
Abstract
Drug-induced liver injury (DILI) complicates safety assessment for new drugs and poses major threats to both patient health and drug development in the pharmaceutical industry. A number of human liver cell-based in vitro models combined with toxicogenomics methods have been developed as an alternative to animal testing for studying human DILI mechanisms. In this review, we discuss the in vitro human liver systems and their applications in omics-based drug-induced hepatotoxicity studies. We furthermore present bioinformatic approaches that are useful for analyzing toxicogenomic data generated from these models and discuss their current and potential contributions to the understanding of mechanisms of DILI. Human pluripotent stem cells, carrying donor-specific genetic information, hold great potential for advancing the study of individual-specific toxicological responses. When co-cultured with other liver-derived non-parenchymal cells in a microfluidic device, the resulting dynamic platform enables us to study immune-mediated drug hypersensitivity and accelerates personalized drug toxicology studies. A flexible microfluidic platform would also support the assembly of a more advanced organs-on-a-chip device, further bridging gap between in vitro and in vivo conditions. The standard transcriptomic analysis of these cell systems can be complemented with causality-inferring approaches to improve the understanding of DILI mechanisms. These approaches involve statistical techniques capable of elucidating regulatory interactions in parts of these mechanisms. The use of more elaborated human liver models, in harmony with causality-inferring bioinformatic approaches will pave the way for establishing a powerful methodology to systematically assess DILI mechanisms across a wide range of conditions.
Similar content being viewed by others


Introduction
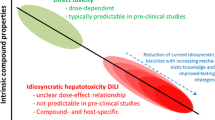
Despite the contributions of modern drug therapy in improving human health and in increasing human lifespan, adverse drug reactions remain a major challenge for healthcare providers, drug developers as well as drug safety regulators. In view of its crucial function in the metabolism of xenobiotic compounds, the liver is particularly prone to injury caused by drugs and other exogenous compounds (Gu and Manautou 2012). Drug-induced liver injury (DILI) refers to any insult inflicted on the liver by a pharmaceutical product that leads to impairment of hepatic function (Leise et al. 2014). Although the incidence is relatively low, DILI is the leading cause of acute liver failure (ALF) in the United States and Europe and the most common reason for drug disapproval and withdrawal from the market (Kullak-Ublick et al. 2017). DILI encompasses a spectrum of clinical manifestations and its features may resemble any known acute and/or chronic liver disease (such as acute hepatitis, hepatic cholestasis and steatosis), or a mixture of different injury types, making the distinction between DILI and other hepatic disorders difficult (Neuman 2019). Remarkably, only a few drugs (e.g. acetaminophen: APAP) are known to cause dose-dependent hepatotoxicity. The majority of DILI cases are idiosyncratic because of its relatively infrequent occurrence and lack of a dose–response. The idiosyncratic nature of DILI increases its unpredictability and uncontrollability (Raschi and De Ponti 2017), thus posing a major threat to public health.
This review is organized in four sections. The introduction briefly summarizes the current challenges and opportunities in DILI mechanistic studies. The second section provides (1) an update on in vitro human liver models used in DILI research, and (2) examples of applying omics techniques to the human in vitro culture platforms and the contributions of the integrated systems to the understanding DILI mechanisms. The third section (1) discusses the bioinformatic approaches that are currently used for analyzing the omics data, and (2) illustrates the state-of-the-art systems biology approaches that can be adopted to investigate drug toxicity. Finally, in the fourth section, we will make suggestions for future studies, aiming to reveal the underlying mechanisms of DILI.
Drug safety assessment: in vitro human-relevant alternatives to animal testing
To control the risk of DILI, animal studies are frequently used to investigate the dose-dependent mechanisms of action of new compounds (Lin and Khetani 2016; Vorrink et al. 2018). The results from such animal tests, however, are poorly correlated with human clinical data as a consequence of interspecies differences in hepatocellular function, pharmacokinetics, drug metabolism and toxicity targets. Hence, the low concordance between drug-induced hepatotoxicity in human and animals results in a low prediction rate (50%) (Lin and Khetani 2016; Vorrink et al. 2018).
In the last few decades, a new strategy, using human hepatic cell-based in vitro culture systems combined with omics-based approaches, is being developed to reduce animal testing as well as to better predict human DILI and other toxic effects of pharmaceuticals (reviewed in (Jiang et al. 2015b)). Compared to the traditional animal studies, these in vitro liver models resemble much better the human physical conditions and provide more human-specific information on drug behavior. In the context of toxicology, omics approaches that measure global alterations in genes, transcripts, proteins and metabolites strive to generate a comprehensive profile of interactions between genetic variability and exposure to exogenous agents (Singh et al. 2010). When used in combination with bioinformatics and systems biology tools, omics-based strategies provide unique opportunities for in-depth understanding of molecular mechanisms of drug-induced toxicity (Singh et al. 2010). The application of omics-based approaches to in vitro human cell-based liver models dissects dynamic changes in intracellular pathways and networks associated with exposure to drugs, thus increasing the chance of identifying more specific and reliable molecular fingerprints capable of improving DILI diagnosis and predicting human in vivo drug responses to new chemicals while generating a plethora of data that can be reused in future studies.
Bottlenecks for understanding the mechanisms of DILI
Although intensive efforts have been made to understand why and how DILI occurs and which individuals may be more susceptible, no specific diagnostic and predictive biomarkers have been identified. Also, the description of possible mechanisms of DILI (e.g. the formation of reactive metabolites, mitochondrial dysfunction, oxidative stress, biliary transport inhibition, lipid accumulation and allergic reactions) is rather incomplete (Kullak-Ublick et al. 2017).
One concern regarding the current in vitro studies on DILI mechanisms is that the exposure time in many human cell-based experiments is limited to a few days due to the rapid dedifferentiation and deterioration of hepatic functions when hepatocytes are cultured in vitro (Jiang et al. 2015b). Thus, these prototypic short-term in vitro studies (24–48 h) predominantly reflect acute toxicity. The development of DILI, however, may take several weeks or months (Tolosa et al. 2019). This delay may be related to drug metabolism, adaptation to drug intake (development of drug tolerance and physical dependence) or chronic liver injury caused by deposition of bilirubin (drug-induced cholestasis) or lipids (steatosis) (Schuemie et al. 2016). For example, fialuridine, an antiviral agent that resulted in 5 deaths and 2 cases of liver transplantation in 15 volunteers, only exhibited severe hepatotoxicity a few weeks after initiating therapy (Vorrink et al. 2018). In fact, in a large prospective study (1257 enrolled subjects with suspected DILI) conducted by the DILI network, 60 of 899 DILI cases (6.7%) had very long latency (over one year) (Chalasani et al. 2015). In addition, in many studies, cell models are exposed to concentrations that are not comparable to relevant therapeutic doses, challenging the translation of results of these in vitro models to the clinic (Atienzar et al. 2016). For instance, APAP, an intrinsic hepatotoxin usually causing acute liver injury after overdose, also demonstrates hepatotoxicity in healthy adults after repeated exposure to therapeutic doses (Mosedale and Watkins 2017). Therefore, a stable long-term human cell-based liver model that allows repeated low-dose exposures and monitoring cellular response to the treatment over time is more representative of the clinical setting and aids the investigation of mechanisms and pathogenesis of DILI. Furthermore, accumulating evidence indicates an essential role of an (idiosyncratic) immune response in the pathogenesis of DILI (Bale et al. 2014). Hepatocytes and their interactions with the resident liver immune cells, stellate cells and endothelial cells both contribute to the lesion development (Bale et al. 2014). Nevertheless, current models, often with low heterogeneity at the level of cellular composition, offer limited physiologically relevant information.
Application of omics-based in vitro human liver systems to DILI mechanistic studies
To this end, a spectrum of more physiologically relevant human liver platforms has been developed. These culture systems include, but are not restricted to, micropatterned co-cultures, three-dimensional (3D) bio-printed cultures, 3D spheroid cultures and microfluidic cultures. These in vitro models have improved control of cellular microenvironments, and liver cells growing on these platforms exhibit extended metabolic stability and a more in vivo-like phenotype, which are unattainable with traditional cell culture methods (Proctor et al. 2017). More recently, these in vitro models have been applied to several long-term toxicity studies. After repeated dosing over longer periods of time, these cultures show potential for distinguishing DILI-inducing from non-DILI-inducing drugs under chronic exposures (Proctor et al. 2017; Tolosa et al. 2019). For instance, when combined with transcriptomic approaches (e.g. deep RNA-sequencing technology), these model systems generate sizable data, depicting longitudinal and genome-wide responses to drug exposure. These data may capture long-term toxic effects of drugs and contain valuable information about delayed reactions to drugs and/or cumulative drug effects following more relevant therapeutic dosing scenarios. Among various layers of omics, transcriptome profiling, capturing genome-wide changes in mRNA expressions in response to drug exposure, has been widely used to derive insight into DILI mechanisms.
Along with the availability of chronic exposure data, bioinformatic tools have been developed aiming to support the analysis of time series data and to infer pathways and gene networks from gene expression profiles. These approaches cover a broad spectrum of topics, ranging from softwares for identifying differentially expressed genes, time series analyses, clustering methods categorizing time-course gene expression data and models analyzing patterns of temporal gene expression. Section “Bioinformatic approaches” provides an overview of bioinformatic methods that can be used to extract pivotal information for the identification of causal mechanisms and the interpretation of toxicological processes involved in DILI.
In vitro cell culture systems for omics-based mechanistic investigation of drug-induced liver disease
In an attempt to advance the understanding of the underlying mechanisms of human DILI, a spectrum of in vitro human liver model systems, ranging from conventionally cultured cancerous hepatic cell lines and primary/renewable human hepatocytes to engineered liver platforms, such as static micropatterned (co-)cultures, bio-printed hepatic models and multicellular perfused systems, has been developed. These human liver cell-derived in vitro models that closely resemble the human physical conditions have moved beyond animal toxicological studies and allow us to investigate drug disposition and hepatic transporter-related drug–drug interactions (Swift et al. 2010). Omics studies, at all levels, including transcriptomics, proteomics and metabolomics, provide information on the interactions between the genome and compounds. In vitro human liver model systems combined with omics-based analyses have great potential for revealing the molecular mechanisms of drug-induced toxicity in humans (Afshari et al. 2011; Cui and Paules 2010).
The characteristics of various culture systems and their application to omics-based mechanistic studies focusing on DILI are discussed below and summarized in Table 1.
Conventional in vitro cultures
Primary human hepatocytes or renewable hepatic cell lines, cultured on adsorbed or between two layers of gelled extracellular matrix (ECM) proteins have been widely used in assessing drug-associated hepatotoxicity and in investigating the mechanisms and pathophysiology of DILI. Since the characteristics of these culture systems have been reviewed extensively (Godoy et al. 2013; Jiang et al. 2015b), we only briefly summarize the advantages and limitations of these conventional culture systems and instead mainly focus on the key findings regarding human DILI mechanisms discovered by applying omics approaches to these models.
Hepatocellular carcinoma cell lines
Immortalized human cell lines have been widely used as cost-effective and sustainable substitutes for primary human hepatocytes (PHH) in initial assessment of DILI risk of candidate compounds prior to preclinical trials (Lin and Khetani 2016). Advantages of using these cancerous hepatic cell lines, including high availability, easy handling, stable phenotype, well-characterized protein expression and long life span (Kuna et al. 2018), have popularized its use in high-throughput toxicity studies and in exploration of mechanisms of liver toxicity. Among various human hepatocellular carcinoma cell lines (i.e. Hep3B, Huh7 and SK-HEP-1), HepG2 and HepaRG constitute routinely used models for hepatotoxicity studies.
HepG2, a perpetual hepatocellular cell line, expresses a variety of liver-specific enzymes and nuclear transcription factors (such as p53 and Nrf2) that play pivotal roles in the development of drug-induced toxicity (Jiang et al. 2015b). Due to their low cost, high reproducibility and unlimited lifespan, HepG2-based in vitro model systems and multiple omics approaches have been extensively exploited in a number of studies for the prediction of drug-induced hepatotoxicity (Ramirez et al. 2018), the classification of cholestatic and necrotic hepatotoxicants (Van den Hof et al. 2014) and the discrimination between genotoxicants and non-genotoxicants (Magkoufopoulou et al. 2011, 2012) as well as to illustrate the corresponding mechanisms. For instance, an in vitro study conducted using HepG2 cells showed that the transcriptome-based classifiers achieved high accuracies (accuracies above 90%) for the discrimination of hepatotoxicants from nonhepatotoxicants and for the separation of cholestatic and non-cholestatic compounds (36-gene and 12-gene classifiers, respectively) (Van den Hof et al. 2014). Aside from the classification power, this in vitro culture system revealed that endoplasmic reticulum (ER) stress and the unfolded protein response are important cellular responses to drug-induced liver toxicity (Van den Hof et al. 2014). In 2018, a research team exposed HepG2 cells to 35 compounds and performed metabolome analyses using mass spectrometry (MS), aiming to develop robust, standardized and reproducible metabolomic systems for prediction of liver toxicity in vitro (Ramirez et al. 2018). The study successfully discovered dose-dependent and compound-specific action modes for the tested hepatotoxins and identified several molecular mechanisms (e.g. liver enzyme induction/inhibition and peroxisome proliferation) responsible for their toxicities (Ramirez et al. 2018). The applications of transcriptomics, proteomics and metabolomics to HepG2-based in vitro models also assist with the identification of key mechanisms underlying drug-induced hepatic steatosis, cholestasis, cytotoxicity and genotoxicity (Chatterjee et al. 2014; Deferme et al. 2015b; Rieswijk et al. 2014; Smit et al. 2017; Van den Hof et al. 2015, 2017; Van Summeren et al. 2011) as well as compounds’ carcinogenicity in humans (Briede et al. 2018; Caiment et al. 2015; Jennen et al. 2011; Lizarraga et al. 2012; Ruiz-Aracama et al. 2011; Souza et al. 2016; van Delft et al. 2012).
In 2012, Van Delft et al. demonstrated the benefits of RNA sequencing (RNA-seq) over microarray in transcriptome profiling of the benzo[a]pyrene (BaP)-exposed HepG2 cells (van Delft et al. 2012). Compared to the microarray technique, RNA-seq detected a larger number of differentially expressed genes (DEGs) (~ 20% more genes) in response to the BaP challenge and the changes of the induced DEGs (fold changes) were more robust (~ threefold) when using RNA-seq (van Delft et al. 2012). Furthermore, the study indicated that RNA-seq enables the investigation of alternative isoform expression in the affected genes and the allele-specific gene expression changes, as illustrated by the identification of BaP-associated alterations in isoform expression in several genotoxic response- or oxidative stress response-regulating genes (i.e. TP53, BCL2, XPA and AKR1B10) (van Delft et al. 2012). Following this research, Caiment and co-authors further studied the BaP-induced carcinogenic processes in HepG2 cells using RNA-seq and small RNA-seq technologies (Caiment et al. 2015). They discovered that the exposure-initiated upregulation of the miR-181a-1_3p expression and the subsequent inhibition of O6-methylguanine DNA methyltransferase were responsible for the BaP-induced carcinogenesis in the liver (Caiment et al. 2015).
Deferme et al. also adopted the HepG2 cells for a thorough study of the development of drug-induced oxidative stress in toxicological phenomena (Deferme et al. 2013, 2015b, 2016). Following exposures of HepG2 cells to several free radical-releasing agents, the authors analyzed the time series multi-omics datasets in combination with functional endpoints measuring oxidative cellular damage (Deferme et al. 2013, 2015b, 2016). Their studies not only elucidate the role of oxidative stress in inducing aberrant DNA methylation and DNA hydroxymethylation modifications, which significantly enhanced the molecular understanding of oxidative stress-induced responses in human hepatocytes (Deferme et al. 2013, 2016), but also provided an oxidative stress-associated gene signature for the prediction of the oxidative stress-inducing ability of a compound, which contributes to improving the drug safety (Deferme et al. 2013, 2015a).
HepG2 cells, due to their high mitochondrial DNA and organelles contents, remain a model of choice for the study of compound-induced mitochondrial dysfunction (Poloznikov et al. 2018). Applied transcriptomic and proteomic analyses to this cell model, Jiang et al. (2015a) and Paemanee et al. (2017) explained the mechanisms of mitochondrial dysfunction associated with APAP and nevirapine (NVP) administrations. Using a HepG2-based in vitro cytokine synergy model, an attempt has been made to investigate the relationship between inflammation and drug idiosyncrasy (Jiang et al. 2017). Through comparing the differences of the transcriptomic and metabolomic profiles of HepG2 cells in response to idiosyncratic and non-idiosyncratic compounds, the study revealed the interaction between inflammatory cytokines and the idiosyncratic drugs and illustrated that the dynamic disequilibrium in ceramides/sphingolipids balance and its coupled ER stress- and JNK-mediated apoptosis could be the common mechanism underlying the inflammation-associated idiosyncratic drug hepatotoxicity (Jiang et al. 2017).
Nonetheless, the lack of activity of several drug transporters (e.g. BSEP, NTCP and OCT-1) and phase I/II enzymes (e.g. GSTA 1/2 and GSTM1) in HepG2 cells (Poloznikov et al. 2018; Van den Hof et al. 2015) challenge the relevance of HepG2-based in vitro models in DILI research (Zeilinger et al. 2016).
Alternative to HepG2 cells, the highly differentiated human hepatoma HepaRG cells have also been used as a surrogate for PHH, especially in in vitro drug metabolism assessment (Le Vee et al. 2013). Recently, studies have demonstrated that compared to HepG2 cells, the baseline transcriptomic (Hart et al. 2010; Jennen et al. 2010; Jetten et al. 2013) and proteomic (Sison-Young et al. 2015) profiling of differentiated HepaRG cells indicates a higher resemblance to PHH. Tascher et al. also showed that the amounts of proteins related to bile acid synthesis, conjugation, detoxification and transport expressed by HepaRG are comparable to PHH, encouraging the use of these cells in studies of drug metabolism, compound-induced liver injury and pathogenesis of cholestatic liver diseases in humans (Tascher et al. 2019). The molecular mechanisms for the hepatic cholestasis (Rodrigues et al. 2018), steatosis (Mesnage et al. 2018) and cytotoxicity have been studied by means of multi-omics-based methods after exposure of HepaRG cells to a number of compounds (Seeger et al. 2019a; Smith et al. 2018). The integrated transcriptomic, proteomic, and metabolomic analyses demonstrated that bosentan-induced cholestatic liver injury was associated with transcriptomic and proteomic alterations in liver cholestasis-, liver necrosis- and liver damage-related pathways and the endogenous metabolite-related mitochondrial impairment (Rodrigues et al. 2018). Using proteomics, Smith et al. revealed that individual or combined administrations to mycotoxins [deoxynivalenol (DON) and zearalenone (ZEA)] affected the production of proteins regulating the cell cycle, cell proliferation and development, as well as DNA metabolic processes in HepaRG cells (Smith et al. 2018).
In addition to the well-known HepG2’s low functionality and HepaRG’s low predictive power of hepatotoxicity (Asplund et al. 2016), none of these human hepatoma cell lines reflect human heterogeneity in response to toxic compounds (Benesic and Gerbes 2015; Jetten et al. 2013). Although a relatively high degree of similarity in gene expression patterns has been found between HepaRG cells and PHH, in general, the phenotypes (Bell et al. 2016) and the basal gene expression profiles of both cell lines still differ substantially from PHH (Harris et al. 2004; Hart et al. 2010). Even though the activity levels of cytochrome oxidase are higher in HepaRG than in HepG2 cells, the levels of their cytochrome activities are markedly decreased in both cell lines compared to those in PHH (reduced by 60% and 90% in HepaRG and HepG2 cells, respectively) (Sison-Young et al. 2015). At the proteome level, there are detectable differences in the proteins regulating cell senescence and proliferation between HepaRG and PHH, probably due to the transdifferentiation features of HepaRG, reminding their similarity to stem cells and liver cancer cells (Tascher et al. 2019), which raise concerns about the applicability of these in vitro models in DILI studies.
Primary human hepatocytes (PHH)
Hepatocytes represent around 60% of the total liver cell population (Poloznikov et al. 2018). Due to their physiological relevance, PHH (freshly isolated or stably cryopreserved) are commonly accepted as the ‘gold standard’ for constructing liver models for in vitro drug testing and DILI mechanistic research (Gerets et al. 2012). In the early 2000s, freshly isolated PHH-derived cell models and transcriptome approaches have been used jointly to evaluate and predict human drug toxicity in vitro. In 2004, Kier and co-authors analyzed the microarray data collected from cultures of PHH (n = 6) maintained on the collagen-coated plates and successfully distinguished troglitazone, a potent hepatotoxin, from its less toxic analogs, rosiglitazone and pioglitazone, using a toxicologically relevant gene set generated from rat in vivo data (Kier et al. 2004). Their research revealed that after 24 h exposure, the hepatotoxic troglitazone, compared to its non-hepatotoxic counterparts, uniquely elevated the expression of genes in several toxicity pathways (e.g. apoptosis and inflammation), but inhibited genes responsible for detoxification pathways (e.g. acute phase proteins and stress‐responsive proteins) (Kier et al. 2004; Liguori et al. 2005), demonstrating the practicality of toxicogenomics for improved understanding of DILI mechanisms. However, PHH maintained in quiescent monolayer cultures rapidly dedifferentiate (4–6 h when kept in suspension and 24–48 h when cultured on plates), resulting in a loss of morphological integrity, liver-specific enzyme activities, hepatic functions and cell viability within days (Bell et al. 2016; Soldatow et al. 2013). Thus, when maintained in suspension or directly seeded onto (collagen-coated) culture plates, the lifespan for PHH is limited and preservation of an in vivo like phenotype of these cells is challenging (Poloznikov et al. 2018).
The optimized culture conditions, such as the use of liver cell co-cultures and sandwich cultures, have been applied to improve cell stability of hepatocytes in conventional cultures. Compared to monolayer cultures, co-cultures of hepatocytes with liver-derived non-parenchymal cells (NPCs) have been known to be beneficial for modulating cell differentiation and prolonging hepatic functions/functionality (Lin and Khetani 2016). When co-cultured with hepatic stellate cells (HSCs), PHH seeded on collagen-coated plates displayed improved hepatic functions in 2-day cultures (Krause et al. 2009). When maintained in a sandwich configuration, PHH demonstrate a prolonged lifespan and delayed dedifferentiation process. It is reported that the viable culture period of the sandwich-cultured PHH may last up to 8 weeks and the cell polarity that enables the development of the canalicular network and the production of bile in the hepatocytes is better preserved (Poloznikov et al. 2018), which favor the use of this model in in vitro investigation of human drug-induced hepatotoxicity.
Using the sandwich-cultured PHH and microarray technology, Kienhuis and Black identified human-specific transcriptome changes in response to 24 h coumarin- (Kienhuis et al. 2009) and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) (Black et al. 2012) treatments. Through analyzing the transcriptome alterations in PHH after exposure to 158 compounds, a research team found that the changes of a set of NRF2-associated genes to a given chemical insult have been found to be indicative of toxicity features of such compound (Copple et al. 2019). At the end of the study, they pointed out that integrating the NRF2 transcriptional network into holistic systems toxicology models could benefit the mechanism studies and preclinical prediction of DILI (Copple et al. 2019).
As a model derived from individuals with different genetic backgrounds, the variability observed in PHH in vitro could better reflect that of the liver in vivo (Gomez-Lechon et al. 2007). Jetten et al. investigated the variability in response to APAP exposures (24 h) in PHH derived from five donors by incorporating multiple omics techniques (transcriptomics and metabolomics). The study found that the variability in levels of APAP metabolites was correlated with the interindividual differences in the expression of genes involved in certain biological processes, such as liver regeneration, inflammatory responses and mitochondrial stress responses, which provided explanations for the observed large variations in susceptibility towards APAP-induced liver injury among humans (Jetten et al. 2016). Due to the physiological relevance of PHH, a research group selected PHH sandwich cultures to understand the hepatic adverse effects of a recombinant human neuregulin-1β (GGF2) observed during a phase I clinical trial (Mosedale et al. 2018). The exposures to GGF2 (up to 72 h) induced an overall decrease (~ 50%) in the expression of protein-coding genes responsible for bilirubin transport and bile acid conjugating without overt cytotoxicity. The treatments also affected genes involved in the acute phase response signaling pathways, mimicing the changes in IL-6 disturbed cells (Mosedale et al. 2018). Subsequently, the GGF2 administrations resulted in a dose- and time-dependent reduction of total endogenous bile acid content and biliary clearance in the treated PHH (n = 5) (Mosedale et al. 2018).
The prolonged stability of PHH attributed to the change of culture configuration also enables the assessment of epigenetic events in DILI development. Recently, sandwich-cultured PHH have been employed to explore the mechanisms of chronic hepatotoxicity following 5-day administrations of aflatoxin B1 (AFB1), a liver carcinogen (Rieswijk et al. 2016), and cyclosporine A (CsA), an inducer of cholestasis (Wolters et al. 2016). Facilitated by transcriptomic and epigenomic approaches, this in vitro culture system revealed interactions between alterations in DNA methylation and mRNA expression changes during the development of AFB1-induced hepatocellular carcinoma (Rieswijk et al. 2016) and revealed the persistent changes in gene expression and microRNA expression in response to CsA-associated cholestasis (Wolters et al. 2016). Wolters et al. (2017, 2018) and Van Breda (van Breda et al. 2018) also used the sandwich-cultured PHH and multiple omics approaches (transcriptomics, epigenomic and proteomics) to examine the roles of epigenetic factors in liver steatosis development induced by repeated exposures (3–5 days) to valproic acid (VPA), a drug to treat epilepsy and bipolar disorders. Through the integrative cross-omics analyses, they discovered several treatment-initiated reactions (e.g. the cross-talk between nuclear DNA and mitochondrial DNA hypermethylation (Wolters et al. 2017), the persistent epigenetic and transcriptomic alterations in the mitochondrial genome coupled with the measurable mitochondrial dysfunction (Wolters et al. 2018) and the inhibition of nuclear receptors at DNA methylation and mRNA levels (van Breda et al. 2018), deepening the understanding of the VPA-induced hepatosteatosis.
Yet, the dedifferentiation process in the sandwich-cultured PHH, although delayed, is unavoidable. It is suggested that when cultured between two collagen layers, PHH only maintain their biotransformation capacity and the ability to induce hepatic phase I/II enzymes close to the in vivo situation for the first 2 weeks (Poloznikov et al. 2018) and sustain their hepatic functions to a level comparable to that observed in the freshly isolated cells for a few days (Lin and Khetani 2016). Additionally, a few other disadvantages, such as the shortage of donated livers and the lack of tissue heterogeneity, limit the potential of 2D or sandwich-cultured PHH in investigating mechanisms of drug-induced hepatotoxicity (Nagamoto et al. 2012).
Human pluripotent and multipotent stem cells
Recently, human pluripotent and multipotent stem cells have gained attention as potentially unlimited cell sources of differentiated hepatocytes because of their pluripotency and proliferative potential (Glicksman 2018). The stem cell-based in vitro liver models carry donor-specific genetic information and may have the potential to contribute to personalized medicine (Ware et al. 2015). The multipotent post-natal stem cells, human skin-derived precursors (hSKP), exhibit the ability to differentiate into hepatocyte-like cells (hSKP-HPC) (Rodrigues et al. 2016). These conversed cells have been shown to express hepatocyte markers (e.g. EPCAM, NCAM2, PROM1, SMAD4 and THY1) as well as phases I and II drug-metabolizing enzymes (e.g. CYP1B1, FMO1, GSTA4, GSTM3, ABCC4, ABCA1, SLC2A5), suggesting the acquisition of hepatic progenitor cell-like properties in hSKP after differentiation. Nevertheless, only a proportion of the hSKP-HPC express adult hepatocyte marker genes or proteins, such as albumin (~ 27% albumin-positive cells), indicating that these transformed cells still retain the characteristics of immaturity of liver progenitor cells (Rodrigues et al. 2014). Microarray-based studies illustrate that the responses of the hSKP-HPC to the APAP treatment were comparable to the reactions in PHH, reflected in the similarity of their functional toxicological analysis results (Rodrigues et al. 2014). Furthermore, among other in vitro models (PHH, HepG2 and HepaRG), hSKP-HPC showed the highest concordance with the clinical liver samples collected from the patients with APAP-induced liver failure (n = 3) at the transcriptome level (Rodrigues et al. 2016). Yet, HepaRG, instead of the hSKP-HPC, demonstrated the highest potential for the prediction of APAP-induced hepatotoxicity when using a prediction diagram consisting of the ‘Damage of Liver’-related genes derived from comparing gene expression profiles between clinical ALF samples and healthy liver tissues (Rodrigues et al. 2016). In contrast, a direct transcriptome comparison between APAP-exposed PHH and hSKP-HPC (5 mM, 24 h) has been made. The results showed that, at the gene and the pathway levels, the treatment-induced alterations in the hSKP-HPC did not always correlate with the changes in PHH (Rodrigues et al. 2014).
Next to hSKP-HPC, human pluripotent stem cells (hPSCs) also show the ability to differentiate into primary hepatic-like cells under culture conditions, providing complementary to PHH for the prediction of drug toxicity and for the investigation of mechanisms of DILI (Kuna et al. 2018; Nagamoto et al. 2012). Human-induced pluripotent stem cells (hiPSCs) and human embryonic stem cells (hESCs) are two types of hPSCs. They both display self-renewal in culture and have the potential to be differentiated into almost all somatic cell types in the body (Zhu and Huangfu 2013). In 2018, Han et al. performed liquid chromatography-tandem mass spectrometry (LC–MS/MS) to assess the idarubicin-induced proteomic changes in basal hiPSCs. After 24 h exposure, they identified over 3000 differentially expressed proteins and revealed the inhibition of the EIF2 signaling pathway and the activation of apoptosis played a key role in idarubicin-related toxicity in hiPSC (Han et al. 2018). Through directed differentiation, hPSCs produce genetically modified cells, tissues and even organoids that mimic the human liver (Apati et al. 2019). hiPSC-derived hepatocytes (hiPSC-Heps) show closer resemblance with PHH at the transcriptome level than the HuH-7, HepG2, and HepG2/C3A cells (Gao and Liu 2017). Lu et al. demonstrate that the genome-wide expression profiles of multiple batches of hiPSC-Heps (proprietary protocol) are more similar to those from PHH isolated from neonatal (n = 1) and adult (n = 4) human livers than those from PHH derived from fetal human hepatocytes (n = 2) (Lu et al. 2015). While, in another study, the differentiated hiPSC-Heps (a 22-day hepatogenic differentiation procedure) expressed high levels of fetal isoforms of cytochrome P450 s (CYPs) (CYP3A5 and CYP3A7) and glutathione S-transferases (GSTP1) as well as transporters associated with dedifferentiation during carcinogenesis (ABCB1 and ABCG2), indicating a lack of maturation of these hiPSC-Heps (Bell et al. 2017). Likewise, other studies have reported that the expression patterns of phase I/II drug-metabolizing enzymes and transporters in the hiPSC- and hESC-differentiated hepatocytes more closely resemble those in fetal livers than in adult livers (Scott et al. 2013). These inconsistent results suggest that the diverse reprogramming techniques can cause highly variable functionality within the produced hepatocyte-like cells (Soldatow et al. 2013). In addition, the expression levels of xenobiotic metabolism-related genes in hiPSC-Heps are still not comparable to those found in freshly isolated PHH or liver tissues (Si-Tayeb et al. 2010) and the transformed cells may lose their liver characteristics and their metabolic activity after a few days when maintained in standard culture conditions (Soldatow et al. 2013). To date, few proof-of-principle toxicity studies carried out with hiPSC-Heps support the concept of using these models for drug toxicity screening (Choi et al. 2013; Szkolnicka et al. 2014; Ware et al. 2015), whereas none of them provided mechanistic insights into the causes and effects of these drugs in liver toxicity at the omics level.
A number of improvements, such as further differentiation and cell culture optimization, are required before stem cell-derived models can be widely used as in vitro adult human liver surrogates for large-scale drug screening or DILI mechanism studies (Scott et al. 2013).
Precision-cut liver slices
Human precision-cut liver slices (hPCLS), often considered as mini livers, better represent the biological organization of the liver (Ijssennagger et al. 2016). Unlike PHH, the preparation of hPCLS does not require the use of any proteolytic enzymes during their preparation, enabling the resultant liver slices to maintain a natural tissue microenvironment and retain intact cell–cell and cell–matrix interactions (Elferink et al. 2011). hPCLS also reflect the liver cell heterogeneity and better preserve liver histoarchitecture, allowing for the interplay between parenchymal and non-parenchymal liver cells (NPCs) as well as liver infiltrating immune cells (Palma et al. 2019). It is reported that, when cultured under proper conditions, hPCLS can preserve their viability for up to 5 days (Paish et al. 2019; Wu et al. 2018), which enhances their utilization for studying the pathophysiological processes in a variety of hepatic diseases, such as alcoholic liver disease, non-alcoholic fatty liver disease (NAFLD), fibrosis and cirrhosis, cholestasis and DILI (Palma et al. 2019). Nowadays, human liver slice-derived cultures combined with omics techniques have been used to study the mechanisms of drug-induced hepatotoxicity (Elferink et al. 2011), steatosis (Ijssennagger et al. 2016) and cholestasis (Vatakuti et al. 2017). In 2016, Vatakuti et al. exposed hPCLS (n = 6) to a number of necrosis- or cholestasis-inducing drugs, aiming to develop a mechanism-based model for the classification and prediction of drug toxicity (Vatakuti et al. 2016). Facilitated with transcriptome analyses, researchers discovered several signaling pathways and gene sets that were commonly affected in the hPCLS treated with the cholestatic drugs and developed a consensus list of marker genes that can be used for hepatotoxicity screening (Vatakuti et al. 2016). More recently, Ščupáková et al. used hPCLS (n = 23) to investigate the aberrant lipid metabolism in NAFLD (Scupakova et al. 2018). Through integrating the lipid mass spectrometry imaging data into pathway analysis, their study demonstrated the lipid–protein interaction networks in nonsteatotic and steatotic regions of the patients, providing insights into the biological processes that direct regional lipid accumulation in NAFLD (Scupakova et al. 2018). The well-preserved cell type diversity and the intercellular interactions also make hPCLS a promising model to recapitulate idiosyncratic drug-induced liver injury (iDILI)-triggering conditions in humans. Vatakuti et al. exposed hPCLS (n = 6) to the iDILI-related (clozapine) or non-IDILI-related (olanzapine) agents in the presence or absence of lipopolysaccharide (LPS) for 24 h (Vatakuti 2016). The presence of LPS exacerbated the toxicity of clozapine but not its non-iDILI-inducing comparator. By analyzing transcriptome changes, they found that co-exposure of hPCLS to clozapine and LPS-induced activation of HMGB1, p38 MAPK, NFkB and NRF2 signaling pathways and upregulated the inflammatory cytokine-coding genes (IFN-γ, IL1A, IL1B), suggesting that mitochondrial dysfunction and inflammation signaling pathways were involved in iDILI in the human liver (Vatakuti 2016).
Although hPCLS serves as a valuable tool for the study of liver disease, the scarcity of donor organs, especially healthy liver materials, is one of the major restrictions that limits the use of human liver slices in high-throughput drug screening (Roth and Lee 2017). Further to this, PCLS maintained in a static culture medium usually display a limited lifespan of 24–48 h (Jiang et al. 2015b). Advanced culture systems such as microfluidic perfusion systems (Khong et al. 2007), bioreactor systems (Paish et al. 2019) and air–liquid interface culture systems (Wu et al. 2018) have been shown to improve their longevity for 3–15 days, while the cultured PCLS still cannot preserve high levels of hepatic function for over 3 days without evident fibrogenesis and necrosis/apoptosis in the model (Lin and Khetani 2016; Paish et al. 2019).
Three-dimensional (3D) spheroids and organoids
The behavior of cells is heavily determined by their microenvironment. Liver cells maintained in the conventional 2D cultures often lack the hierarchy and structural components of the liver, resulting in poor stability of such models (Bale et al. 2014). Due to the enhanced heterotypic cell–cell contacts, 3D cultured human hepatocytes retain their periportal and perivenous phenotypes and exhibit superior activity of drug-metabolizing enzymes and transporter proteins as compared to 2D monolayer cultured PHH in extended culture periods (Foster et al. 2019). Results from transcriptomic, proteomic and metabolomic studies have demonstrated that the 3D (co-)cultured PHH-derived liver microtissues have reached long-term functional, phenotypical and metabolic stability (Messner et al. 2018; Vorrink et al. 2017) and remain sensitive to chronic hepatotoxin exposures during the culture period (up to 5 weeks) (Bell et al. 2016). Moreover, the PHH-derived liver spheroids displayed detectable pharmacokinetic differences among donors, which enable studies of interindividual variability in response to drugs and provide options for the investigation of genotype‐specific mechanisms for drug-induced hepatotoxicity in humans (Vorrink et al. 2017). When co-cultured with NPCs, the PHH-based 3D in vitro system further mimics in vivo hepatic histoarchitecture, rationalizing the use of this model in long-term (idiosyncratic) DILI research (Bell et al. 2016; Kuna et al. 2018).
A few studies have applied omics approaches to the PHH/Kupffer cells (KC) co-cultured 3D liver microtissues, aiming at improving the understanding of the mechanisms of acute (24 h) and chronic (3–14 days) drug-induced hepatotoxicity under normal or inflammatory conditions (Bell et al. 2017; Bruderer et al. 2015; Jiang et al. 2019; Sarkar et al. 2017). To better understand the pathogenesis of fatty liver disease, a research group recently exposed liver spheroids, containing PHH and detectable amounts of KC and stellate cells, to oleic and palmitic acid (Kozyra et al. 2018). High-resolution MS was used to analyze the long-term (21 days) metabolomic responses to the disturbances and identified the most upregulated lipid classes following the lipid administrations (Kozyra et al. 2018).
In addition to the PHH liver spheroids, hiPSCs have been used to create liver organoids. A study showed that the human self-aggregated PSCs spheroids exhibited a stable hepatic phenotype and remained modest liver function for over 1 year in culture (Rashidi et al. 2018). The whole-genome profiling of hiPSC organoids co-cultured with human stromal cells or with human bile duct-derived bipotent progenitor cells displayed a close resemblance to in vivo liver-bud-derived cells (Takebe et al. 2013) as well as chromosome and structural stability of up to 2 months (Huch et al. 2015). In another study, researchers used single-cell RNA sequencing (scRNA-seq) to decipher the path of hiPS-Hep lineage progression and discovered that the 3D cultured liver organoids were more closely correlated with fetal liver cells than with adult hepatocytes (Camp et al. 2017).
More recently, bio-printing technology has been applied to produce 3D multicellular hepatic spheroids. Compared to the randomly distributed heterotypic liver cells in self-assembled spheroids/organoids, bio-printing enables more precise control over the spatial arrangement of different cell populations so that sophisticated homotypic and heterotypic cell–cell interactions can be further simulated (Underhill and Khetani 2018). Kizawa et al. used the bio-printing technology to create a 3D liver tissue consisting of PHH and mouse fibroblasts and applied microarrays to assess the resulting model (Kizawa et al. 2017). Their study demonstrated that the 3D bio-printed liver tissue maintained long-term expression of liver function-related genes (2 weeks) and formed bile duct and sinusoid-like structures during an extended culture period of 50 days (Kizawa et al. 2017). Other research groups also reported their successfulness in establishing multicellular liver microtissues using PHH (Norona et al. 2016), HepG2 cells (Jeon et al. 2017) or hiPS-Hep (Ma et al. 2016) with or without adding the NPCs (such as hepatic stellate cells, endothelial cells). These studies also showed prolonged cell viability, increased drug sensitivity to methotrexate and thioacetamide and stable liver-specific gene expression in the long-term in vitro cultures (21–32 days) (Jeon et al. 2017; Ma et al. 2016; Norona et al. 2016). However, additional studies are still needed to further understand the characteristics of these in vitro models at the omics level.
Overall, 3D cultured liver tissues appeared recently as a promising way to evaluate human drug toxicity in vitro, yet so far the currently available technologies are still unable to produce standardized and validated 3D liver models with stable readouts. Hence, further studies are urgently needed to evaluate the sensitivity and accuracy of these 3D liver cultures proposed for detecting DILI. Besides, further adaptation is still required for adjusting different 3D culture systems for high-throughput drug-screening applications.
Micropatterned (co-)culture systems
In situ, mammalian cells integrate and actively respond to mechanical cues of their microenvironment. Under conventional culture conditions, however, uncontrolled parameters, such as mechanical and geometrical properties, form artificial environments for cells to proliferate and differentiate, which affect the architecture, mechanics, polarity and function of cells in such cultures (Thery 2010). Combined with photolithographic, microfluidic and microgrooving techniques, recent advances in cell micropatterning enable the establishment of in vitro microenvironments that are similar to cells’ in vivo situations, specifically via adjusting surface topographies (Beijer et al. 2017; Leuning et al. 2018) or chemistries (Hui and Bhatia 2007) of the culture materials, leading to an overall improvement of the physiological relevance of the cell models (Thery 2010). Pioneer studies on micropatterned human hepatocyte islands stabilized by fibroblasts, stromal cells or primary human liver sinusoidal endothelial cells demonstrated that the PHH maintained in these co-cultures could preserve their hepatic functions for up to 6 weeks, as demonstrated by the stable albumin secretion and urea synthesis, active phase I/II drug metabolism and canalicular transport (Khetani and Bhatia 2008; Khetani et al. 2013; Wang et al. 2010; Ware et al. 2018). To capture the aspects of the processes related to immune-mediated drug-induced hepatotoxicity, researchers have established micropatterned co-cultures incorporating the donor-matched primary rat hepatocytes and rat KC and exposed the models to LPS for 48 h (Rose et al. 2016). Following the treatment, they detected amplified inflammatory response and increased metabolic rate, represented with the elevated cytokine response and increased CYP3A concentration in rat hepatocyte/KC micropatterned co-cultures, but not in the hepatocyte monocultures (Rose et al. 2016). Using the LPS-sensitized rat micropatterned co-culture system, the authors demonstrated that APAP amplified immune-mediated liver toxicity similar to that of trovafloxacin, a known iDILI-inducing compound. (Rose et al. 2016). To date, however, a corresponding micropatterned human cell-based co-culture system (containing PHH and human KC) has not been reported yet.
Nowadays, studies have demonstrated the utility of human liver cell-derived micropatterned co-cultures in drug development, including DILI assessment (Khetani et al. 2013; Trask et al. 2014), yet only few studies have attempted to apply omics technologies to these in vitro culture platforms (Ware et al. 2017). Using a micropatterned co-culture model containing PHH and mouse 3T3-J2 fibroblasts, Ware et al. explored the hepatotoxic effects of chronic (up to 14 days) and low-dose exposure to several DILI-inducing drugs (Ware et al. 2017). The authors found that these hepatotoxins led to a greater number of DEGs in the co-culture modes compared to their non-liver-toxic analogs. They also pointed out the transcriptome changes in fatty acid- and drug metabolism-related pathways after troglitazone exposure were responsible for its hepatotoxicity (Ware et al. 2017).
More recently, the micropatterning technology has also been adapted for long-term cultures of hiPS-Hep (Berger et al. 2015; Davidson et al. 2015; Ware et al. 2015), providing the opportunity for the sustainable evaluation of responses of hepatocytes to hepatotoxins with consideration of human genetic diversity (Underhill and Khetani 2018). Promising evidences regarding the use of these in vitro platforms for DILI detection have been described (around 65–85% sensitivity) (Berger et al. 2015; Davidson et al. 2015; Ware et al. 2015), though detailed data originating from omics studies of these hiPS-Hep-based micropatterned co-cultures are still lacking.
Overall, the micropatterning methods have shown the importance of geometry of the cellular microenvironment in regulating cell physiology. The aforementioned studies have demonstrated the feasibility of enhancing the reconstitution of tissue-like conditions for the growth of liver cells in vitro and illustrated the use of micropatterned (co-)culture models in drug toxicity studies. Nevertheless, to accurately simulate liver environment in vitro, these developed (co-)culture platforms usually require the fabrication of complex scaffolds, which may hinder omics data collection from these liver cultures. More recently, a surface topography that enables simplified long-term maintenance of human hepatocytes (up to 30 days) has been reported (Beijer et al. 2017), yet a mature industrial product using such topography is still under development. Therefore, future progresses of the micropatterning technologies as well as applications of micropatterned (co-)culture models for integrated multi-omics approaches in drug toxicity studies are anticipated.
Liver-on-a-chip devices
In vivo, cells acquire oxygen, nutrients and hormones as well as receive physical/chemical stimulation via blood flow (Kimura et al. 2018). Flow shear stress induced by blood or other fluids is another feature of the liver microenvironment that is essential for organ development and plays important roles in both disease and health states (Aziz et al. 2017; Kimura et al. 2018). When cultured in a static platform (e.g. the conventional 2D cultures), the experimental compounds enter the cells only via diffusion, which differs from the dynamic in vivo environments (Kimura et al. 2018). In contrast, the perfusion culture technique allows precise control over architecture, medium PH and flow rate, mass transport, oxygen and nutrient gradients (zonation) as well as cell shear stress (Bale et al. 2014; Underhill and Khetani 2018). When applied to develop hepatic cellular models, the resulting culture systems not only facilitate better nutrient/waste exchange but also enable studying effects of hepatic zonation on toxicity exposures, further closing the gap between in vivo and in vitro conditions (Kimura et al. 2018). Evolved from tissue engineering, organs-on-a-chip models culture human cells in tissue-specific 3D settings in attempts to recapitulate the multifaceted (extra)cellular and molecular cues to biological function of a given organ system (Ronaldson-Bouchard and Vunjak-Novakovic 2018). Among various on-chip systems, liver-on-a-chip platforms that integrate microfluidics and micro-sized human liver spheroids/organoids have been actively pursued (Foster et al. 2019).
Liver-on-a-chip
Early attempts at combining cell culture with microfluidics led to the precursors of today’s on-chip liver tissue models (Ronaldson-Bouchard and Vunjak-Novakovic 2018). Investigators have pioneered the development of 3D microfabricated bioreactors using primary rat hepatocytes with or without rat fibroblasts co-cultures and showed that the liver organoids cultured under perfusion conditions maintained the metabolic capacities and stable functional viability over longer periods (~ 2 weeks), as evidenced by the steady albumin and urea production and the formation of the bile canaliculi along the hepatic cord-like structures (Nakao et al. 2011; Powers et al. 2002a, b).
Transcriptomic analyses of HepG2/C3A cells maintained in the poly(dimethylsiloxane) (PDMS) microfluidic chips coupled with a perfusion system showed upregulated Phase I/II enzymes (e.g. CYPs, SULT1A1 and SULT1A2), transporters (MDR1 and MRP2), hepatocyte markers (e.g. albumin, A1AT, transferrin, and ceruloplasmin), the cyotoplasmic filament protein (i.e. cytokeratin 18) and the tight junction protein (i.e. ZO-1) at the levels of transcription and protein accumulation in long-term cultures (up to 30 days) (Bhise et al. 2016; Prot et al. 2011). A comparison between biochip- and Petri dish-cultured HepG2/C3A cells was made at the transcriptomic and proteomic levels (Cheng et al. 2012). The study revealed that in the dynamic cultures the expression of CYP-coding genes was activated, indicating the improved metabolic capacity of the cells (Cheng et al. 2012). In contrast to the outstanding cancer-specific profiles and the perivenous-like phenotype observed in the static cultures, the dynamic cultured-hepatocytes demonstrated a predominant periportal-like hepatocyte (Cheng et al. 2012). In 2016, HepG2/C3A-based microfluidic platform has been used to explore the DILI mechanisms (Bavli et al. 2016). Bavli et al. real-time monitored the dose-dependent effects of troglitazone on metabolic fluxes and quantified the exposure-resulted mitochondrial damage, providing a mechanism for the observed idiosyncrasy of the troglitazone-induced hepatotoxicity (Bavli et al. 2016), whereas no omics data were provided with these treated liver devices. In 2012, Shintu et al. performed the nuclear magnetic resonance (NMR)-based metabolomic analyses on the microfluidic bio-artificial organs derived from monocultured HepG2/C3A or co-cultures containing HepG2/C3A and Madin-Darby canine kidney tubular epithelial cells (MDCK) (Shintu et al. 2012). Their study identified the system-specific metabolic signatures in response to the same insults and discovered that the synergistic metabolic responses to the stressors were detected only in the HepG2/C3A-MDCK co-cultures, but such findings were not detected in the monocultured liver models (Shintu et al. 2012). In the same year, Prot et al. integrated transcriptomics, proteomics and metabolomics to elaborately investigate the APAP-induced hepatotoxicity using a HepG2/C3A cultivated in the microfluidic biochips (Prot et al. 2012). Their multi-omics results demonstrated that APAP toxicity was associated with alterations in the expression of genes involved in the pathways related to DNA damage, cell cycle arrest and apoptosis- and necrosis-mediated cell death. Notably, the data from the biochip-cultured human hepatocytes allowed the identification of two additional APAP-affected pathways, namely “lipid metabolism and peroxidation” and “calcium homeostasis” and the formation of APAP-GSH adducts, which were not discovered in the Petri dish-cultured HepG2/C3A (Prot et al. 2012).
Researchers have coupled the multicellular co-cultures, containing human hepatocytes (PHH or HepaRG) and various NPCs (e.g. fibroblasts, stellate cells, KCs, endothelial cells monocytes and macrophages), with different perfusion apparatuses to create more sophisticated liver chips (Esch et al. 2015; Prodanov et al. 2016; Rennert et al. 2015; Vernetti et al. 2016), which even reflect zone-specific functions in human liver (Allen et al. 2005; Lee-Montiel et al. 2017; Lee et al. 2007; Prodanov et al. 2016). Compared to the static controls, the established perfused liver organoids demonstrated enhanced albumin synthesis and urea excretion, due to the presence of shear forces and the improved spatial arrangement of the cells (Esch et al. 2015; Prodanov et al. 2016; Rennert et al. 2015) and exhibited potentials to study physiologic zonal responses to hepatotoxins, such as diclofenac (DCF) and APAP (Allen et al. 2005; Lee-Montiel et al. 2017; Lee et al. 2007; Prodanov et al. 2016). Additionally, the hepatocyte-based liver chips incorporated with liver immune cells were able to detect increased trovafloxacin toxicity in the presence of LPS (Vernetti et al. 2016), inferring these biomimetic hepatic models are more physiologically accurate and are useful for studying the immune-mediated drug toxicity. The PHH/KC aggregated liver perfusion models have been reported to maintain long-term (over 42 weeks) stability of albumin and urea production and consistent bioactivity of IL-6, and yielded reliable in vitro drug metabolism data (6 compounds), which were highly correlated with the observed in vivo values (Long et al. 2016; Tsamandouras et al. 2017). The Tannenbaum group further investigated the adverse immune-mediated drug reactions using the PHH/KC-generated liver chips at the metabolome level (Sarkar et al. 2015, 2017). This co-culture platform elaborately depicted the LPS-evoked inflammatory response as confirmed by the elevated overall number of acute phase proteins and increased release of KC-mediated cytokines (e.g. IL-1β, IL-1Ra, IL-6, IL-8 and TNFα), recapitulating an in vivo pro-inflammatory response (Sarkar et al. 2015, 2017). When exposed to DCF, the immunocompetent co-culture systems successfully captured DCF metabolites and identified glycine-conjugated bile acid as a sensitive marker indicating the dose-dependent DCF toxicity (Sarkar et al. 2015, 2017).
Even though the human liver cell-based on-chip liver platforms look promising, the difficulty to obtain human liver materials limits the potential use of this platform to study patient-specific pharmacological and toxicological responses. The development of perfusable liver platforms, encapsulated hiPSC and NPCs, could provide an unlimited supply of human liver cells with diverse genetic backgrounds, which would add opportunity to assess patient-specific drug responses (Schepers et al. 2016). Recently, Schepers et al. (2016) described promising results towards the establishment of the hiPSC-Heps co-culture microfluidic tissue models. These platforms not only showed stable on-chip functionality as evidenced by robust albumin production for 28 days, but also developed a mixed population of differentiated cell types resembling hepatocytes and biliary cells (Schepers et al. 2016), yet thorough assessment at the omics level is still needed to further evaluate these newly developed liver microtissues.
Multi-organs-on-a-chip
The success of the on-chip liver platforms has stimulated investigators to achieve a more systemic level of simulating the physiology of the entire human body in vitro (Bhise et al. 2014). The so-called “body-on-a-chip” system aims to encompass multiple human organ equivalents within one dynamically changing environment, where various cell/tissue culture compartments are connected with an on-chip micro-pump (Atac et al. 2013; Bhise et al. 2014). Compared to the individual organ-on-a-chip, when wrapped with the liver compartments, the multiple organ-on-a-chip systems, taking into account the interactions between organs and tissues, could better reflect the complex organ functions and would ultimately improve the accuracy for understanding and predicting human response to drugs in vivo (Kimura et al. 2018).
Early attempts to create organs-on-a-chip devices have been made by connecting several single organ-on-a-chip platforms. Back to 2004, Lau et al. developed the gut-liver-on-a-chip models using the Caco-2/PHH indirect co-cultures to investigate the human oral bioavailability of 24 randomly chosen marketed drugs (Lau et al. 2004). Based on the “wells within a well” concept, Viravaidya et al. (2004) and Lau et al. (2004) also built multiple-chamber cell culture devices, encapsulating cells derived from liver (PHH or HepG2/C3A), kidney (renal proximal tubule cells), lung (human small airway epithelial cells or rat lung Type II epithelial cells), central nervous system (astrocytes), blood vessels (human aortic or vein endothelial cells) as well as intestine (primary human jejunal enteroids), and conducted proof-of-principle studies in drug toxicity testing. Their successes have encouraged other researchers to design the liver compartment (built with human hepatic cell lines and PHH)-containing multi-organ-chips and to apply the resulting platforms in drug toxicity studies (Bauer et al. 2017; Choucha-Snouber et al. 2013; Esch et al. 2014; Li et al. 2012; Ma et al. 2012; Maschmeyer et al. 2015; Materne et al. 2015a, b; Sung et al. 2010; Vernetti et al. 2017; Wagner et al. 2013; Zhang et al. 2009). These biochips have exhibited functional and metabolic stability during long-term cultures (up to 28 days), enabling the repeated exposures to toxic substances. Besides, these devices have demonstrated cross-talk among different cell culture units, making them more closely resemble the in vivo situation. In general, the application of these multi-organ systems have largely accelerated the generation of physiologically relevant data regarding drug metabolism pharmacokinetics, and disposition as well as their organ-specific toxicities in humans. In 2016, Miller and Shuler (2016) described the development of a 13-organ culture platform, consisting of 14 separable culture chambers that represent both barrier (lung, skin and gastrointestinal tract) and non-barrier tissues (fat, kidney, heart, adrenal glands, liver, spleen, pancreas, bone marrow, brain, muscle). The long-lasting viability of the five cultured human cell lines (including the human hepatocytes, HepG2/C3A) observed in their study has proved the feasibility of constructing a sophisticated in vitro multi-organ platform that allows monitoring of liver-specific functions such as the CYP450 activities and the albumin and urea production (Miller and Shuler 2016). Recently, the multi-organ devices, containing hiPSC, have also been developed by assembling hiPSC differentiated cardiomyocytes (hiPSC-CM), human hepatocytes (HepG2/C3A or PHH organoids) and cells from other tissues (e.g. human skeletal myofibers and iPSC-derived cortical-like neurons) (Oleaga et al. 2016; Zhang et al. 2017). Connecting microfluidic channels with gravity-driven flow, Oleaga et al. (2016) assessed the pharmacological activity of five drugs (doxorubicin, atorvastatin, statins, VPA, APAP and AMAP) in a cardiac-muscle-neuronal-liver platform. Using their four-organ system, the team observed multi-organ toxicity responses to the toxic compounds in line with the published in vivo human toxicity data, indicating the possibility of conducting systemic toxicity screening in vitro. Nevertheless, none of the abovementioned studies have evaluated their models or investigated the drug-associated responses with their multi-organ chips at the omics level.
Bioinformatic approaches
A cell model and cultivation technique that mimic the in vivo toxicity response can be used to gain mechanistic insight in DILI. Mechanistic elements can be measured on different levels using techniques from proteomics, metabolomics as well as transcriptomics. Since proteomic and metabolomic approaches have been reviewed elsewhere, we focus on transcriptomics. Moreover, the transcriptomic level covers regulatory interactions on a genome scale and the technology becomes more and more standardized and affordable. Advances have been made to interpret these transcriptomic profile changes. This section focuses on the interpretation of transcriptomic data and how a toxicity mechanism can be derived from it.
Finding compound-induced transcriptomic changes
The first step in interpreting a transcriptomic profile for elucidating a mechanism is selecting the most relevant genes. Due to the large variability in gene expression measurements, most methods used to identify differentially expressed genes select not only based on the fold change, but also on a significance estimate based on a distribution specifically selected for microarray data or RNA-seq data. These tools compare two conditions, such as with and without the presence of a toxic compound.
Different approaches can be taken regarding transcriptomic datasets of similar toxic compounds. Vatakuti et al. searched for a mechanism that is common among a range of drugs with similar effects. In this case, DEGs were selected only when they were differentially expressed for at least two out of five cholestasis-causing compounds (Vatakuti et al. 2017). The DEGs have been found by performing unpaired moderated t tests using the limma R package (Ritchie et al. 2015). On the contrary, to find a compound-specific mechanism, the DEGs unique for that compound can be used to formulate a specific mechanism. Liguori et al. used PHH from four donors and exposed them to seven quinolone compounds. Trovafloxacin led to the highest number of DEGs in every donor, as found by the software Rosetta Resolver (Weng et al. 2006). From the set of DEGs uniquely found for trovafloxacin, it was deduced which process changes are specific to this exposure, which led to new suggestions for mechanisms (Liguori et al. 2005). Transcriptomic differences can be found that explain interindividual differences in susceptibility to APAP. A correlation analysis per gene among individuals can be used to find genes with low correlation that explain these interindividual differences (Jetten et al. 2016).
DEG analysis can help to fill gaps in a compound’s mechanism of action. When a pathway is known to be activated by a compound, suggestions for the direct downstream targets of this pathway can be generated by finding the most highly DEGs. IJssennagger et al. found targets for the FXR pathway that were differentially expressed upon obeticholic acid exposure in human livers slices using a regularized paired t test (Ijssennagger et al. 2016).
Interpreting gene expression changes
The interpretation of gene expression profiles has become the key activity in toxicological transcriptomics, now that their acquisition, storage and comparison have been greatly facilitated. Functional interpretation of the narrowed-down group of genes is typically done by checking how many genes of a functional gene set are differentially expressed, which is referred to as a gene set enrichment analysis. A functional gene set constitutes of genes that execute the same function or are related to a disease. A function in this sense can be a metabolic activity, signaling transduction activity or any other general function such as apoptosis or cell cycle regulation. Such a function is represented by a gene set. There are varieties in gene set definitions, but Gene Ontology (GO) is the most common (Ashburner et al. 2000; The Gene Ontology Consortium 2019). In case the data are linked to a certain phenotype or disease, a gene set enrichment analysis is useful (Subramanian et al. 2005). However, the genes are not necessarily mechanistically linked to each other in a causal way and are, therefore, not always useful for finding a toxicity mechanism. Indeed, gene set enrichment analyses are competitive, meaning that the whole gene set is considered a good marker of a phenotype, without implying that all genes in the set are related as a mechanism (Goeman and Buhlmann 2007). An enrichment analysis consists of a statistical test that checks for which functions the gene expressions are significantly different. Similar to DEG analysis, a gene set enrichment analysis can find functions that are likely to be altered after exposure to similar toxic compounds. Bell et al. show that the three tested DILI compounds cause significantly many genes in the ribosome biogenesis, calcium signaling and MAPK signaling gene sets to be differentially expressed (Bell et al. 2017). The statistical testing was done using WebGestalt, which employs two-tailed unpaired t tests (Wang et al. 2017). This approach can also be used to get an indication of the effect of different combinations of exposures. For example, when exposing 3D multicellular microtissues with APAP, the amount of DEGs that have mitochondria-related GO terms is greatly increased in the presence of lipopolysaccharides, which suggests an interplay between lipopolysaccharides and APAP in mitochondria-related processes (Jiang et al. 2019).
In the identification of a mechanism of toxicity, using pathways in an enrichment analysis is much more valuable than using gene sets that correlate with a phenotype of toxicity. The causal structure of a pathway helps to interpret the transcriptomic effects caused by a toxic compound. Wolters et al. generated a hypothesis for the effect of valproic acid on mitochondria after the pathway-enrichment analysis from ConsensusPathDB highlighted the mitochondria–nucleus signaling pathway (Kamburov et al. 2013; Wolters et al. 2018).
Using pathways in enrichment analyses leads to hypotheses with suggested causal interactions between genes. These interactions simplify the design of validation experiments. By intervening in pathways that are known to be activated by a toxic compound, it can be checked whether a pathway is crucial for the toxic phenotype. For example, Fredriksson et al. validated whether lysosome rupture causes oxidative stress (Fredriksson et al. 2014). Silencing key lysosome rupture genes using RNA interference and checking whether the oxidative level changes in the predicted direction validates the relevance and direction of the effect of these genes. The order of the gene cascade can be validated by silencing the upstream gene and measuring the downstream transcript expression or protein expression or activity. If the measured gene is affected by the silencing, it is downstream of the silenced gene. For HepG2 cells, activated PERK was found to be upstream of EIF4A1, whereas CHOP was found to be downstream (Fredriksson et al. 2014).
Time series can improve the resolution of mechanistic findings
Time series gene expression data can be used to put transcriptomic profile changes of different cell models, compounds and compound concentrations into perspective. The temporal changes of the transcriptomic profile of a sample can be well displayed in a plot following from a Principle Component Analysis (PCA). A PCA is a reduction technique that searches for a linear combination of, in this case, gene expressions that represent the differences between samples. Elferink et al. showed the effect of APAP on human liver slices, compared with the transcriptomic changes of degrading liver slices without exposure (Elferink et al. 2011). Such an analysis can be a starting point for determining a useful compound concentration for a toxicity study, especially when using a degrading cell model. A manual inspection of a time series can be performed when a certain process is of interest. For example, the gene expression of human drug transporters was stable during APAP exposure (Elferink et al. 2011). Unexpected results can be neglected when they are negative. A good way to investigate time series data in detail is to visualize a pathway that is expected to be changed by mapping the data onto it (Elferink et al. 2011; Verheijen et al. 2018).
Interpreting transcriptomic data as gene expression patterns is more indicative of the whole toxicity development than comparing single time points. Gene expression patterns can be used to divide genes into early- and late-responding genes. Van Delft et al. have used HepG2 cells to investigate the toxicity responses to benzo[a]pyrene on transcriptomic level in time series. By clustering the differentially expressed gene expression patterns using the tool Short Time-series Expression Miner, they were able to categorize genes into four groups according to their response time (Ernst et al. 2005). Subsequent interpretation of these groups of genes helped to generate hypotheses regarding the toxicity development: DNA adducts form early on and changes in cell cycle regulations follow (van Delft et al. 2010). Further analysis can be done on a significant cluster with an interesting pattern, aiming at, for example, finding a process that only happens at a certain time point, but seems to be crucial for toxicity development. For example, by focusing on one cluster, Caiment et al. found that only around 24 h of benzo[a]pyrene exposure, many genes related to transmembrane transporter activity were upregulated (Caiment et al. 2015).
The selection of time points for RNA collection is crucial for the gene expression patterns to provide sufficient extra information on the toxicity mechanism. The range of time points should preferably cover the primary toxicity response. A good way of determining the range of time points is selection based on phenotypic changes (van Delft et al. 2010; Waters et al. 2003). Gene expression patterns not showing much alteration typically add little information (Van Delft et al. 2010). Even when the time points have been selected carefully, it is possible that a time series is not reproduced. For this purpose, dynamic time warping has been developed for omics data specifically (Cavill et al. 2013). Dynamic time warping looks for shifts in the sequence when comparing two time series. The shifts can reveal that time series are more similar in respect to the sequence of events, than when comparing single time points only. This way, HepG2 and Caco-2 cells have been found to be dissimilar in the sequence of their gene expression in addition to the obvious differences per time point (Deferme et al. 2015a).
Time series have the potential of increasing the sensitivity of DEG finding. Genes that have a slight but consistently altered expression pattern can be identified as differentially expressed when taking into account several time points. For this reason, the tool maSigPro has been developed for microarray data (Conesa et al. 2006) and Next maSigPro as its adaptation to work with RNA-Seq data (Nueda et al. 2014). However, this tool has not been used in DILI research before. Using this tool, Ishimoto et al. have found a set of genes that were consistently upregulated to a small extent in a coculture of macrophages and intestinal epithelial cells, which probably would not have been found using methods that focus on single time points only (Ishimoto et al. 2010). The gene TNFR1, which only had a fold change of more than 1.5 for 2 of the 5 time points, was suggested to make the cells more sensitive to TNF-α. Such a weak effect could have been missed in a single cell culture. The sensitization is suspected to be a coculturing effect, which is a finding that should not be missed due to the use of tools not capable of analyzing time series.
Selecting genes using multiple time points can be done using linear regression, in which the size of the linear regression parameters indicates to what extent the gene expression changes steadily upon compound exposure. maSigPro uses this method to filter genes of small expression changes (Conesa et al. 2006). Subsequently, it tries to find significantly differentially expressed genes using the Lasso regularization method. Its extension to generalized linear models can be used for discrete gene expression data, allowing the analysis of RNA-Seq data (Nueda et al. 2014). Hannibal et al. have found the gene SVEP1 to show increasing expression over the course of pregnancy using maSigPro on RNA-Seq data from 24 samples (Hannibal et al. 2018). Gene expression time series therefore have the potential of improving the sensitivity of transcriptome analyses to single-gene resolution.
Revealing causal toxicity mechanisms
Generating hypotheses for an overarching mechanism is facilitated greatly when based on a series of activated pathways, compared to using a list of DEGs. However, events happening in sequence are not necessarily caused one after another. Importantly, a validation in different cell models does not validate causality either. Hence, a workflow is required to determine the network structure giving rise to correlating gene expressions. Time series gene expression data allow to focus on parts of the toxicity-causing mechanism separately by proper time point selection. Testing mechanisms at different time points can verify mechanistic links that are supported by biological or pathway-specific knowledge. A method for revealing causal relationships between genes requires more than simple correlation. The two methods that will be discussed here are graphical Gaussian networks, which are based on calculating partial correlations and Bayesian networks. Although these methods could be very useful in finding causal mechanisms of DILI, these methods have not been used in DILI studies so far.
A graphical Gaussian network can be a representation of causal links between genes (Bühlmann et al. 2014). In this context, genes are believed to have a regulatory connection when their partial correlation is nonzero. The partial correlation is the correlation between the expression levels of two genes after removing effects of other genes. By removing the effect of other genes, some correlations are explained through indirect effects (Fig. 1, left boxes). In practice, the estimation of the partial correlations becomes less reliable when the number of samples is much lower than the number of genes. Therefore, it is advised to construct a graphical Gaussian network from a small subset of genes. In addition, the partial correlations will have to be estimated by analyzing subsets of three genes at once. For example, Wille et al. focused on two isoprenoid pathways in Arabidopsis thaliana and constructed a graphical Gaussian network to find cross-talk between the pathways (Wille et al. 2004). The network suggested regulatory mechanisms on the single gene level. Therefore, graphical Gaussian networks can be used to identify regulatory mechanisms between pathways, resulting in a more complete picture of the pathways involved in mechanism causing toxicity.

Visualizations of networks that can be used in revealing toxicity mechanisms. The correlation network can be constructed by calculating the correlation between the time series of different genes and picking a threshold. The other networks are explained in the text
A graphical Gaussian network can therefore hint towards interaction between pathways. The interactions between pathways that were found using graphical Gaussian networks can be validated by silencing the effectors of the one pathway and measuring the effect on the other and vice versa (Fredriksson et al. 2014). For example, the protective effect of Nrf2 in the HepG2 diclofenac toxicity mechanism has been found not to interact with the PERK-CHOP apoptosis pathway, but these mechanisms appear to be different (Fredriksson et al. 2014).
Next to verifying hypotheses, a graphical Gaussian network can also be used to generate hypotheses. Ma et al. demonstrated how a graphical Gaussian network can be used to link pathways to previously unassociated genes (Ma et al. 2007). Their aim was to construct a genome-scale graphical Gaussian network of the A. thaliana transcriptome. They circumvented the network size limitation by constructing many networks of size 2000 and combining them by picking the lowest partial correlation values per gene pair. Genes without functional annotation were linked to transcription factors in the plant’s cold response (Ma et al. 2007). These findings open new leads for a causal mechanism. It should be noted that the results emerging from graphical Gaussian networks are more reliable as compared to simple correlation. These genes are found not only to act in a similar or closely associated process, but also directly impose (or are imposed by) a regulatory effect. A striking example for this network is genes encoding ribosomal proteins, which are among the genes with the highest Pearson correlation (Ma et al. 2007). However, the regulatory effect on the genes they are correlated with is doubtful. The partial correlations for these genes were much lower and, therefore, more in accordance with what is expected. Partial correlation is a more reliable measure of functional and mechanistic relatedness than simple correlation.
Bayesian networks depict directed causal relationships between genes. More precisely, a Bayesian network describes the probabilistic relation between gene expressions (Fig. 1, top right). An optimization procedure is commonly employed to find a network that fits the data. During the optimization, it is determined which genes affect which genes and what the parameters of the distributions are. To restrict the number of possible networks and distributions, a small number of genes is selected to construct the network and a maximum number of incoming edges for a node is set (Husmeier 2003). Still, finding the optimal network is ambiguous, since different networks could generate the same data. Generating an ensemble of networks and checking for consistency in the gene interactions poses a remedy for this problem. Gendelman et al. have used such an ensemble of networks to find which gene in the model has the largest impact on the phenotype. It was found that in silico up- or downregulation of TRIB1 led to the most significant change in the G1-S phase transition. They had found that TRIB1 affects the MEK/ERK pathway via NFκB (Gendelman et al. 2017). In brief, performing simulations with optimized Bayesian networks can help to find pathway regulators important for phenotype development. A prediction regarding the importance of a gene for a phenotype can be validated using RNA interference. Genes found to cause the toxic effect can be silenced to check how important they are for the toxic phenotype. By performing a silencing experiment for every important gene in the mechanism, the TRIB1 gene was confirmed to cause an increase in the amount of cells in the G1 state (Gendelman et al. 2017). The Bayesian network had been able to simulate and predict the silencing effect successfully.
Dynamic Bayesian networks can be used to disentangle the genetic interactions among a set of genes in a pathway using the whole time series at once (Fig. 1, bottom right). It describes how the elements in a network influence the same elements at the next time point. This way, the network is allowed to have cycles, whereas static Bayesian networks do not. Fröhlich et al. have optimized a dynamic Bayesian network with protein and gene expression data to find the interactions between and within the MEK/ERK and Hedgehog pathway (Fröhlich et al. 2015). The MEK/ERK edges found in the literature have been confirmed with high accuracy. For the Hedgehog pathway, it remained unclear whether it is activated via HHIP or PTCH1. The optimized network after robustness check suggested that a mechanism via HHIP is more probable (Fröhlich et al. 2015). This approach can be used to select one hypothesis over the other.
A Bayesian network can be constructed to find genes that are likely to be directly affected by a compound, which can be considered the start of the mechanism. Jennen et al. have optimized a Bayesian network by searching for links between blood cotinine levels and leukocytes gene expression (Jennen et al. 2015). Blood cotinine levels are a proxy for cigarette smoke exposure. The network was created using the expression of all differentially expressed genes and used to determine which genes are most directly affected by the cotinine level. Genes found to interact with cotinine form the so-called Markov blanket around cotinine. The genes in the Markov blanket were shown to fit in the mechanistic framework for understanding the effects of smoking (Jennen et al. 2015). Investigating the genes in the Markov blanket of the toxic compound in a Bayesian network can help finding the initial responding pathways upon exposure. In brief, causality-inferring methods such as graphical Gaussian networks and Bayesian networks are useful in assessing causality.
Conclusions and future perspectives
DILI is the most prominent cause of ALF (around 50% of the ALF cases) in the United States and Western Europe (Biolato et al. 2017). Also, it is a primary reason for drug regulatory actions and market withdrawal (Davern et al. 2011). Over the past 50 years, compound-induced hepatotoxicity caused 15 of the 47 drugs (32%) withdrawn from the market (Onakpoya et al. 2018), making DILI a major problem for both patient health and drug development in pharmaceutical industry (Regev 2014).
In the past 2 decades, human toxicogenomics-based in vitro assays have been developed as an alternative to animal testing for assessing chemical toxicity. In this review, we have discussed the in vitro human liver models derived from immortal cell lines (HepG2 and HepaRG), PHH and hSKP- or iPSC-derived hepatocyte-like cells, and their applications in omics-based drug-induced hepatotoxicity studies. Additionally, we have introduced the available bioinformatic approaches that have the potential for analyzing the toxicogenomic data collected from these in vitro liver models and their contributions to the understanding of mechanisms of DILI at the omics level.
The significant progress in the development of human liver cell-based in vitro models in combination with applications of omics and bioinformatics tools deepen our understanding of molecular mechanisms of human DILI. In comparison to human hepatocellular carcinoma cell lines, PHH are considered as the ‘gold standard’ for drug toxicity studies (Gerets et al. 2012). The PHH-derived in vitro liver models are capable of detecting interindividual differences in the sensitivity towards DILI outcomes due to the heterogeneous genetic makeup of human population (Lin and Khetani 2016). As an alternative to PHH, hiPSC-Heps, derived from different donors, also capture the genetic background of individuals, and therefore, reflect interindividual variability in susceptibility to drugs (Lin and Khetani 2016) without raising ethical concerns as do the hESC-Heps (Volarevic et al. 2018). Thus, when properly induced, the iPSC-Hep-based liver models seem to hold a great potential for advancing the study of individual-specific toxicological responses (Matsa et al. 2016). Compared to the monocultures, in vitro human liver systems that incorporate human hepatocytes with NPCs replicate human immunity-associated variances in drug responsiveness, facilitating the study of immune-mediated drug hypersensitivity or idiosyncrasy. In contrast to conventional 2D cultures, sophisticated engineering techniques, such as micropatterning, bio-printing and microfluidic devices, allow more precise control over the spatial arrangement and the microenvironment of liver cells, leading to stabilized hepatic functions over longer time periods and enabling the long-term chronic toxicity studies in vitro (Underhill and Khetani 2018). Recent advances in the development of organs-on-a-chip devices, assembling multiple functionally activated tissue models in vitro, further improve accuracy of simulating the complexities of the human body by taking into account the interactions between organs and tissues (Kimura et al. 2018). Since multiple factors are involved in the onset and the development of (idiosyncratic) DILI in humans, further progress in the organs-on-chip platforms and iPSC technology are anticipated for the development of highly biomimetic in vitro culture systems that allow us to use toxicogenomic analysis to investigate DILI mechanisms in physiologically relevant conditions, moving us closer to bridge the in vitro–in vivo gap and helping to accelerate personalized drug toxicology studies.
Transcriptomic data analysis has made successful use of gene expression interpretation tools such as pathway enrichment analysis and GO term enrichment analysis. However, when the aim is to find a mechanism, such as a non-acute toxicity mechanism, tools are required to find causality between biological events, such as pathway activation or between differentially expressed genes. Two tools that serve this purpose are graphical Gaussian network and (dynamic) Bayesian networks. Graphical Gaussian networks can be used to remove correlation that arises from indirect interactions between genes and Bayesian networks aim at finding a directed network that would depict dependencies between genes. Ideas have been presented to extend upon functional enrichment analyses using these causality-revealing tools to find pathway cross-talk, direct compound effects, toxicity effectors and hypothesis selection.
The discovery and development of new cell models and cultivation techniques can improve the lifespan and in vivo resemblance, allowing for more relevant toxicity studies. As omics analysis methods become more widely available, opportunities arise for mechanistic toxicity studies. A proper understanding of toxicity mechanisms will strengthen in vitro approaches aiming at predicting and preventing compound-induced hepatotoxicity, drug-induced liver failure, and more specifically, acute liver failure.
References
Afshari CA, Hamadeh HK, Bushel PR (2011) The evolution of bioinformatics in toxicology: advancing toxicogenomics. Toxicol Sci 120(Suppl 1):S225–S237. https://doi.org/10.1093/toxsci/kfq373
Allen JW, Khetani SR, Bhatia SN (2005) In vitro zonation and toxicity in a hepatocyte bioreactor. Toxicol Sci 84(1):110–119. https://doi.org/10.1093/toxsci/kfi052
Apati A, Varga N, Berecz T, Erdei Z, Homolya L, Sarkadi B (2019) Application of human pluripotent stem cells and pluripotent stem cell-derived cellular models for assessing drug toxicity. Expert Opin Drug Metab Toxicol 15(1):61–75. https://doi.org/10.1080/17425255.2019.1558207
Ashburner M, Ball CA, Blake JA et al (2000) Gene ontology: tool for the unification of biology. Nat Genet 25(1):25–29. https://doi.org/10.1038/75556
Asplund A, Pradip A, van Giezen M et al (2016) One standardized differentiation procedure robustly generates homogenous hepatocyte cultures displaying metabolic diversity from a large panel of human pluripotent stem cells. Stem Cell Rev 12(1):90–104. https://doi.org/10.1007/s12015-015-9621-9
Atac B, Wagner I, Horland R et al (2013) Skin and hair on-a-chip: in vitro skin models versus ex vivo tissue maintenance with dynamic perfusion. Lab Chip 13(18):3555–3561. https://doi.org/10.1039/c3lc50227a
Atienzar FA, Blomme EA, Chen M et al (2016) Key challenges and opportunities associated with the use of in vitro models to detect human DILI: integrated risk assessment and mitigation plans. Biomed Res Int 2016:9737920. https://doi.org/10.1155/2016/9737920
Aziz AUR, Geng C, Fu M, Yu X, Qin K, Liu B (2017) The role of microfluidics for organ on chip simulations. Bioengineering (Basel). https://doi.org/10.3390/bioengineering4020039
Bale SS, Vernetti L, Senutovitch N et al (2014) In vitro platforms for evaluating liver toxicity. Exp Biol Med (Maywood) 239(9):1180–1191. https://doi.org/10.1177/1535370214531872
Bauer S, Wennberg Huldt C, Kanebratt KP et al (2017) Functional coupling of human pancreatic islets and liver spheroids on-a-chip: towards a novel human ex vivo type 2 diabetes model. Sci Rep 7(1):14620. https://doi.org/10.1038/s41598-017-14815-w
Bavli D, Prill S, Ezra E et al (2016) Real-time monitoring of metabolic function in liver-on-chip microdevices tracks the dynamics of mitochondrial dysfunction. Proc Natl Acad Sci USA 113(16):E2231–E2240. https://doi.org/10.1073/pnas.1522556113
Beijer NR, Vasilevich AS, Pilavci B et al (2017) TopoWellPlate: a well-plate-based screening platform to study cell–surface topography interactions. Adv Biosyst 1(4):1700002
Bell CC, Hendriks DF, Moro SM et al (2016) Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Sci Rep 6:25187. https://doi.org/10.1038/srep25187
Bell CC, Lauschke VM, Vorrink SU et al (2017) Transcriptional, functional, and mechanistic comparisons of stem cell-derived hepatocytes, HepaRG cells, and three-dimensional human hepatocyte spheroids as predictive in vitro systems for drug-induced liver injury. Drug Metab Disposition Biol Fate Chem 45(4):419–429. https://doi.org/10.1124/dmd.116.074369
Bell CC, Dankers ACA, Lauschke VM et al (2018) Comparison of hepatic 2D sandwich cultures and 3D spheroids for long-term toxicity applications: a multicenter study. Toxicol Sci 162(2):655–666. https://doi.org/10.1093/toxsci/kfx289
Benesic A, Gerbes AL (2015) Drug-induced liver injury and individual cell models. Dig Dis 33(4):486–491. https://doi.org/10.1159/000374094
Berger DR, Ware BR, Davidson MD, Allsup SR, Khetani SR (2015) Enhancing the functional maturity of induced pluripotent stem cell-derived human hepatocytes by controlled presentation of cell-cell interactions in vitro. Hepatology 61(4):1370–1381. https://doi.org/10.1002/hep.27621
Bhise NS, Ribas J, Manoharan V et al (2014) Organ-on-a-chip platforms for studying drug delivery systems. J Control Release 190:82–93. https://doi.org/10.1016/j.jconrel.2014.05.004
Bhise NS, Manoharan V, Massa S et al (2016) A liver-on-a-chip platform with bioprinted hepatic spheroids. Biofabrication 8(1):014101. https://doi.org/10.1088/1758-5090/8/1/014101
Biolato M, Araneo C, Marrone G et al (2017) Liver transplantation for drug-induced acute liver failure. Eur Rev Med Pharmacol Sci 21(1 Suppl):37–45
Black MB, Budinsky RA, Dombkowski A et al (2012) Cross-species comparisons of transcriptomic alterations in human and rat primary hepatocytes exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Sci 127(1):199–215. https://doi.org/10.1093/toxsci/kfs069
Briede JJ, Deferme L, Wolters JEJ et al (2018) A cross-omics approach to investigate temporal gene expression regulation by 5-hydroxymethylcytosine via TBH-derived oxidative stress showed involvement of different regulatory kinases. Toxicol In Vitro 48:318–328. https://doi.org/10.1016/j.tiv.2018.02.006
Bruderer R, Bernhardt OM, Gandhi T et al (2015) Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol Cell Proteomics 14(5):1400–1410. https://doi.org/10.1074/mcp.M114.044305
Bühlmann P, Kalisch M, Meier L (2014) High-dimensional statistics with a view toward applications in biology. Annu Rev Stat Appl 1(1):255–278. https://doi.org/10.1146/annurev-statistics-022513-115545
Caiment F, Gaj S, Claessen S, Kleinjans J (2015) High-throughput data integration of RNA-miRNA-circRNA reveals novel insights into mechanisms of benzo[a]pyrene-induced carcinogenicity. Nucleic Acids Res 43(5):2525–2534. https://doi.org/10.1093/nar/gkv115
Camp JG, Sekine K, Gerber T et al (2017) Multilineage communication regulates human liver bud development from pluripotency. Nature 546(7659):533–538. https://doi.org/10.1038/nature22796
Cavill R, Kleinjans J, Briede J (2013) DTW4OmicsL comparing patterns in biological time series. PLoS One. https://doi.org/10.1371/journal.pone.0071823.g001
Chalasani N, Bonkovsky HL, Fontana R et al (2015) Features and outcomes of 899 patients with drug-induced liver injury: the DILIN prospective study. Gastroenterology 148(7):1340.e7–1352.e7. https://doi.org/10.1053/j.gastro.2015.03.006
Chatterjee N, Eom HJ, Choi J (2014) A systems toxicology approach to the surface functionality control of graphene-cell interactions. Biomaterials 35(4):1109–1127. https://doi.org/10.1016/j.biomaterials.2013.09.108
Cheng S, Prot JM, Leclerc E, Bois FY (2012) Zonation related function and ubiquitination regulation in human hepatocellular carcinoma cells in dynamic vs. static culture conditions. BMC Genom 13:54. https://doi.org/10.1186/1471-2164-13-54
Choi SM, Kim Y, Shim JS et al (2013) Efficient drug screening and gene correction for treating liver disease using patient-specific stem cells. Hepatology 57(6):2458–2468. https://doi.org/10.1002/hep.26237
Choucha-Snouber L, Aninat C, Grsicom L et al (2013) Investigation of ifosfamide nephrotoxicity induced in a liver-kidney co-culture biochip. Biotechnol Bioeng 110(2):597–608. https://doi.org/10.1002/bit.24707
Conesa A, Nueda MJ, Ferrer A, Talon M (2006) maSigPro: a method to identify significantly differential expression profiles in time-course microarray experiments. Bioinformatics 22(9):1096–1102. https://doi.org/10.1093/bioinformatics/btl056
Copple IM, den Hollander W, Callegaro G et al (2019) Characterisation of the NRF2 transcriptional network and its response to chemical insult in primary human hepatocytes: implications for prediction of drug-induced liver injury. Arch Toxicol 93(2):385–399. https://doi.org/10.1007/s00204-018-2354-1
Cui Y, Paules RS (2010) Use of transcriptomics in understanding mechanisms of drug-induced toxicity. Pharmacogenomics 11(4):573–585. https://doi.org/10.2217/pgs.10.37
Davern TJ, Chalasani N, Fontana RJ et al (2011) Acute hepatitis E infection accounts for some cases of suspected drug-induced liver injury. Gastroenterology 141(5):1665–1672. https://doi.org/10.1053/j.gastro.2011.07.051 (e1-9)
Davidson MD, Ware BR, Khetani SR (2015) Stem cell-derived liver cells for drug testing and disease modeling. Discov Med 19(106):349–358
Deferme L, Briede JJ, Claessen SM, Jennen DG, Cavill R, Kleinjans JC (2013) Time series analysis of oxidative stress response patterns in HepG2: a toxicogenomics approach. Toxicology 306:24–34. https://doi.org/10.1016/j.tox.2013.02.001
Deferme L, Briede JJ, Claessen SM, Cavill R, Kleinjans JC (2015a) Cell line-specific oxidative stress in cellular toxicity: a toxicogenomics-based comparison between liver and colon cell models. Toxicol In Vitro 29(5):845–855. https://doi.org/10.1016/j.tiv.2015.03.007
Deferme L, Wolters J, Claessen S, Briede J, Kleinjans J (2015b) Oxidative stress mechanisms do not discriminate between genotoxic and nongenotoxic liver carcinogens. Chem Res Toxicol 28(8):1636–1646. https://doi.org/10.1021/acs.chemrestox.5b00222
Deferme L, Wolters JE, Claessen SM et al (2016) Dynamic interplay between the transcriptome and methylome in response to oxidative and alkylating stress. Chem Res Toxicol 29(9):1428–1438. https://doi.org/10.1021/acs.chemrestox.6b00090
Elferink MG, Olinga P, van Leeuwen EM et al (2011) Gene expression analysis of precision-cut human liver slices indicates stable expression of ADME-Tox related genes. Toxicol Appl Pharmacol 253(1):57–69. https://doi.org/10.1016/j.taap.2011.03.010
Ernst J, Nau GJ, Bar-Joseph Z (2005) Clustering short time series gene expression data. Bioinformatics 21(Suppl 1):i159–i168. https://doi.org/10.1093/bioinformatics/bti1022
Esch MB, Mahler GJ, Stokol T, Shuler ML (2014) Body-on-a-chip simulation with gastrointestinal tract and liver tissues suggests that ingested nanoparticles have the potential to cause liver injury. Lab Chip 14(16):3081–3092. https://doi.org/10.1039/c4lc00371c
Esch MB, Prot JM, Wang YI et al (2015) Multi-cellular 3D human primary liver cell culture elevates metabolic activity under fluidic flow. Lab Chip 15(10):2269–2277. https://doi.org/10.1039/c5lc00237k
Foster AJ, Chouhan B, Regan SL et al (2019) Integrated in vitro models for hepatic safety and metabolism: evaluation of a human liver-chip and liver spheroid. Arch Toxicol 93(4):1021–1037. https://doi.org/10.1007/s00204-019-02427-4
Fredriksson L, Wink S, Herpers B et al (2014) Drug-induced endoplasmic reticulum and oxidative stress responses independently sensitize toward TNFα-mediated hepatotoxicity. Toxicol Sci 140(1):144–159. https://doi.org/10.1093/toxsci/kfu072
Fröhlich H, Bahamondez G, Götschel F, Korf U (2015) Dynamic Bayesian network modeling of the interplay between EGFR and Hedgehog Signaling. PLoS ONE 10(11):e0142646. https://doi.org/10.1371/journal.pone.0142646
Gao X, Liu Y (2017) A transcriptomic study suggesting human iPSC-derived hepatocytes potentially offer a better in vitro model of hepatotoxicity than most hepatoma cell lines. Cell Biol Toxicol 33(4):407–421. https://doi.org/10.1007/s10565-017-9383-z
Gendelman R, Xing H, Mirzoeva OK et al (2017) Bayesian network inference modeling identifies TRIB1 as a novel regulator of cell-cycle progression and survival in cancer cells. Cancer Res 77(7):1575–1585. https://doi.org/10.1158/0008-5472.CAN-16-0512
Gerets HH, Tilmant K, Gerin B et al (2012) Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol Toxicol 28(2):69–87. https://doi.org/10.1007/s10565-011-9208-4
Glicksman MA (2018) Induced pluripotent stem cells: the most versatile source for stem cell therapy. Clin Ther 40(7):1060–1065. https://doi.org/10.1016/j.clinthera.2018.06.004
Godoy P, Hewitt NJ, Albrecht U et al (2013) Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch Toxicol 87(8):1315–1530. https://doi.org/10.1007/s00204-013-1078-5
Goeman JJ, Buhlmann P (2007) Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics 23(8):980–987. https://doi.org/10.1093/bioinformatics/btm051
Goetz AK, Dix DJ (2009) Toxicogenomic effects common to triazole antifungals and conserved between rats and humans. Toxicol Appl Pharmacol 238(1):80–89. https://doi.org/10.1016/j.taap.2009.04.016
Gomez-Lechon MJ, Castell JV, Donato MT (2007) Hepatocytes—the choice to investigate drug metabolism and toxicity in man: in vitro variability as a reflection of in vivo. Chem Biol Interact 168(1):30–50. https://doi.org/10.1016/j.cbi.2006.10.013
Gu X, Manautou JE (2012) Molecular mechanisms underlying chemical liver injury. Expert Rev Mol Med 14:e4. https://doi.org/10.1017/S1462399411002110
Han Y, Zhao J, Huang R, Xia M, Wang D (2018) Omics-based platform for studying chemical toxicity using stem cells. J Proteome Res 17(1):579–589. https://doi.org/10.1021/acs.jproteome.7b00693
Hannibal RL, Cardoso-Moreira M, Chetty SP et al (2018) Investigating human placentation and pregnancy using first trimester chorionic villi. Placenta 65:65–75. https://doi.org/10.1016/j.placenta.2018.03.005
Harris AJ, Dial SL, Casciano DA (2004) Comparison of basal gene expression profiles and effects of hepatocarcinogens on gene expression in cultured primary human hepatocytes and HepG2 cells. Mutat Res 549(1–2):79–99. https://doi.org/10.1016/j.mrfmmm.2003.11.014
Hart SN, Li Y, Nakamoto K, Subileau EA, Steen D, Zhong XB (2010) A comparison of whole genome gene expression profiles of HepaRG cells and HepG2 cells to primary human hepatocytes and human liver tissues. Drug Metab Disposition Biol Fate Chem 38(6):988–994. https://doi.org/10.1124/dmd.109.031831
Huch M, Gehart H, van Boxtel R et al (2015) Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 160(1–2):299–312. https://doi.org/10.1016/j.cell.2014.11.050
Hui EE, Bhatia SN (2007) Micromechanical control of cell-cell interactions. Proc Natl Acad Sci USA 104(14):5722–5726. https://doi.org/10.1073/pnas.0608660104
Husmeier D (2003) Sensitivity and specificity of inferring genetic regulatory interactions from microarray experiments with dynamic Bayesian networks. Bioinformatics 19(17):2271–2282. https://doi.org/10.1093/bioinformatics/btg313
Ijssennagger N, Janssen AWF, Milona A et al (2016) Gene expression profiling in human precision cut liver slices in response to the FXR agonist obeticholic acid. J Hepatol 64(5):1158–1166. https://doi.org/10.1016/j.jhep.2016.01.016
Ishimoto Y, Nakai Y, Satsu H, Totsuka M, Shimizu M (2010) Transient up-regulation of immunity- and apoptosis-related genes in Caco-2 cells cocultured with THP-1 cells evaluated by DNA microarray analysis. Biosci Biotechnol Biochem 74(2):437–439. https://doi.org/10.1271/bbb.90732
Jennen DG, Magkoufopoulou C, Ketelslegers HB, van Herwijnen MH, Kleinjans JC, van Delft JH (2010) Comparison of HepG2 and HepaRG by whole-genome gene expression analysis for the purpose of chemical hazard identification. Toxicol Sci 115(1):66–79. https://doi.org/10.1093/toxsci/kfq026
Jennen D, Ruiz-Aracama A, Magkoufopoulou C et al (2011) Integrating transcriptomics and metabonomics to unravel modes-of-action of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in HepG2 cells. BMC Syst Biol 5:139. https://doi.org/10.1186/1752-0509-5-139
Jennen DGJ, van Leeuwen DM, Hendrickx DM, Gottschalk RWH, van Delft JHM, Kleinjans JCS (2015) Bayesian network inference enables unbiased phenotypic anchoring of transcriptomic responses to cigarette smoke in humans. Chem Res Toxicol 28(10):1936–1948. https://doi.org/10.1021/acs.chemrestox.5b00145
Jeon H, Kang K, Park SA et al (2017) Generation of multilayered 3D structures of HepG2 cells using a bio-printing technique. Gut Liver 11(1):121–128. https://doi.org/10.5009/gnl16010
Jetten MJ, Kleinjans JC, Claessen SM, Chesne C, van Delft JH (2013) Baseline and genotoxic compound induced gene expression profiles in HepG2 and HepaRG compared to primary human hepatocytes. Toxicol In Vitro 27(7):2031–2040. https://doi.org/10.1016/j.tiv.2013.07.010
Jetten MJ, Ruiz-Aracama A, Coonen ML et al (2016) Interindividual variation in gene expression responses and metabolite formation in acetaminophen-exposed primary human hepatocytes. Arch Toxicol 90(5):1103–1115. https://doi.org/10.1007/s00204-015-1545-2
Jiang J, Briede JJ, Jennen DG et al (2015a) Increased mitochondrial ROS formation by acetaminophen in human hepatic cells is associated with gene expression changes suggesting disruption of the mitochondrial electron transport chain. Toxicol Lett 234(2):139–150. https://doi.org/10.1016/j.toxlet.2015.02.012
Jiang J, Wolters JE, van Breda SG, Kleinjans JC, de Kok TM (2015b) Development of novel tools for the in vitro investigation of drug-induced liver injury. Expert Opin Drug Metab Toxicol 11(10):1523–1537. https://doi.org/10.1517/17425255.2015.1065814
Jiang J, Mathijs K, Timmermans L et al (2017) Omics-based identification of the combined effects of idiosyncratic drugs and inflammatory cytokines on the development of drug-induced liver injury. Toxicol Appl Pharmacol 332:100–108. https://doi.org/10.1016/j.taap.2017.07.014
Jiang J, Messner S, Kelm JM et al (2019) Human 3D multicellular microtissues: an upgraded model for the in vitro mechanistic investigation of inflammation-associated drug toxicity. Toxicol Lett 312:34–44. https://doi.org/10.1016/j.toxlet.2019.05.004
Kamburov A, Stelzl U, Lehrach H, Herwig R (2013) The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res 41(Database issue):D793–D800. https://doi.org/10.1093/nar/gks1055
Khetani SR, Bhatia SN (2008) Microscale culture of human liver cells for drug development. Nat Biotechnol 26(1):120–126. https://doi.org/10.1038/nbt1361
Khetani SR, Kanchagar C, Ukairo O et al (2013) Use of micropatterned cocultures to detect compounds that cause drug-induced liver injury in humans. Toxicol Sci 132(1):107–117. https://doi.org/10.1093/toxsci/kfs326
Khong YM, Zhang J, Zhou S et al (2007) Novel intra-tissue perfusion system for culturing thick liver tissue. Tissue Eng 13(9):2345–2356. https://doi.org/10.1089/ten.2007.0040
Kienhuis AS, van de Poll MC, Dejong CH et al (2009) A toxicogenomics-based parallelogram approach to evaluate the relevance of coumarin-induced responses in primary human hepatocytes in vitro for humans in vivo. Toxicol In Vitro 23(6):1163–1169. https://doi.org/10.1016/j.tiv.2009.06.005
Kier LD, Neft R, Tang L et al (2004) Applications of microarrays with toxicologically relevant genes (tox genes) for the evaluation of chemical toxicants in Sprague Dawley rats in vivo and human hepatocytes in vitro. Mutat Res 549(1–2):101–113. https://doi.org/10.1016/j.mrfmmm.2003.11.015
Kimura H, Sakai Y, Fujii T (2018) Organ/body-on-a-chip based on microfluidic technology for drug discovery. Drug Metab Pharmacokinet 33(1):43–48. https://doi.org/10.1016/j.dmpk.2017.11.003
Kizawa H, Nagao E, Shimamura M, Zhang G, Torii H (2017) Scaffold-free 3D bio-printed human liver tissue stably maintains metabolic functions useful for drug discovery. Biochem Biophys Rep 10:186–191. https://doi.org/10.1016/j.bbrep.2017.04.004
Kozyra M, Johansson I, Nordling A, Ullah S, Lauschke VM, Ingelman-Sundberg M (2018) Human hepatic 3D spheroids as a model for steatosis and insulin resistance. Sci Rep 8(1):14297. https://doi.org/10.1038/s41598-018-32722-6
Krause P, Saghatolislam F, Koenig S, Unthan-Fechner K, Probst I (2009) Maintaining hepatocyte differentiation in vitro through co-culture with hepatic stellate cells. Vitro Cell Dev Biol Anim 45(5–6):205–212. https://doi.org/10.1007/s11626-008-9166-1
Kullak-Ublick GA, Andrade RJ, Merz M et al (2017) Drug-induced liver injury: recent advances in diagnosis and risk assessment. Gut 66(6):1154–1164. https://doi.org/10.1136/gutjnl-2016-313369
Kuna L, Bozic I, Kizivat T et al (2018) Models of drug induced liver injury (DILI)—current issues and future perspectives. Curr Drug Metab 19(10):830–838. https://doi.org/10.2174/1389200219666180523095355
Lau YY, Chen YH, Liu TT et al (2004) Evaluation of a novel in vitro Caco-2 hepatocyte hybrid system for predicting in vivo oral bioavailability. Drug Metab Disposition Biol Fate Chem 32(9):937–942
Le Vee M, Noel G, Jouan E, Stieger B, Fardel O (2013) Polarized expression of drug transporters in differentiated human hepatoma HepaRG cells. Toxicol In Vitro 27(6):1979–1986. https://doi.org/10.1016/j.tiv.2013.07.003
Lee PJ, Hung PJ, Lee LP (2007) An artificial liver sinusoid with a microfluidic endothelial-like barrier for primary hepatocyte culture. Biotechnol Bioeng 97(5):1340–1346. https://doi.org/10.1002/bit.21360
Lee-Montiel FT, George SM, Gough AH et al (2017) Control of oxygen tension recapitulates zone-specific functions in human liver microphysiology systems. Exp Biol Med (Maywood) 242(16):1617–1632. https://doi.org/10.1177/1535370217703978
Leise MD, Poterucha JJ, Talwalkar JA (2014) Drug-induced liver injury. Mayo Clinic proceedings 89(1):95–106. https://doi.org/10.1016/j.mayocp.2013.09.016
Leuning DG, Beijer NRM, du Fosse NA et al (2018) The cytokine secretion profile of mesenchymal stromal cells is determined by surface structure of the microenvironment. Sci Rep 8(1):7716. https://doi.org/10.1038/s41598-018-25700-5
Li AP, Uzgare A, LaForge YS (2012) Definition of metabolism-dependent xenobiotic toxicity with co-cultures of human hepatocytes and mouse 3T3 fibroblasts in the novel integrated discrete multiple organ co-culture (IdMOC) experimental system: results with model toxicants aflatoxin B1, cyclophosphamide and tamoxifen. Chem Biol Interact 199(1):1–8. https://doi.org/10.1016/j.cbi.2012.05.003
Liguori MJ, Anderson MG, Bukofzer S et al (2005) Microarray analysis in human hepatocytes suggests a mechanism for hepatotoxicity induced by trovafloxacin. Hepatology 41(1):177–186. https://doi.org/10.1002/hep.20514
Lin C, Khetani SR (2016) Advances in engineered liver models for investigating drug-induced liver injury. Biomed Res Int 2016:1829148. https://doi.org/10.1155/2016/1829148
Lizarraga D, Gaj S, Brauers KJ, Timmermans L, Kleinjans JC, van Delft JH (2012) Benzo[a]pyrene-induced changes in microRNA-mRNA networks. Chem Res Toxicol 25(4):838–849. https://doi.org/10.1021/tx2003799
Long TJ, Cosgrove PA, Dunn RT 2nd et al (2016) Modeling therapeutic antibody-small molecule drug-drug interactions using a three-dimensional perfusable human liver coculture platform. Drug Metab Disposition Biol Fate chem 44(12):1940–1948. https://doi.org/10.1124/dmd.116.071456
Lu J, Einhorn S, Venkatarangan L et al (2015) Morphological and functional characterization and assessment of iPSC-derived hepatocytes for in vitro toxicity testing. Toxicol Sci 147(1):39–54. https://doi.org/10.1093/toxsci/kfv117
Ma S, Gong Q, Bohnert HJ (2007) An Arabidopsis gene network based on the graphical Gaussian model. Genome Res 17(11):1614–1625. https://doi.org/10.1101/gr.6911207
Ma L, Barker J, Zhou C et al (2012) Towards personalized medicine with a three-dimensional micro-scale perfusion-based two-chamber tissue model system. Biomaterials 33(17):4353–4361. https://doi.org/10.1016/j.biomaterials.2012.02.054
Ma X, Qu X, Zhu W et al (2016) Deterministically patterned biomimetic human iPSC-derived hepatic model via rapid 3D bioprinting. Proc Natl Acad Sci U S A 113(8):2206–2211. https://doi.org/10.1073/pnas.1524510113
Magkoufopoulou C, Claessen SM, Jennen DG, Kleinjans JC, van Delft JH (2011) Comparison of phenotypic and transcriptomic effects of false-positive genotoxins, true genotoxins and non-genotoxins using HepG2 cells. Mutagenesis 26(5):593–604. https://doi.org/10.1093/mutage/ger021
Magkoufopoulou C, Claessen SM, Tsamou M, Jennen DG, Kleinjans JC, van Delft JH (2012) A transcriptomics-based in vitro assay for predicting chemical genotoxicity in vivo. Carcinogenesis 33(7):1421–1429. https://doi.org/10.1093/carcin/bgs182
Maschmeyer I, Lorenz AK, Schimek K et al (2015) A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab Chip 15(12):2688–2699. https://doi.org/10.1039/c5lc00392j
Materne EM, Maschmeyer I, Lorenz AK et al (2015a) The multi-organ chip—a microfluidic platform for long-term multi-tissue coculture. J Vis Exp 98:e52526. https://doi.org/10.3791/52526
Materne EM, Ramme AP, Terrasso AP et al (2015b) A multi-organ chip co-culture of neurospheres and liver equivalents for long-term substance testing. J Biotechnol 205:36–46. https://doi.org/10.1016/j.jbiotec.2015.02.002
Matsa E, Burridge PW, Yu KH et al (2016) Transcriptome profiling of patient-specific human iPSC-cardiomyocytes predicts individual drug safety and efficacy responses in vitro. Cell Stem Cell 19(3):311–325. https://doi.org/10.1016/j.stem.2016.07.006
Mesnage R, Biserni M, Balu S et al (2018) Integrated transcriptomics and metabolomics reveal signatures of lipid metabolism dysregulation in HepaRG liver cells exposed to PCB 126. Arch Toxicol 92(8):2533–2547. https://doi.org/10.1007/s00204-018-2235-7
Messner S, Fredriksson L, Lauschke VM et al (2018) Transcriptomic, proteomic, and functional long-term characterization of multicellular three-dimensional human liver microtissues. Appl Toxicol 4(1):1–12
Miller PG, Shuler ML (2016) Design and demonstration of a pumpless 14 compartment microphysiological system. Biotechnol Bioeng 113(10):2213–2227. https://doi.org/10.1002/bit.25989
Mosedale M, Watkins PB (2017) Drug-induced liver injury: advances in mechanistic understanding that will inform risk management. Clin Pharmacol Ther 101(4):469–480. https://doi.org/10.1002/cpt.564
Mosedale M, Button D, Jackson JP et al (2018) Transient changes in hepatic physiology that alter bilirubin and bile acid transport may explain elevations in liver chemistries observed in clinical trials of GGF2 (Cimaglermin Alfa). Toxicol Sci 161(2):401–411. https://doi.org/10.1093/toxsci/kfx222
Nagamoto Y, Tashiro K, Takayama K et al (2012) The promotion of hepatic maturation of human pluripotent stem cells in 3D co-culture using type I collagen and Swiss 3T3 cell sheets. Biomaterials 33(18):4526–4534. https://doi.org/10.1016/j.biomaterials.2012.03.011
Nakao Y, Kimura H, Sakai Y, Fujii T (2011) Bile canaliculi formation by aligning rat primary hepatocytes in a microfluidic device. Biomicrofluidics 5(2):22212. https://doi.org/10.1063/1.3580753
Neuman MG (2019) Biomarkers of drug-induced liver toxicity. Ther Drug Monit 41(2):227–234. https://doi.org/10.1097/FTD.0000000000000610
Norona LM, Nguyen DG, Gerber DA, Presnell SC, LeCluyse EL (2016) Editor’s highlight: modeling compound-induced fibrogenesis in vitro using three-dimensional bioprinted human liver tissues. Toxicol Sci 154(2):354–367. https://doi.org/10.1093/toxsci/kfw169
Nueda MJ, Tarazona S, Conesa A (2014) Next maSigPro: updating maSigPro bioconductor package for RNA-seq time series. Bioinformatics 30(18):2598–2602. https://doi.org/10.1093/bioinformatics/btu333
Oleaga C, Bernabini C, Smith AS et al (2016) Multi-Organ toxicity demonstration in a functional human in vitro system composed of four organs. Sci Rep 6:20030. https://doi.org/10.1038/srep20030
Onakpoya IJ, Heneghan CJ, Aronson JK (2018) Post-marketing withdrawal of analgesic medications because of adverse drug reactions: a systematic review. Expert Opin Drug Saf 17(1):63–72. https://doi.org/10.1080/14740338.2018.1398232
Paemanee A, Sornjai W, Kittisenachai S et al (2017) Nevirapine induced mitochondrial dysfunction in HepG2 cells. Sci Rep 7(1):9194. https://doi.org/10.1038/s41598-017-09321-y
Paish HL, Reed LH, Brown H et al (2019) A bioreactor technology for modeling fibrosis in human and rodent precision-cut liver slices. Hepatology. https://doi.org/10.1002/hep.30651
Palma E, Doornebal EJ, Chokshi S (2019) Precision-cut liver slices: a versatile tool to advance liver research. Hepatol Int 13(1):51–57. https://doi.org/10.1007/s12072-018-9913-7
Poloznikov A, Gazaryan I, Shkurnikov M et al (2018) In vitro and in silico liver models: current trends, challenges and opportunities. Altex 35(3):397–412. https://doi.org/10.14573/altex.1803221
Powers MJ, Domansky K, Kaazempur-Mofrad MR et al (2002a) A microfabricated array bioreactor for perfused 3D liver culture. Biotechnol Bioeng 78(3):257–269
Powers MJ, Janigian DM, Wack KE, Baker CS, Beer Stolz D, Griffith LG (2002b) Functional behavior of primary rat liver cells in a three-dimensional perfused microarray bioreactor. Tissue Eng 8(3):499–513. https://doi.org/10.1089/107632702760184745
Proctor WR, Foster AJ, Vogt J et al (2017) Utility of spherical human liver microtissues for prediction of clinical drug-induced liver injury. Arch Toxicol 91(8):2849–2863. https://doi.org/10.1007/s00204-017-2002-1
Prodanov L, Jindal R, Bale SS et al (2016) Long-term maintenance of a microfluidic 3D human liver sinusoid. Biotechnol Bioeng 113(1):241–246. https://doi.org/10.1002/bit.25700
Prot JM, Aninat C, Griscom L et al (2011) Improvement of HepG2/C3a cell functions in a microfluidic biochip. Biotechnol Bioeng 108(7):1704–1715. https://doi.org/10.1002/bit.23104
Prot JM, Bunescu A, Elena-Herrmann B et al (2012) Predictive toxicology using systemic biology and liver microfluidic “on chip” approaches: application to acetaminophen injury. Toxicol Appl Pharmacol 259(3):270–280. https://doi.org/10.1016/j.taap.2011.12.017
Ramirez T, Strigun A, Verlohner A et al (2018) Prediction of liver toxicity and mode of action using metabolomics in vitro in HepG2 cells. Arch Toxicol 92(2):893–906. https://doi.org/10.1007/s00204-017-2079-6
Raschi E, De Ponti F (2017) Drug-induced liver injury: towards early prediction and risk stratification. World J Hepatol 9(1):30–37. https://doi.org/10.4254/wjh.v9.i1.30
Rashidi H, Luu NT, Alwahsh SM et al (2018) 3D human liver tissue from pluripotent stem cells displays stable phenotype in vitro and supports compromised liver function in vivo. Arch Toxicol 92(10):3117–3129. https://doi.org/10.1007/s00204-018-2280-2
Regev A (2014) Drug-induced liver injury and drug development: industry perspective. Semin Liver Dis 34(2):227–239. https://doi.org/10.1055/s-0034-1375962
Rennert K, Steinborn S, Groger M et al (2015) A microfluidically perfused three dimensional human liver model. Biomaterials 71:119–131. https://doi.org/10.1016/j.biomaterials.2015.08.043
Rieswijk L, Lizarraga D, Brauers KJ, Kleinjans JC, van Delft JH (2014) Characterisation of cisplatin-induced transcriptomics responses in primary mouse hepatocytes, HepG2 cells and mouse embryonic stem cells shows conservation of regulating transcription factor networks. Mutagenesis 29(1):17–26. https://doi.org/10.1093/mutage/get055
Rieswijk L, Claessen SM, Bekers O et al (2016) Aflatoxin B1 induces persistent epigenomic effects in primary human hepatocytes associated with hepatocellular carcinoma. Toxicology 350–352:31–39. https://doi.org/10.1016/j.tox.2016.05.002
Ritchie ME, Phipson B, Wu D et al (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43(7):e47. https://doi.org/10.1093/nar/gkv007
Rodrigues RM, De Kock J, Branson S et al (2014) Human skin-derived stem cells as a novel cell source for in vitro hepatotoxicity screening of pharmaceuticals. Stem Cells Dev 23(1):44–55. https://doi.org/10.1089/scd.2013.0157
Rodrigues RM, Heymans A, De Boe V et al (2016) Toxicogenomics-based prediction of acetaminophen-induced liver injury using human hepatic cell systems. Toxicol Lett 240(1):50–59. https://doi.org/10.1016/j.toxlet.2015.10.014
Rodrigues RM, Kollipara L, Chaudhari U et al (2018) Omics-based responses induced by bosentan in human hepatoma HepaRG cell cultures. Arch Toxicol 92(6):1939–1952. https://doi.org/10.1007/s00204-018-2214-z
Ronaldson-Bouchard K, Vunjak-Novakovic G (2018) Organs-on-a-Chip: a fast track for engineered human tissues in drug development. Cell Stem Cell 22(3):310–324. https://doi.org/10.1016/j.stem.2018.02.011
Rose KA, Holman NS, Green AM, Andersen ME, LeCluyse EL (2016) Co-culture of hepatocytes and kupffer cells as an in vitro model of inflammation and drug-induced hepatotoxicity. J Pharm Sci 105(2):950–964. https://doi.org/10.1016/S0022-3549(15)00192-6
Roth AD, Lee MY (2017) Idiosyncratic drug-induced liver injury (IDILI): potential mechanisms and predictive assays. Biomed Res Int 2017:9176937. https://doi.org/10.1155/2017/9176937
Ruiz-Aracama A, Peijnenburg A, Kleinjans J et al (2011) An untargeted multi-technique metabolomics approach to studying intracellular metabolites of HepG2 cells exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. BMC Genomics 12:251. https://doi.org/10.1186/1471-2164-12-251
Sarkar U, Rivera-Burgos D, Large EM et al (2015) Metabolite profiling and pharmacokinetic evaluation of hydrocortisone in a perfused three-dimensional human liver bioreactor. Drug Metab Disposition Biol Fate Chem 43(7):1091–1099. https://doi.org/10.1124/dmd.115.063495
Sarkar U, Ravindra KC, Large E et al (2017) Integrated assessment of diclofenac biotransformation, pharmacokinetics, and omics-based toxicity in a three-dimensional human liver-immunocompetent coculture system. Drug metabolism and disposition: the biological fate of chemicals 45(7):855–866. https://doi.org/10.1124/dmd.116.074005
Schepers A, Li C, Chhabra A, Seney BT, Bhatia S (2016) Engineering a perfusable 3D human liver platform from iPS cells. Lab Chip 16(14):2644–2653. https://doi.org/10.1039/c6lc00598e
Schuemie MJ, Trifiro G, Coloma PM, Ryan PB, Madigan D (2016) Detecting adverse drug reactions following long-term exposure in longitudinal observational data: the exposure-adjusted self-controlled case series. Stat Methods Med Res 25(6):2577–2592. https://doi.org/10.1177/0962280214527531
Scott CW, Peters MF, Dragan YP (2013) Human induced pluripotent stem cells and their use in drug discovery for toxicity testing. Toxicol Lett 219(1):49–58. https://doi.org/10.1016/j.toxlet.2013.02.020
Scupakova K, Soons Z, Ertaylan G et al (2018) Spatial systems lipidomics reveals nonalcoholic fatty liver disease heterogeneity. Anal Chem 90(8):5130–5138. https://doi.org/10.1021/acs.analchem.7b05215
Seeger B, Mentz A, Knebel C et al (2019a) Assessment of mixture toxicity of (tri)azoles and their hepatotoxic effects in vitro by means of omics technologies. Arch Toxicol. https://doi.org/10.1007/s00204-019-02502-w
Seeger B, Mentz A, Knebel C et al (2019b) Assessment of mixture toxicity of (tri)azoles and their hepatotoxic effects in vitro by means of omics technologies. Arch Toxicol 93(8):2321–2333. https://doi.org/10.1007/s00204-019-02502-w
Shintu L, Baudoin R, Navratil V et al (2012) Metabolomics-on-a-chip and predictive systems toxicology in microfluidic bioartificial organs. Anal Chem 84(4):1840–1848. https://doi.org/10.1021/ac2011075
Singh S, Singhal NK, Srivastava G, Singh MP (2010) Omics in mechanistic and predictive toxicology. Toxicol Mech Methods 20(7):355–362. https://doi.org/10.3109/15376510903559976
Sison-Young RL, Mitsa D, Jenkins RE et al (2015) Comparative proteomic characterization of 4 human liver-derived single cell culture models reveals significant variation in the capacity for drug disposition, bioactivation, and detoxication. Toxicol Sci 147(2):412–424. https://doi.org/10.1093/toxsci/kfv136
Si-Tayeb K, Noto FK, Nagaoka M et al (2010) Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 51(1):297–305. https://doi.org/10.1002/hep.23354
Smit E, Souza T, Jennen DGJ, Kleinjans JCS, van den Beucken T (2017) Identification of essential transcription factors for adequate DNA damage response after benzo(a)pyrene and aflatoxin B1 exposure by combining transcriptomics with functional genomics. Toxicology 390:74–82. https://doi.org/10.1016/j.tox.2017.09.002
Smith MC, Timmins-Schiffman E, Coton M et al (2018) Differential impacts of individual and combined exposures of deoxynivalenol and zearalenone on the HepaRG human hepatic cell proteome. J Proteomics 173:89–98. https://doi.org/10.1016/j.jprot.2017.11.025
Soldatow VY, Lecluyse EL, Griffith LG, Rusyn I (2013) In vitro models for liver toxicity testing. Toxicol Res 2(1):23–39. https://doi.org/10.1039/C2TX20051A
Souza T, Jennen D, van Delft J, van Herwijnen M, Kyrtoupolos S, Kleinjans J (2016) New insights into BaP-induced toxicity: role of major metabolites in transcriptomics and contribution to hepatocarcinogenesis. Arch Toxicol 90(6):1449–1458. https://doi.org/10.1007/s00204-015-1572-z
Subramanian A, Tamayo P, Mootha VK et al (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102(43):15545–15550. https://doi.org/10.1073/pnas.0506580102
Sung JH, Kam C, Shuler ML (2010) A microfluidic device for a pharmacokinetic-pharmacodynamic (PK-PD) model on a chip. Lab Chip 10(4):446–455. https://doi.org/10.1039/b917763a
Swift B, Pfeifer ND, Brouwer KL (2010) Sandwich-cultured hepatocytes: an in vitro model to evaluate hepatobiliary transporter-based drug interactions and hepatotoxicity. Drug Metab Rev 42(3):446–471. https://doi.org/10.3109/03602530903491881
Szkolnicka D, Farnworth SL, Lucendo-Villarin B et al (2014) Accurate prediction of drug-induced liver injury using stem cell-derived populations. Stem Cells Transl Med 3(2):141–148. https://doi.org/10.5966/sctm.2013-0146
Takebe T, Sekine K, Enomura M et al (2013) Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 499(7459):481–484. https://doi.org/10.1038/nature12271
Tascher G, Burban A, Camus S et al (2019) In-depth proteome analysis highlights HepaRG cells as a versatile cell system surrogate for primary human hepatocytes. Cells. https://doi.org/10.3390/cells8020192
The Gene Ontology Consortium (2019) The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res 47(D1):D330–D338. https://doi.org/10.1093/nar/gky1055
Thery M (2010) Micropatterning as a tool to decipher cell morphogenesis and functions. J Cell Sci 123(Pt 24):4201–4213. https://doi.org/10.1242/jcs.075150
Tolosa L, Jimenez N, Pelecha M, Castell JV, Gomez-Lechon MJ, Donato MT (2019) Long-term and mechanistic evaluation of drug-induced liver injury in Upcyte human hepatocytes. Arch Toxicol 93(2):519–532. https://doi.org/10.1007/s00204-018-2349-y
Trask OJ Jr, Moore A, LeCluyse EL (2014) A micropatterned hepatocyte coculture model for assessment of liver toxicity using high-content imaging analysis. Assay Drug Dev Technol 12(1):16–27. https://doi.org/10.1089/adt.2013.525
Tsamandouras N, Kostrzewski T, Stokes CL, Griffith LG, Hughes DJ, Cirit M (2017) Quantitative assessment of population variability in hepatic drug metabolism using a perfused three-dimensional human liver microphysiological system. J Pharmacol Exp Ther 360(1):95–105. https://doi.org/10.1124/jpet.116.237495
Underhill GH, Khetani SR (2018) Bioengineered liver models for drug testing and cell differentiation studies. Cell Mol Gastroenterol Hepatol 5(3):426–439. https://doi.org/10.1016/j.jcmgh.2017.11.012
van Breda SGJ, Claessen SMH, van Herwijnen M et al (2018) Integrative omics data analyses of repeated dose toxicity of valproic acid in vitro reveal new mechanisms of steatosis induction. Toxicology 393:160–170. https://doi.org/10.1016/j.tox.2017.11.013
van Delft JH, Mathijs K, Staal YC et al (2010) Time series analysis of benzo[A]pyrene-induced transcriptome changes suggests that a network of transcription factors regulates the effects on functional gene sets. Toxicol Sci 117(2):381–392. https://doi.org/10.1093/toxsci/kfq214
van Delft J, Gaj S, Lienhard M et al (2012) RNA-Seq provides new insights in the transcriptome responses induced by the carcinogen benzo[a]pyrene. Toxicol Sci 130(2):427–439. https://doi.org/10.1093/toxsci/kfs250
Van den Hof WF, Coonen ML, van Herwijnen M et al (2014) Classification of hepatotoxicants using HepG2 cells: a proof of principle study. Chem Res Toxicol 27(3):433–442. https://doi.org/10.1021/tx4004165
Van den Hof WF, Ruiz-Aracama A, Van Summeren A et al (2015) Integrating multiple omics to unravel mechanisms of Cyclosporin A induced hepatotoxicity in vitro. Toxicol In Vitro 29(3):489–501. https://doi.org/10.1016/j.tiv.2014.12.016
Van den Hof W, Coonen MLJ, van Herwijnen M et al (2017) Validation of gene expression profiles from cholestatic hepatotoxicants in vitro against human in vivo cholestasis. Toxicol In Vitro 44:322–329. https://doi.org/10.1016/j.tiv.2017.07.024
Van Summeren A, Renes J, Bouwman FG et al (2011) Proteomics investigations of drug-induced hepatotoxicity in HepG2 cells. Toxicol Sci 120(1):109–122. https://doi.org/10.1093/toxsci/kfq380
Vatakuti S (2016) Toxicogenomics of precision-cut liver slices for prediction of human liver toxicity. University of Groningen
Vatakuti S, Pennings JL, Gore E, Olinga P, Groothuis GM (2016) Classification of cholestatic and necrotic hepatotoxicants using transcriptomics on human precision-cut liver slices. Chem Res Toxicol 29(3):342–351. https://doi.org/10.1021/acs.chemrestox.5b00491
Vatakuti S, Olinga P, Pennings JLA, Groothuis GMM (2017) Validation of precision-cut liver slices to study drug-induced cholestasis: a transcriptomics approach. Arch Toxicol 91(3):1401–1412. https://doi.org/10.1007/s00204-016-1778-8
Verheijen M, Schrooders Y, Gmuender H et al (2018) Bringing in vitro analysis closer to in vivo: studying doxorubicin toxicity and associated mechanisms in 3D human microtissues with PBPK-based dose modelling. Toxicol Lett 294:184–192. https://doi.org/10.1016/j.toxlet.2018.05.029
Vernetti LA, Senutovitch N, Boltz R et al (2016) A human liver microphysiology platform for investigating physiology, drug safety, and disease models. Exp Biol Med (Maywood) 241(1):101–114. https://doi.org/10.1177/1535370215592121
Vernetti L, Gough A, Baetz N et al (2017) Functional coupling of human microphysiology systems: intestine, liver, kidney proximal tubule, blood-brain barrier and skeletal muscle. Sci Rep 7:42296. https://doi.org/10.1038/srep42296
Viravaidya K, Sin A, Shuler ML (2004) Development of a microscale cell culture analog to probe naphthalene toxicity. Biotechnol Prog 20(1):316–323. https://doi.org/10.1021/bp0341996
Volarevic V, Markovic BS, Gazdic M et al (2018) Ethical and safety issues of stem cell-based therapy. Int J Med Sci 15(1):36–45. https://doi.org/10.7150/ijms.21666
Vorrink SU, Ullah S, Schmidt S et al (2017) Endogenous and xenobiotic metabolic stability of primary human hepatocytes in long-term 3D spheroid cultures revealed by a combination of targeted and untargeted metabolomics. FASEB J 31(6):2696–2708. https://doi.org/10.1096/fj.201601375R
Vorrink SU, Zhou Y, Ingelman-Sundberg M, Lauschke VM (2018) Prediction of drug-induced hepatotoxicity using long-term stable primary hepatic 3D spheroid cultures in chemically defined conditions. Toxicol Sci 163(2):655–665. https://doi.org/10.1093/toxsci/kfy058
Wagner I, Materne EM, Brincker S et al (2013) A dynamic multi-organ-chip for long-term cultivation and substance testing proven by 3D human liver and skin tissue co-culture. Lab Chip 13(18):3538–3547. https://doi.org/10.1039/c3lc50234a
Wang WW, Khetani SR, Krzyzewski S, Duignan DB, Obach RS (2010) Assessment of a micropatterned hepatocyte coculture system to generate major human excretory and circulating drug metabolites. Drug metabolism and disposition: the biological fate of chemicals 38(10):1900–1905. https://doi.org/10.1124/dmd.110.034876
Wang J, Vasaikar S, Shi Z, Greer M, Zhang B (2017) WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res 45(W1):W130–W137. https://doi.org/10.1093/nar/gkx356
Ware BR, Berger DR, Khetani SR (2015) Prediction of drug-induced liver injury in micropatterned co-cultures containing iPSC-derived human hepatocytes. Toxicol Sci 145(2):252–262. https://doi.org/10.1093/toxsci/kfv048
Ware BR, McVay M, Sunada WY, Khetani SR (2017) Exploring chronic drug effects on microengineered human liver cultures using global gene expression profiling. Toxicol Sci 157(2):387–398. https://doi.org/10.1093/toxsci/kfx059
Ware BR, Durham MJ, Monckton CP, Khetani SR (2018) A Cell culture platform to maintain long-term phenotype of primary human hepatocytes and endothelial cells. Cell Mol Gastroenterol Hepatol 5(3):187–207. https://doi.org/10.1016/j.jcmgh.2017.11.007
Waters MD, Olden K, Tennant RW (2003) Toxicogenomic approach for assessing toxicant-related disease. Mutat Res Rev Mutat Res 544(2–3):415–424. https://doi.org/10.1016/j.mrrev.2003.06.014
Weng L, Dai H, Zhan Y, He Y, Stepaniants SB, Bassett DE (2006) Rosetta error model for gene expression analysis. Bioinformatics 22(9):1111–1121. https://doi.org/10.1093/bioinformatics/btl045
Wille A, Zimmermann P, Vranova E et al (2004) Sparse graphical Gaussian modeling of the isoprenoid gene network in Arabidopsis thaliana. Genome Biol 5(11):R92. https://doi.org/10.1186/gb-2004-5-11-r92
Wolters JE, van Herwijnen MH, Theunissen DH et al (2016) Integrative “-Omics” analysis in primary human hepatocytes unravels persistent mechanisms of cyclosporine a-induced cholestasis. Chem Res Toxicol 29(12):2164–2174. https://doi.org/10.1021/acs.chemrestox.6b00337
Wolters JEJ, van Breda SGJ, Caiment F, Claessen SM, de Kok T, Kleinjans JCS (2017) Nuclear and mitochondrial DNA methylation patterns induced by valproic acid in human hepatocytes. Chem Res Toxicol 30(10):1847–1854. https://doi.org/10.1021/acs.chemrestox.7b00171
Wolters JEJ, van Breda SGJ, Grossmann J, Fortes C, Caiment F, Kleinjans JCS (2018) Integrated ‘omics analysis reveals new drug-induced mitochondrial perturbations in human hepatocytes. Toxicol Lett 289:1–13. https://doi.org/10.1016/j.toxlet.2018.02.026
Wu X, Roberto JB, Knupp A et al (2018) Precision-cut human liver slice cultures as an immunological platform. J Immunol Methods 455:71–79. https://doi.org/10.1016/j.jim.2018.01.012
Zeilinger K, Freyer N, Damm G, Seehofer D, Knospel F (2016) Cell sources for in vitro human liver cell culture models. Exp Biol Med (Maywood) 241(15):1684–1698. https://doi.org/10.1177/1535370216657448
Zhang C, Zhao Z, Abdul Rahim NA, van Noort D, Yu H (2009) Towards a human-on-chip: culturing multiple cell types on a chip with compartmentalized microenvironments. Lab Chip 9(22):3185–3192. https://doi.org/10.1039/b915147h
Zhang YS, Aleman J, Shin SR et al (2017) Multisensor-integrated organs-on-chips platform for automated and continual in situ monitoring of organoid behaviors. Proc Natl Acad Sci USA 114(12):E2293–E2302. https://doi.org/10.1073/pnas.1612906114
Zhu Z, Huangfu D (2013) Human pluripotent stem cells: an emerging model in developmental biology. Development 140(4):705–717. https://doi.org/10.1242/dev.086165
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jiang, J., Pieterman, C.D., Ertaylan, G. et al. The application of omics-based human liver platforms for investigating the mechanism of drug-induced hepatotoxicity in vitro. Arch Toxicol 93, 3067–3098 (2019). https://doi.org/10.1007/s00204-019-02585-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-019-02585-5