347–371. https://doi.org/10.1146/annurev-marine-120709-142811
Gole J, Gore A, Richards A, Chiu YJ, Fung HL, Bushman D, Chiang HI, Chun J, Lo YH, Zhang K (2013) Massively parallel polymerase cloning and genome sequencing of single cells using nanoliter microwells. Nat Biotechnol 31:1126–1132. https://doi.org/10.1038/nbt.2720
González-Cabaleiro R, Mitchell AM, Smith W, Wipat A, Ofiteru ID (2017) Heterogeneity in pure microbial systems: experimental measurements and modeling. Front Microbiol 8:1813
Grieb A, Bowers RM, Oggerin M, Goudeau D, Lee J, Malmstrom RR, Woyke T, Fuchs BM (2020) A pipeline for targeted metagenomics of environmental bacteria. Microbiome 8:21. https://doi.org/10.1186/s40168-020-0790-7
Grindberg RV, Ishoey T, Brinza D, Esquenazi E, Coates RC, Ting LW, Gerwick L, Dorrestein PC, Pevzner P, Lasken R, Gerwick WH (2011) Single cell genome amplification accelerates identification of the apratoxin biosynthetic pathway from a complex microbial assemblage. PLoS One 6:e18565. https://doi.org/10.1371/journal.pone.0018565
Hall E, Kim S, Appadoo V, Zare R (2013) Lysis of a single Cyanobacterium for whole genome amplification. Micromachines 4:321–332. https://doi.org/10.3390/mi4030321
Haroon MF, Skennerton CT, Steen JA, Lachner N, Hugenholtz P, Tyson GW (2013) In-solution fluorescence in situ hybridization and fluorescence-activated cell sorting for single cell and population genome recovery, 1st edn. Elsevier Inc.
Hatzenpichler R, Scheller S, Tavormina PL, Babin BM, Tirrell DA, Orphan VJ (2014) In situ visualization of newly synthesized proteins in environmental microbes using amino acid tagging and click chemistry. Environ Microbiol 16:2568–2590. https://doi.org/10.1111/1462-2920.12436
Hatzenpichler R, Connon SA, Goudeau D, Malmstrom RR, Woyke T, Orphan VJ (2016) Visualizing in situ translational activity for identifying and sorting slow-growing archaeal - bacterial consortia. Proc Natl Acad Sci U S A 113:E4069–E4078. https://doi.org/10.1073/pnas.1603757113
Hatzenpichler R, Krukenberg V, Spietz RL, Jay ZJ (2020) Next-generation physiology approaches to study microbiome function at single cell level. Nat Rev Microbiol 1–16
He J, Du S, Tan X, Arefin A, Han CS (2016) Improved lysis of single bacterial cells by a modified alkaline-thermal shock procedure. Biotechniques 60:60–135. https://doi.org/10.2144/000114389
Hedlund BP, Dodsworth JA, Murugapiran SK, Rinke C, Woyke T (2014) Impact of single-cell genomics and metagenomics on the emerging view of extremophile microbial dark matter. Extremophiles 18:865–875
Ho SS, Urban AE, Mills RE (2020) Structural variation in the sequencing era. Nat Rev Genet 21:171–189
Huang WE, Griffiths RI, Thompson IP, Bailey MJ, Whiteley AS (2004) Raman microscopic analysis of single microbial cells. Anal Chem 76:4452–4458. https://doi.org/10.1021/ac049753k
Huang WE, Li M, Jarvis RM, Goodacre R, Banwart SA (2010) Shining light on the microbial world the application of Raman microspectroscopy. Adv Appl Microbiol 70:153–186
Huang L, Ma F, Chapman A, Lu S, Xie XS (2015) Single-cell whole-genome amplification and sequencing: methodology and applications. Annu Rev Genomics Hum Genet 16:79–102. https://doi.org/10.1146/annurev-genom-090413-025352
Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, Castelle CJ, Butterfield CN, Hernsdorf AW, Amano Y, Ise K (2016) A new view of the tree of life. Nat Microbiol 1:16048
Iverson V, Morris RM, Frazar CD, Berthiaume CT, Morales RL, Armbrust EV (2012) Untangling genomes from metagenomes: revealing an uncultured class of marine euryarchaeota. Science 335:587–590. https://doi.org/10.1126/science.1212665
Kang Y, Norris MH, Zarzycki-Siek J, Nierman WC, Donachie SP, Hoang TT (2011) Transcript amplification from single bacterium for transcriptome analysis. Genome Res 21:925–935. https://doi.org/10.1101/gr.116103.110
Kang Y, McMillan I, Norris MH, Hoang TT (2015) Single prokaryotic cell isolation and total transcript amplification protocol for transcriptomic analysis. Nat Protoc 10:974–984. https://doi.org/10.1038/nprot.2015.058
Kanter I, Kalisky T (2015) Single cell transcriptomics: methods and applications. Front Oncol 5
Kaster AK, Mayer-Blackwell K, Pasarelli B, Spormann AM (2014) Single cell genomic study of dehalococcoidetes species from deep-sea sediments of the peruvian margin. ISME J 8:1831–1842. https://doi.org/10.1038/ismej.2014.24
Katz L, Baltz RH (2016) Natural product discovery: past, present, and future. J Ind Microbiol Biotechnol 43:155–176
Kim S, De Jonghe J, Kulesa AB, Feldman D, Vatanen T, Bhattacharyya RP, Berdy B, Gomez J, Nolan J, Epstein S, Blainey PC (2017) High-throughput automated microfluidic sample preparation for accurate microbial genomics. Nat Commun 8:1–10. https://doi.org/10.1038/ncomms13919
Kolb S, Knief C, Dunfield PF, Conrad R (2005) Abundance and activity of uncultured methanotrophic bacteria involved in the consumption of atmospheric methane in two forest soils. Environ Microbiol 7:1150–1161. https://doi.org/10.1111/j.1462-2920.2005.00791.x
Kumar R, Kumar P (2017) Future microbial applications for bioenergy production: a perspective. Front Microbiol 8:450. https://doi.org/10.3389/fmicb.2017.00450
Labonté JM, Field EK, Lau M, Chivian D, Van Heerden E, Wommack KE, Kieft TL, Onstott TC, Stepanauskas R (2015) Single cell genomics indicates horizontal gene transfer and viral infections in a deep subsurface Firmicutes population. Front Microbiol 6:349. https://doi.org/10.3389/fmicb.2015.00349
Lan F, Demaree B, Ahmed N, Abate AR (2017) Single-cell genome sequencing at ultra-high-throughput with microfluidic droplet barcoding. Nat Biotechnol 35:640–646. https://doi.org/10.1038/nbt.3880
Landry ZC, Giovanonni SJ, Quake SR, Blainey PC (2013) Optofluidic cell selection from complex microbial communities for single-genome analysis. In: Methods in Enzymology. Academic Press Inc., pp 61–90
Landry Z, Swan BK, Herndl GJ, Stepanauskas R, Giovannoni SJ (2017) SAR202 genomes from the dark ocean predict pathways for the oxidation of recalcitrant dissolved organic matter. MBio 8:e00413–e00417. https://doi.org/10.1128/MBIO.00413-17
Lasken RS (2009) Genomic DNA amplification by the multiple displacement amplification (MDA) method. Biochem Soc Trans 37:450–453. https://doi.org/10.1042/BST0370450
Lasken RS, Stockwell TB (2007) Mechanism of chimera formation during the multiple displacement amplification reaction. BMC Biotechnol 7:19. https://doi.org/10.1186/1472-6750-7-19
Lee PKH, Men Y, Wang S, He J, Alvarez-Cohen L (2015) Development of a fluorescence-activated cell sorting method coupled with whole genome amplification to analyze minority and trace Dehalococcoides genomes in microbial communities. Environ Sci Technol 49:1585–1593. https://doi.org/10.1021/es503888y
León-Zayas R, Peoples L, Biddle JF, Podell S, Novotny M, Cameron J, Lasken RS, Bartlett DH (2017) The metabolic potential of the single cell genomes obtained from the Challenger Deep, Mariana Trench within the candidate superphylum Parcubacteria (OD1). Environ Microbiol 19:2769–2784. https://doi.org/10.1111/1462-2920.13789
Letunic I, Bork P (2019) Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47:W256–W259. https://doi.org/10.1093/nar/gkz239
Leung K, Klaus A, Lin BK, Laks E, Biele J, Lai D, Bashashati A, Huang YF, Aniba R, Moksa M, Steif A, Mes-Masson AM, Hirst M, Shah SP, Aparicio S, Hansen CL (2016) Robust high-performance nanoliter-volume single-cell multiple displacement amplification on planar substrates. Proc Natl Acad Sci U S A 113:8484–8489. https://doi.org/10.1073/pnas.1520964113
Li M, Xu J, Romero-Gonzalez M, Banwart SA, Huang WE (2012) Single cell Raman spectroscopy for cell sorting and imaging. Curr Opin Biotechnol 23:56–63
Liu Y, Jeraldo P, Jang JS, Eckloff B, Jen J, Walther-Antonio M (2019) Bacterial single cell whole transcriptome amplification in microfluidic platform shows putative gene expression heterogeneity. Anal Chem 91:8036–8044. https://doi.org/10.1021/acs.analchem.8b04773
Lloyd KG, Steen AD, Ladau J, Yin J, Crosby L (2018) Phylogenetically novel uncultured microbial cells dominate earth microbiomes. mSystems 3, 3. https://doi.org/10.1128/msystems.00055-18
Mahdavi A, Szychowski J, Ngo JT, Sweredoski MJ, Graham RLJ, Hess S, Schneewind O, Mazmanian SK, Tirrell DA (2014) Identification of secreted bacterial proteins by noncanonical amino acid tagging. Proc Natl Acad Sci U S A 111:433–438. https://doi.org/10.1073/pnas.1301740111
Marcy Y, Ishoey T, Lasken RS, Stockwell TB, Walenz BP, Halpern AL, Beeson KY, Goldberg SMD, Quake SR (2007a) Nanoliter reactors improve multiple displacement amplification of genomes from single cells. PLoS Genet 3:1702–1708. https://doi.org/10.1371/journal.pgen.0030155
Marcy Y, Ouverney C, Bik EM, Lösekann T, Ivanova N, Martin HG, Szeto E, Platt D, Hugenholtz P, Relman DA, Quake SR (2007b) Dissecting biological “dark matter” with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proc Natl Acad Sci U S A 104:11889–11894. https://doi.org/10.1073/pnas.0704662104
Martinez-Garcia M, Brazel DM, Swan BK, Arnosti C, Chain PSG, Reitenga KG, Xie G, Poulton NJ, Gomez ML, Masland DED, Thompson B, Bellows WK, Ziervogel K, Lo C-C, Ahmed S, Gleasner CD, Detter CJ, Stepanauskas R (2012a) Capturing single cell genomes of active polysaccharide degraders: an unexpected contribution of Verrucomicrobia. PLoS One 7:e35314. https://doi.org/10.1371/journal.pone.0035314
Martinez-Garcia M, Swan BK, Poulton NJ, Gomez ML, Masland D, Sieracki ME, Stepanauskas R (2012b) High-throughput single-cell sequencing identifies photoheterotrophs and chemoautotrophs in freshwater bacterioplankton. ISME J 6:113–123. https://doi.org/10.1038/ismej.2011.84
Martinez-Hernandez F, Fornas O, Lluesma Gomez M, Bolduc B, De La Cruz Peña MJ, Martínez JM, Anton J, Gasol JM, Rosselli R, Rodriguez-Valera F, Sullivan MB, Acinas SG, Martinez-Garcia M (2017) Single-virus genomics reveals hidden cosmopolitan and abundant viruses. Nat Commun 8:1–13. https://doi.org/10.1038/ncomms15892
Mason OU, Hazen TC, Borglin S, Chain PSG, Dubinsky EA, Fortney JL, Han J, Holman HYN, Hultman J, Lamendella R, MacKelprang R, Malfatti S, Tom LM, Tringe SG, Woyke T, Zhou J, Rubin EM, Jansson JK (2012) Metagenome, metatranscriptome and single-cell sequencing reveal microbial response to Deepwater Horizon oil spill. ISME J 6:1715–1727. https://doi.org/10.1038/ismej.2012.59
McDonald D, Price MN, Goodrich J, Nawrocki EP, Desantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P (2012) An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6:610–618. https://doi.org/10.1038/ismej.2011.139
McLean JS, Lombardo MJ, Badger JH, Edlund A, Novotny M, Yee-Greenbaum J, Vyahhi N, Hall AP, Yang Y, Dupont CL, Ziegler MG, Chitsaz H, Allen AE, Yooseph S, Tesler G, Pevzner PA, Friedman RM, Nealson KH, Venter JC, Lasken RS (2013) Candidate phylum TM6 genome recovered from a hospital sink biofilm provides genomic insights into this uncultivated phylum. Proc Natl Acad Sci U S A 110:E2390–E2399. https://doi.org/10.1073/pnas.1219809110
Mukherjee S, Stamatis D, Bertsch J, Ovchinnikova G, Katta HY, Mojica A, Chen I-MA, Kyrpides NC, Reddy T (2019) Genomes OnLine database (GOLD) v.7: updates and new features. Nucleic Acids Res 47:D649–D659. https://doi.org/10.1093/nar/gky977
Müller S, Nebe-Von-Caron G (2010) Functional single-cell analyses: flow cytometry and cell sorting of microbial populations and communities. FEMS Microbiol Rev 34:554–587
Mullis MM, Rambo IM, Baker BJ, Reese BK (2019) Diversity, ecology, and prevalence of antimicrobials in nature. Front Microbiol 10:2518. https://doi.org/10.3389/fmicb.2019.02518
Munson-Mcgee JH, Peng S, Dewerff S, Stepanauskas R, Whitaker RJ, Weitz JS, Young MJ (2018) A virus or more in (nearly) every cell: ubiquitous networks of virus-host interactions in extreme environments. ISME J 12:1706–1714. https://doi.org/10.1038/s41396-018-0071-7
Narasingarao P, Podell S, Ugalde JA, Brochier-Armanet C, Emerson JB, Brocks JJ, Heidelberg KB, Banfield JF, Allen EE (2012) De novo metagenomic assembly reveals abundant novel major lineage of Archaea in hypersaline microbial communities. ISME J 6:81–93. https://doi.org/10.1038/ismej.2011.78
Nichols D, Cahoon N, Trakhtenberg EM, Pham L, Mehta A, Belanger A, Kanigan T, Lewis K, Epstein SS (2010) Use of ichip for high-throughput in situ cultivation of uncultivable microbial species▽. Appl Environ Microbiol 76:2445–2450. https://doi.org/10.1128/AEM.01754-09
Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, Prjibelski AD, Pyshkin A, Sirotkin A, Sirotkin Y, Stepanauskas R, Clingenpeel SR, Woyke T, Mclean JS, Lasken R, Tesler G, Alekseyev MA, Pevzner PA (2013) Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol 20:714–737. https://doi.org/10.1089/cmb.2013.0084
Pachiadaki MG, Brown JM, Brown J, Bezuidt O, Berube PM, Biller SJ, Poulton NJ, Burkart MD, La Clair JJ, Chisholm SW, Stepanauskas R (2019) Charting the complexity of the marine microbiome through single-cell genomics. Cell 179:1623-1635.e11. https://doi.org/10.1016/j.cell.2019.11.017
Pernthaler A, Pernthaler J, Amann R (2002) Fluorescence in situ hybridization and catalyzed reporter deposition for the identification of marine bacteria. Appl Environ Microbiol 68:3094–3101. https://doi.org/10.1128/AEM.68.6.3094-3101.2002
Picelli S (2017) Single-cell RNA-sequencing: the future of genome biology is now. RNA Biol 14:637–650. https://doi.org/10.1080/15476286.2016.1201618
Piel J, Cahn J (2019) Opening up the single-cell toolbox for microbial natural products research. Angew Chemie Int Ed. https://doi.org/10.1002/anie.201900532
Podar M, Abulencia CB, Walcher M, Hutchison D, Zengler K, Garcia JA, Holland T, Cotton D, Hauser L, Keller M (2007) Targeted access to the genomes of low-abundance organisms in complex microbial communities. Appl Environ Microbiol 73:3205–3214. https://doi.org/10.1128/AEM.02985-06
Pratscher J, Stichternoth C, Fichtl K, Schleifer KH, Braker G (2009) Application of recognition of individual genes-fluorescence in situ hybridization (RING-FISH) to detect nitrite reductase genes (nirK) of denitrifiers in pure cultures and environmental samples. Appl Environ Microbiol 75:802–810. https://doi.org/10.1128/AEM.01992-08
Pratscher J, Vollmers J, Wiegand S, Dumont MG, Kaster A-K (2018) Unravelling the identity, metabolic potential and global biogeography of the atmospheric methane-oxidizing upland soil cluster α. Environ Microbiol 20:1016–1029. https://doi.org/10.1111/1462-2920.14036
Quince C, Walker AW, Simpson JT, Loman NJ, Segata N (2017) Shotgun metagenomics, from sampling to analysis. Nat Biotechnol 35:833–844
Raghunathan A, Ferguson HR, Bornarth CJ, Song W, Driscoll M, Lasken RS (2005) Genomic DNA amplification from a single bacterium. Appl Environ Microbiol 71:3342–3347. https://doi.org/10.1128/AEM.71.6.3342-3347.2005
Riba J, Gleichmann T, Zimmermann S, Zengerle R, Koltay P (2016) Label-free isolation and deposition of single bacterial cells from heterogeneous samples for clonal culturing. Sci Rep 6:1–9. https://doi.org/10.1038/srep32837
Rinke C, Lee J, Nath N, Goudeau D, Thompson B, Poulton N, Dmitrieff E, Malmstrom R, Stepanauskas R, Woyke T (2014) Obtaining genomes from uncultivated environmental microorganisms using FACS-based single-cell genomics. Nat Protoc 9:1038–1048. https://doi.org/10.1038/nprot.2014.067
Roberfroid S, Vanderleyden J, Steenackers H (2016) Gene expression variability in clonal populations: causes and consequences. Crit Rev Microbiol 42:969–984
Rodrigue S, Malmstrom RR, Berlin AM, Birren BW, Henn MR, Chisholm SW (2009) Whole genome amplification and de novo assembly of single bacterial cells. PLoS One 4:e6864. https://doi.org/10.1371/journal.pone.0006864
Rusch DB, Martiny AC, Dupont CL, Halpern AL, Venter JC (2010) Characterization of Prochlorococcus clades from iron-depleted oceanic regions. Proc Natl Acad Sci U S A 107:16184–16189. https://doi.org/10.1073/pnas.1009513107
Sabina J, Leamon JH (2015) Bias in whole genome amplification: causes and considerations. In: Kroneis T (ed) Methods in molecular biology. Humana press Inc., pp 15–41
Sayers EW, Beck J, Brister JR, Bolton EE, Canese K, Comeau DC, Funk K, Ketter A, Kim S, Kimchi A, Kitts PA, Kuznetsov A, Lathrop S, Lu Z, McGarvey K, Madden TL, Murphy TD, O’Leary N, Phan L, Schneider VA, Thibaud-Nissen F, Trawick BW, Pruitt KD, Ostell J (2020) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 48:D9–D16. https://doi.org/10.1093/nar/gkz899
Schulz F, Alteio L, Goudeau D, Ryan EM, Yu FB, Malmstrom RR, Blanchard J, Woyke T (2018) Hidden diversity of soil giant viruses. Nat Commun 9:4881. https://doi.org/10.1038/s41467-018-07335-2
Sekar R, Fuchs BM, Amann R, Pernthaler J (2004) Flow sorting of marine bacterioplankton after fluorescence in situ hybridization. Appl Environ Microbiol 70:6210–6219. https://doi.org/10.1128/AEM.70.10.6210-6219.2004
Sewell HL, Kaster A-K, Spormann AM (2017) Homoacetogenesis in Deep-Sea Chloroflexi, as inferred by single-cell genomics, provides a link to reductive dehalogenation in terrestrial Dehalococcoidetes. MBio 8:e02022–e02017. https://doi.org/10.1128/mBio.02022-17
Shapiro BJ, Friedman J, Cordero OX, Preheim SP, Timberlake SC, Szabó G, Polz MF, Alm EJ (2012) Population genomics of early events in the ecological differentiation of bacteria. Science (80- ) 335:48–51. https://doi.org/10.1126/science.1218198
Sherpa R, Reese C, Aliabadi HM (2015) Application of iChip to grow “uncultivable” microorganisms and its impact on antibiotic discovery. Pharm Fac Artic Res
Sherratt AR, Rouleau Y, Luebbert C, Strmiskova M, Veres T, Bidawid S, Corneau N, Pezacki JP (2017) Rapid screening and identification of living pathogenic organisms via optimized bioorthogonal non-canonical amino acid tagging. Cell Chem Biol 24:1048-1055.e3. https://doi.org/10.1016/j.chembiol.2017.06.016
Shi Y, Tyson GW, Eppley JM, DeLong EF (2011) Integrated metatranscriptomic and metagenomic analyses of stratified microbial assemblages in the open ocean. ISME J 5:999–1013. https://doi.org/10.1038/ismej.2010.189
Singh R, Singh P, Sharma R (2014) Microorganism as a tool of bioremediation technology for cleaning environment: a review
Song Y, Cui L, López JÁS, Xu J, Zhu YG, Thompson IP, Huang WE (2017a) Raman-deuterium isotope probing for in-situ identification of antimicrobial resistant bacteria in Thames River. Sci Rep 7:1–10. https://doi.org/10.1038/s41598-017-16898-x
Song Y, Kaster AK, Vollmers J, Song Y, Davison PA, Frentrup M, Preston GM, Thompson IP, Murrell JC, Yin H, Hunter CN, Huang WE (2017b) Single-cell genomics based on Raman sorting reveals novel carotenoid-containing bacteria in the Red Sea. Microb Biotechnol 10:125–137. https://doi.org/10.1111/1751-7915.12420
Stepanauskas R (2012) Single cell genomics: an individual look at microbes. Curr Opin Microbiol 15:613–620
Stepanauskas R, Sieracki ME (2007) Matching phylogeny and metabolism in the uncultured marine bacteria, one cell at a time. Proc Natl Acad Sci U S A 104:9052–9057. https://doi.org/10.1073/pnas.0700496104
Stepanauskas R, Fergusson EA, Brown J, Poulton NJ, Tupper B, Labonté JM, Becraft ED, Brown JM, Pachiadaki MG, Povilaitis T, Thompson BP, Mascena CJ, Bellows WK, Lubys A (2017) Improved genome recovery and integrated cell-size analyses of individual uncultured microbial cells and viral particles. Nat Commun 8:8. https://doi.org/10.1038/s41467-017-00128-z
Stincone P, Brandelli A (2020) Marine bacteria as source of antimicrobial compounds. Crit Rev Biotechnol 40:306–319
Swan BK, Martinez-Garcia M, Preston CM, Sczyrba A, Woyke T, Lamy D, Reinthaler T, Poulton NJ, Masland EDP, Gomez ML, Sieracki ME, DeLong EF, Herndl GJ, Stepanauskas R (2011) Potential for chemolithoautotrophy among ubiquitous bacteria lineages in the dark ocean. Science 333:1296–1300. https://doi.org/10.1126/science.1203690
Temperton B, Giovannoni SJ (2012) Metagenomics: microbial diversity through a scratched lens. Curr Opin Microbiol 15:605–612
Tsang JCH, Yu Y, Burke S, Buettner F, Wang C, Kolodziejczyk AA, Teichmann SA, Lu L, Liu P (2015) Single-cell transcriptomic reconstruction reveals cell cycle and multi-lineage differentiation defects in Bcl11a-deficient hematopoietic stem cells. Genome Biol 16:178. https://doi.org/10.1186/s13059-015-0739-5
Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, Wu D, Paulsen I, Nelson KE, Nelson W, Fouts DE, Levy S, Knap AH, Lomas MW, Nealson K, White O, Peterson J, Hoffman J, Parsons R, Baden-Tillson H, Pfannkoch C, Rogers YH, Smith HO (2004) Environmental genome shotgun sequencing of the Sargasso Sea. Science (80- ) 304:66–74. https://doi.org/10.1126/science.1093857
Vergin KL, Tripp HJ, Wilhelm LJ, Denver DR, Rappé MS, Giovannoni SJ (2007) High intraspecific recombination rate in a native population of Candidatus Pelagibacter ubique (SAR11). Environ Microbiol 9:2430–2440. https://doi.org/10.1111/j.1462-2920.2007.01361.x
Vollmers J, Frentrup M, Rast P, Jogler C, Kaster AK (2017a) Untangling genomes of novel Planctomycetal and Verrucomicrobial species from monterey bay kelp forest metagenomes by refined binning. Front Microbiol 8:8. https://doi.org/10.3389/fmicb.2017.00472
Vollmers J, Wiegand S, Kaster AK (2017b) Comparing and evaluating metagenome assembly tools from a microbiologist’s perspective - not only size matters!
Wakimoto T, Egami Y, Nakashima Y, Wakimoto Y, Mori T, Awakawa T, Ito T, Kenmoku H, Asakawa Y, Piel J, Abe I (2014) Calyculin biogenesis from a pyrophosphate protoxin produced by a sponge symbiont. Nat Chem Biol 10:648–655. https://doi.org/10.1038/nchembio.1573
Wallner G, Fuchs B, Spring S, Beisker W, Amann R (1997) Flow sorting of microorganisms for molecular analysis. Appl Environ Microbiol 63:4223–4231. https://doi.org/10.1128/aem.63.11.4223-4231.1997
Wang Y, Navin NE (2015) Advances and applications of single-cell sequencing technologies. Mol Cell 58:598–609
Wang J, Chen L, Chen Z, Zhang W (2015) RNA-seq based transcriptomic analysis of single bacterial cells. Integr Biol 7:1466–1476. https://doi.org/10.1039/C5IB00191A
Wiegand S, Jogler M, Boedeker C, Pinto D, Vollmers J, Rivas-Marín E, Kohn T, Peeters SH, Heuer A, Rast P, Oberbeckmann S, Bunk B, Jeske O, Meyerdierks A, Storesund JE, Kallscheuer N, Lücker S, Lage OM, Pohl T, Merkel BJ, Hornburger P, Müller RW, Brümmer F, Labrenz M, Spormann AM, Op den Camp HJM, Overmann J, Amann R, Jetten MSM, Mascher T, Medema MH, Devos DP, Kaster AK, Øvreås L, Rohde M, Galperin MY, Jogler C (2020) Cultivation and functional characterization of 79 planctomycetes uncovers their unique biology. Nat Microbiol 5:126–140. https://doi.org/10.1038/s41564-019-0588-1
Woyke T, Jarett J (2015) Function-driven single-cell genomics. Microb Biotechnol 8:38–39. https://doi.org/10.1111/1751-7915.12247
Woyke T, Xie G, Copeland A, González JM, Han C (2009) Assembling the marine metagenome, one cell at a time. PLoS One 4:5299. https://doi.org/10.1371/journal.pone.0005299
Woyke T, Tighe D, Mavromatis K, Clum A, Copeland A, Schackwitz W, Lapidus A, Wu D, Mccutcheon JP, Mcdonald BR, Moran NA, Bristow J, Cheng JF (2010) One bacterial cell, one complete genome. PLoS One 5:e10314. https://doi.org/10.1371/journal.pone.0010314
Woyke T, Doud DFR, Schulz F (2017) The trajectory of microbial single-cell sequencing. Nat Methods 14:1045–1054. https://doi.org/10.1038/nmeth.4469
Woyke T, Doud DF, Eloe-Fadrosh EA (2018) Genomes from uncultivated microorganisms a brief history of microbial genomics technical aspects for accessing genomes from uncultivated microorganisms. https://doi.org/10.1016/B978-0-12-809633-8.90682-4
Wu D, Raymond J, Wu M, Chatterji S, Ren Q, Graham JE, Bryant DA, Robb F, Colman A, Tallon LJ, Badger JH, Madupu R, Ward NL, Eisen JA (2009) Complete genome sequence of the aerobic CO-oxidizing thermophile Thermomicrobium roseum. PLoS One 4:e4207. https://doi.org/10.1371/journal.pone.0004207
Xu Y, Zhao F (2018) Single-cell metagenomics: challenges and applications. Protein Cell 9:501–510
Xu L, Brito IL, Alm EJ, Blainey PC (2016) Virtual microfluidics for digital quantification and single-cell sequencing. Nat Methods 13:759–762. https://doi.org/10.1038/nmeth.3955
Yamaguchi T, Fuchs BM, Amann R, Kawakami S, Kubota K, Hatamoto M, Yamaguchi T (2015a) Rapid and sensitive identification of marine bacteria by an improved in situ DNA hybridization chain reaction (quickHCR-FISH). Syst Appl Microbiol 38:400–405. https://doi.org/10.1016/j.syapm.2015.06.007
Yamaguchi T, Kawakami S, Hatamoto M, Imachi H, Takahashi M, Araki N, Yamaguchi T, Kubota K (2015b) In situ DNA-hybridization chain reaction (HCR): a facilitated in situ HCR system for the detection of environmental microorganisms. Environ Microbiol 17:2532–2541. https://doi.org/10.1111/1462-2920.12745
Yilmaz S, Haroon MF, Rabkin BA, Tyson GW, Hugenholtz P (2010) Fixation-free fluorescence in situ hybridization for targeted enrichment of microbial populations. ISME J 4:1352–1356. https://doi.org/10.1038/ismej.2010.73
Yu FB, Blainey PC, Schulz F, Woyke T, Horowitz MA, Quake SR (2017) Microfluidic-based mini-metagenomics enables discovery of novel microbial lineages from complex environmental samples. elifesciences.org. https://doi.org/10.7554/eLife.26580.001
Zhang K, Martiny AC, Reppas NB, Barry KW, Malek J, Chisholm SW, Church GM (2006) Sequencing genomes from single cells by polymerase cloning. Nat Biotechnol 24:680–686. https://doi.org/10.1038/nbt1214
Zhang Y, Gao J, Huang Y, Wang J (2018) Biophysical perspective recent developments in single-cell RNA-seq of microorganisms. Biophysj 115:173–180. https://doi.org/10.1016/j.bpj.2018.06.008
Acknowledgements
We would also like to thank Dr. John Vollmers for assisting us in collecting data for Figure 2.
Funding
Open Access funding provided by Projekt DEAL. This work was supported by the German Research Foundation (DFG) (Grant No. 320579085).
Author information
Authors and Affiliations
Contributions
AK conceived and designed the manuscript. AK and MS wrote the manuscript and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kaster, AK., Sobol, M.S. Microbial single-cell omics: the crux of the matter. Appl Microbiol Biotechnol 104, 8209–8220 (2020). https://doi.org/10.1007/s00253-020-10844-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-020-10844-0