
Abstract
Aims/hypothesis
Excessive production of reactive oxygen species (ROS) plays a detrimental role in the progression of diabetic kidney disease (DKD). Renal oxidative stress activates proinflammatory cytokines, chemokines and profibrotic factors in DKD. Increased expression of the prooxidant enzyme NADPH oxidase (NOX) 5 in kidneys of diabetic individuals has been hypothesised to correlate with renal injury and progression of DKD. Since the gene encoding NOX5 is not expressed in the mouse genome, we examined the effect of inducible human NOX5 expression in renal cells, selectively in either endothelial cells or vascular smooth muscle cells (VSMCs)/mesangial cells in a model of insulin-deficient diabetes, the Akita mouse.
Methods
Renal structural injury, including glomerulosclerosis, mesangial expansion and extracellular matrix protein accumulation, as well as renal inflammation, ROS formation and albuminuria, were examined in the NOX5 transgenic Akita mouse model of DKD.
Results
Expression of NOX5 in either endothelial cells or VSMCs/mesangial cells in diabetic Akita mice was associated with increased renal inflammation (monocyte chemoattractant protein-1, NF-κB and toll-like receptor-4) and glomerulosclerosis, as well as upregulation of protein kinase C-α and increased expression of extracellular matrix genes (encoding collagen III, fibronectin and α-smooth muscle actin) and proteins (collagen IV), most likely mediated via enhanced renal ROS production. The effect of VSMC/mesangial cell-specific NOX5 expression resulted in more pronounced renal fibrosis in comparison with endothelial cell-specific NOX5 expression in diabetic mice. In addition, albuminuria was significantly increased in diabetic VEcad+NOX5+ mice (1192 ± 194 μg/24 h) when compared with diabetic VEcad+NOX5− mice (770 ± 98 μg/24 h). Furthermore, the regulatory components of NOX5 activation, including heat shock protein 90 and transient receptor potential cation channel subfamily C member 6, were upregulated only in the presence of both NOX5 and diabetes.
Conclusions/interpretation
The findings from this study highlight the importance of NOX5 in promoting diabetes-related renal injury and provide the rationale for the development of a selective NOX5 inhibitor for the prevention and/or treatment of DKD.
Similar content being viewed by others


Introduction
Diabetes is the most common cause of chronic kidney disease, which increases the risk of cardiovascular disease leading to premature death [1]. The unique structural arrangement of distinct glomerular cell types, including podocytes, endothelial cells and mesangial cells, creates a highly specialised microvascular bed: the filtration barrier. Tightly coordinated movement and crosstalk between these cell types is required for the formation of a functional glomerular filtration barrier which filters blood to form urine [2, 3]. Indeed, failure to adequately maintain glomerular barrier function in conditions such as diabetic kidney disease (DKD) results in the excretion of critical blood components including albumin into the urine [2, 4].
The prooxidant enzyme NADPH oxidase (NOX), particularly the NOX4 isoform, generates reactive oxygen species (ROS), which contribute to renal pathology in a range of experimental models of DKD [5,6,7,8]. However, another isoform of NOX, NOX5, has attracted increasing attention in the context of human DKD [9,10,11]. Indeed, previous studies including our own have shown increased expression of NOX5 protein in human kidney biopsies/nephrectomy samples from diabetic individuals [10, 11]. Importantly, the NOX5 gene present in humans is absent from the mouse and rat genome and therefore it has been difficult to explore the functional importance of this specific isoform in experimental diabetes. However, a study in an inducible human transgenic NOX5 mouse model with selective expression of NOX5 in podocytes has shown not only podocyte injury but also albuminuria and renal inflammation in association with enhanced ROS production, further accelerated in the presence of streptozotocin (STZ)-induced diabetes [10].
Glomerular endothelial and mesangial cells are key components of the glomeruli and damage of these cells at a structural and functional level in diabetes leads to the progression of DKD. Thus, in this study, we examined the effect of inducible human NOX5 expression either in endothelial cells or in vascular smooth muscle cells (VSMCs; representing mesangial cells in the kidney) in transgenic mice, in the presence and absence of diabetes. Rather than using a chemical model of insulin-deficient diabetes (i.e. STZ-induced), we chose to study transgenic expression of NOX5 in the Akita mouse model, a genetic model of insulin-deficient diabetes with more advanced renal fibrosis, inflammation and albuminuria.
Methods
Animals
The NOX5 transgenic mice were generated by C. Kennedy at the University of Ottawa and R. Touyz, at the University of Glasgow as described previously [10, 11]. Briefly, the purified human NOX5β gene coding region was ligated with the Tet-responsive promoter Pbi-1 (Clontech, Mountain View, CA, USA) to generate the NOX5β founder mouse on the FVB/N background. The SM22-tTA-FVB/N mouse and VEcad-tTA-FVB/N mouse strains were generated by introducing the VSMC-specific promoter transgelin (SM22; encoded by Sm22, also known as Tagln) or the endothelial cell-specific promoter vascular endothelial cadherin (VEcad; encoded by VEcad, also known as Cdh5) to the tetracycline-regulated transcriptional activator (tTA-Off) gene. Subsequently, to generate VSMC- and endothelial cell-specific NOX5 transgenic mice, the SM22-tTA-FVB/N strain or VEcad-tTA-FVB/N strain were crossed with the NOX5β FVB/N strain to produce either Sm22+NOX5+ and Sm22+NOX5− mice or VEcad+NOX5+ and VEcad+NOX5− mice at the animal facility at Monash University. To subsequently generate insulin-deficient diabetic Sm22+NOX5+ and Sm22+NOX5− mice or VEcad+NOX5+ and VEcad+NOX5− mice, SM22-tTA-FVB/N or VEcad-tTA-FVB/N strains were crossed with Akita/NOX5β FVB/N mice. FVB-Akita mice were sourced from The Jackson Laboratory (Bar Harbor, ME, USA). Six-week-old male, heterozygous Akita mice with and without NOX5 expression (Sm22+NOX5+ and Sm22+NOX5− or VEcad+NOX5+ and VEcad+NOX5−) were used for the start of the study at which time point they had already developed hyperglycaemia. The non-Akita littermate Sm22+NOX5+ and Sm22+NOX5− mice or VEcad+NOX5+ and VEcad+NOX5− mice were used as controls. All animal studies were approved by the Alfred Medical Research & Education Precinct (AMREP) Animal Ethics Committee under guidelines laid down by the National Health and Medical Research Council of Australia.
Mice were placed individually into metabolic cages (Iffa Credo, Lyon, France) for 24 h at 15 weeks of age. Blood glucose, HbA1c and systolic blood pressure were measured, as described previously [6, 12]. Urinary albumin concentration was measured by using a mouse albumin ELISA quantification kit (Bethyl Laboratories, Montgomery, TX, USA). Urinary and plasma creatinine were determined by using the Cobas Integra 400 Plus analyser (Roche Diagnostics, Indianapolis, IN, USA). After 10 weeks (at 16 weeks of age), mice were anaesthetised by i.p. injection of sodium pentobarbital (100 mg/kg body weight; Euthatal; Sigma-Aldrich, Castle Hill, NSW, Australia). As there is no specific intervention and mice develop diabetes spontaneously they were not randomised. Only mice with blood glucose levels ≥15 mmol/l have been included in the experiments; mice with blood glucose <15 mmol/l and with polycystic kidney were excluded from the study (<5% of the total number of mice).
Histological assessment of renal pathology
Paraffin kidney sections (3 μm) were stained with Periodic Acid–Schiff for the measurement of mesangial expansion, glomerulosclerotic index (GSI) and tubulointerstitial injury (TII), as described previously [6, 11, 13]. Immunostaining for nitrotyrosine (rabbit anti-nitrotyrosine, catalogue no. ab5411; Millipore, Billerica, MA, USA), collagen IV (goat anti-type IV collagen, catalogue no. 1340-01; Southern Biotech, Birmingham, AL, USA), protein kinase C-α (PKC-α) (rabbit polyclonal, catalogue no. sc-208; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and toll-like receptor-4 (TLR-4) (rabbit polyclonal, catalogue no. bs-1021R; Bioss Antibodies, Woburn, MA, USA) were performed as described previously [6, 11]. Mesangial area and quantification of glomerular collagen IV, nitrotyrosine, PKC-α and TLR-4 were analysed from digital pictures of glomeruli (20 glomeruli per mouse) and tubules (10 tubulointerstitial fields per mouse for TII) using Image-Pro plus 7.0 software (Media Cybernetics, Bethesda, MD, USA). GSI and tubulointerstitial injury were graded, as described previously [6, 11, 13]. All assessments were performed in a blinded manner.
Immunofluorescence
Double immunostaining of mouse frozen kidney sections (5 μm) was performed as described previously [11]. Briefly, double staining for either NOX5 and SM22-α or NOX5 and CD31 in mouse kidney tissue was performed by incubation with primary antibody to NOX5 (rabbit polyclonal, catalogue no. ab191010; Abcam, Cambridge, MA, USA) and SM22-α (goat polyclonal, catalogue no. ab10135; Abcam) or CD31 (goat polyclonal, catalogue no. AF3628; R&D Systems, Minneapolis, MN, USA), followed by incubation with secondary antibody Alexa Fluor 568 (donkey anti-rabbit; Invitrogen, Eugene, OR, USA) and Alexa Fluor 488 (donkey anti-goat; Invitrogen). Dihydroethidium (DHE) staining was performed as described previously [11]. All stained sections were examined and images were captured using a Nikon eclipse-Ni (Tokyo, Japan) fluorescence microscope.
Quantitative RT-PCR
Total RNA from the renal cortex was extracted with Trizol and analysed, and cDNA generated as described previously [6, 7]. Gene expression using probes and primers as described in electronic supplementary material (ESM) Table 1 were analysed quantitatively and reported relative to the expression of the housekeeping gene 18S (18S ribosomal RNA Taqman Control Reagent kit) using the Taqman system (ABI Prism 7500; Perkin-Elmer, Poster City, CA, USA) [6, 12]. Results were expressed relative to respective non-diabetic Sm22+NOX5− mice or VEcad+NOX5− mice, which were arbitrarily assigned a value of 1.
Western blot
Western blot analysis of frozen kidney was performed as described previously [11]. Briefly, protein extracts from the renal cortex were electrophoresed on 7.5–10% acrylamide gels under non-reducing conditions. Western blot analysis was then performed after incubation with primary antibodies to the following: NOX5 (rabbit polyclonal, catalogue no. ab191010; Abcam); nitrotyrosine (rabbit polyclonal, catalogue no. N0409; Sigma-Aldrich-Merck, Castle Hill, NSW, Australia), nuclear factor erythroid 2-related factor 2 (NRF2) (goat polyclonal, catalogue no. SAB2501713; Sigma-Aldrich-Merck); monocyte chemoattractant protein-1 (MCP-1) (rabbit polyclonal, catalogue no. ab25124; Abcam); NF-κB-p65 (rabbit polyclonal, catalogue no. 51-0500; Invitrogen); TLR-4 (rabbit polyclonal, catalogue no. bs-1021R; Bioss Antibodies); PKC-α (rabbit polyclonal, catalogue no. sc-208; Santa Cruz Biotechnology); heat shock protein 90 (HSP90) (rat monoclonal, catalogue no. ab13494; Abcam) and transient receptor potential cation channel subfamily C member 6 (TRPC-6) (rabbit polyclonal, catalogue no. ab62461; Abcam). Blots were then incubated with secondary antibodies as follows: goat anti-rabbit for NOX5, nitrotyrosine, MCP-1, NF-κB-p65, TLR-4, PKC-α and TRPC-6; rabbit anti-goat for NRF2 and rabbit anti-rat for HSP90 (Dako Corp., Carpinteria, CA, USA). Membranes were subsequently probed for β-actin (42 kDa; Sigma-Aldrich) to confirm equal loading of samples. Blots were detected using the ECL detection kit (Sigma-Aldrich) and densitometry was performed using Quantity-One software (Bio-Rad Laboratories, Richmond, CA, USA).
Statistical analysis
All variables were analysed by one-way ANOVA using GraphPad Prism 7 (San Diego, CA, USA) for multiple comparison of the means followed by Tukey’s post hoc test or analysed by the two-tailed unpaired Mann–Whitney U test when required. A p value <0.05 was considered to be statistically significant. Results are expressed as mean ± SEM, unless otherwise specified.
Results
Characterisation of human NOX5 expression in VSMCs or endothelial cells of transgenic mice
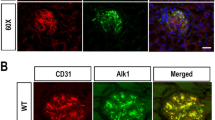
NOX5 protein expression in mesangial cells was confirmed by co-localisation of NOX5 and SM22-α (a marker of smooth muscle cells) (Fig. 1a) and in endothelial cells by co-localisation of NOX5 and CD31 (a marker of endothelial cells) (Fig. 1d) in the glomeruli of respective Sm22+NOX5+ and VEcad+NOX5+ transgenic mice. Both Sm22+NOX5+ and VEcad+NOX5+diabetic transgenic Akita mice demonstrated increased renal NOX5 protein levels when compared with non-diabetic control mice (Fig. 1b,c,e,f).

Expression of human NOX5 in mouse glomerular cells. NOX5 transgenic mice expressing NOX5β either in VSMCs/mesangial cells (Sm22+NOX5+) or in endothelial cells (VEcad+NOX5+) in the glomerulus were used. (a, d) Co-localisation of NOX5 (red staining), SM22-α (a marker of smooth muscle cells; green staining in a) and CD31 (a marker of endothelial cells; green staining in d) in frozen kidney sections of respective non-diabetic control Sm22+NOX5+ (a) and VEcad+NOX5+ (d) transgenic mice. Scale bar, 20 μm in all photomicrographs. (b, e) Western blots showing expression of NOX5 (86 kDa) in the renal cortex of the control and Akita diabetic Sm22+NOX5+ (b) and VEcad+NOX5+ (e) transgenic mice; (c, f) quantification of (b) and (e), respectively (n=6/group). β-Actin (42 kDa) serves as a housekeeping protein. Data are shown as mean ± SEM. †p<0.05 vs respective control Sm22+NOX5+ or VEcad+NOX5+ mice. C, non-diabetic control mice; D, diabetic mice
Metabolic variables
Both Sm22+NOX5− and Sm22+NOX5+ diabetic Akita mice displayed reduced body weight, increased kidney weight/body weight ratio, elevated plasma glucose and HbA1c levels, increased food and water intake and enhanced urine output (Table 1) when compared with their respective non-diabetic control mice. Similar findings with respect to metabolic variables were observed in VEcad+NOX5− and VEcad+NOX5+ Akita diabetic vs non-diabetic mice (Table 2). Expression of NOX5 in VSMCs or in endothelial cells had no effect on hyperglycaemia-induced changes in these metabolic variables (Tables 1 and 2). In addition, systolic blood pressure was similar in all groups (Tables 1 and 2).
Renal oxidative stress
Expression levels of renal nitrotyrosine (Fig. 2a–d) and intensity of DHE fluorescence (Fig. 2e) were increased by diabetes in Sm22+NOX5− Akita mice; these diabetes-induced changes were more marked in Sm22+NOX5+ diabetic Akita mice. Similar findings were seen in endothelial cell-specific NOX5-expressing Akita mice (Fig. 2h–l). Expression of both Nox2 (also known as Cybb) and Nox4 mRNA were increased in Sm22+NOX5− (Fig. 2f,g) and VEcad+NOX5− diabetic Akita mice (Fig. 2m,n). The presence of NOX5, either in VSMCs or endothelial cells, did not have any effect on Nox2 mRNA expression in both non-diabetic and diabetic mice (Fig. 2f,m); however, downregulation of Nox4 was observed in the presence or absence of diabetes in both Sm22+NOX5+ (Fig. 2g) and VEcad+NOX5+ mice (Fig. 2n).

Expression of NOX5 either in VSMCs/mesangial cells (a–e) or in endothelial cells (h–l) enhances renal ROS production in Akita diabetic mice. (a, b, h, i) Immunostaining of glomerular nitrotyrosine (a, h) and its quantification (b, i) (n=6–9/group) in control and Akita diabetic Sm22+NOX5− and Sm22+NOX5+ or VEcad+NOX5− and VEcad+NOX5+ transgenic mice, respectively. (c, d, j, k) Western blots showing the expression of renal cortical nitrotyrosine (68 kDa) (c, j) and its quantification (d, k) (n=3/group) in control and Akita diabetic Sm22+NOX5− and Sm22+NOX5+ or VEcad+NOX5− and VEcad+NOX5+ transgenic mice, respectively. (e–g, l–n) Immunofluorescence staining for glomerular DHE (e, l) and gene expression of Nox2 and Nox4 in the renal cortex (f, g, m, n) of respective control and Akita diabetic Sm22+NOX5− and Sm22+NOX5+ or VEcad+NOX5− and VEcad+NOX5+ transgenic mice. 18s RNA and β-actin (42 kDa) serve as housekeeping gene and protein, respectively. Scale bar, 20 μm in all photomicrographs. Data are shown as mean ± SEM. *p<0.05 vs respective control Sm22+NOX5− or VEcad+NOX5− mice; †p<0.05 vs respective control Sm22+NOX5+ or VEcad+NOX5+ mice; ‡p<0.05 vs respective diabetic Sm22+NOX5− or VEcad+NOX5− mice; C, non-diabetic control mice; D, diabetic mice; NT, nitrotyrosine
NRF2
The expression levels of the key transcription factor linked to antioxidant–redox signalling, NRF2, were increased by diabetes alone or by expression of the human NOX5 gene in both mesangial cells (Fig. 3a,c) and endothelial cells (Fig. 3b,d) of NOX5 transgenic mice. The raised mRNA expression levels of Nrf2 (also known as Nfe2l2) in diabetic mice were further increased by the expression of NOX5 in these renal cells.

Expression of NRF2 protein in the kidney. Western blots showing the expression of NRF2 (61 kDa) in the renal cortex of the control and Akita diabetic Sm22+NOX5− and Sm22+NOX5+ (a) or VEcad+NOX5− and VEcad+NOX5+ (b) transgenic mice and quantification of the respective NRF2 expression levels (c, d) (n=3/group). β-Actin (42 kDa) serves as a housekeeping protein. Data are shown as mean ± SEM. *p<0.05 vs respective control Sm22+NOX5− or VEcad+NOX5− mice; †p<0.05 vs respective control Sm22+NOX5+ or VEcad+NOX5+ mice; ‡p<0.05 vs respective diabetic Sm22+NOX5− or VEcad+NOX5− mice; ¶p=0.06 vs control VEcad+NOX5− mice. C, non-diabetic control mice; D, diabetic mice
Renal inflammation
NF-κB and MCP-1
The gene and protein expression of the key proinflammatory transcription factor NF-κB (gene also known as Rela) (Fig. 4a–c) and the NF-κB-dependent chemokine MCP-1 (gene also known as Ccl2) (Fig. 5a–c) were increased in the renal cortex of Sm22+NOX5− diabetic Akita mice. Renal NF-κB and MCP-1 expression was further increased in diabetic Akita mice expressing NOX5 in VSMCs/mesangial cells when compared with diabetic Akita Sm22+NOX5− mice (Figs 4a–c, 5a–c). In addition, renal expression of both NF-κB and MCP-1 were upregulated even in the absence of diabetes in Sm22+NOX5+ mice (Figs 4a–c, 5a–c). Similar findings were observed in endothelial cell-specific NOX5-expressing transgenic Akita mice (Figs 4d–f, 5d–f).

Gene and protein expression of the p65 subunit of NF-κB in the kidney. (a, c, d, f) Western blots showing the expression of NF-κB (68 kDa) protein in the renal cortex of the respective control and Akita diabetic Sm22+NOX5− and Sm22+NOX5+ (a) or VEcad+NOX5− and VEcad+NOX5+ (d) transgenic mice, and quantification of the expression levels (c, f) (n=6/group). (b, e) Renal Nfκb mRNA expression in control and Akita diabetic Sm22+NOX5− and Sm22+NOX5+ (b) or VEcad+NOX5− and VEcad+NOX5+ (e) transgenic mice (n=8–10/group). 18s RNA and β-actin (42 kDa) serve as housekeeping gene and protein, respectively. Data are shown as mean ± SEM. *p<0.05 vs respective control Sm22+NOX5− or VEcad+NOX5− mice; †p<0.05 vs respective control Sm22+NOX5+ or VEcad+NOX5+ mice; ‡p<0.05 vs respective diabetic Sm22+NOX5− or VEcad+NOX5− mice; ¶p=0.055 vs diabetic VEcad+NOX5− mice. C, non-diabetic control mice; D, diabetic mice

Gene and protein expression of MCP-1 in the kidney. (a, c, d, f) Western blots showing the expression of MCP-1 (25 kDa) in the renal cortex of the respective control and Akita diabetic Sm22+NOX5− and Sm22+NOX5+ (a) or VEcad+NOX5− and VEcad+NOX5+ (d) transgenic mice and quantification of the expression levels (c, f) (n=6 or 7/group). (b, e) Renal Mcp-1 mRNA expression in control and Akita diabetic Sm22+NOX5− and Sm22+NOX5+ (b) or VEcad+NOX5− and VEcad+NOX5+ (e) transgenic mice (n=8–10/group). 18s RNA and β-actin (42 kDa) serve as housekeeping gene and protein, respectively. Data are shown as mean ± SEM. *p<0.05 vs respective control Sm22+NOX5− or VEcad+NOX5− mice; †p<0.05 vs respective control Sm22+NOX5+ or VEcad+NOX5+ mice; ‡p<0.05 vs respective diabetic Sm22+NOX5− or VEcad+NOX5− mice; ¶p=0.055 vs diabetic VEcad+NOX5− mice. C, non-diabetic control mice; D, diabetic mice
TLR-4 and PKC-α
Both TLR-4 and PKC-α have been associated with increased expression of MCP-1 via activation of NF-κB and promote renal inflammation in DKD. The expression levels of TLR-4 (Fig. 6a–e) and PKC-α (Fig. 7a–e) gene and protein were increased in the renal cortex of Sm22+NOX5− diabetic Akita mice and were further increased in diabetic Akita mice expressing NOX5 in VSMCs/mesangial cells (Figs 6a–e, 7a–e). In addition, PKC-α expression was increased even in the absence of diabetes in Sm22+NOX5+ mice (Fig. 7a–e). Similar findings with regards to TLR-4 and PKC-α expression were also observed in endothelial cell-specific NOX5-expressing Akita mice (Figs 6f–j, 7f–j).

Gene and protein expression of TLR-4 in the kidney. (a, c, f, h) Western blots showing the expression of TLR-4 (120 kDa) in the renal cortex of the respective control and Akita diabetic Sm22+NOX5− and Sm22+NOX5+ (a) or VEcad+NOX5− and VEcad+NOX5+ (f) transgenic mice and quantification of the expression levels (n=3/group) (c, h). (b, g) Gene expression of Tlr4 in control and Akita diabetic Sm22+NOX5− and Sm22+NOX5+ (b) or VEcad+NOX5− and VEcad+NOX5+ (g) transgenic mice (n=8–10/group). (d, e, i, j) Immunostaining of glomerular TLR-4 in control and Akita diabetic Sm22+NOX5− and Sm22+NOX5+ (d) or VEcad+NOX5− and VEcad+NOX5+ (i) transgenic mice and quantification (e, j) (n=5 or 6/group). 18s RNA and β-actin (42 kDa) serve as housekeeping gene and protein, respectively. Scale bar, 20 μm in all photomicrographs. Data are shown as mean ± SEM. *p<0.05 vs respective control Sm22+NOX5− or VEcad+NOX5− mice; †p<0.05 vs respective control Sm22+NOX5+ or VEcad+NOX5+ mice; ‡p<0.05 vs respective diabetic Sm22+NOX5− or VEcad+NOX5− mice and ¶p=0.055 vs diabetic VEcad+NOX5− mice. C, non-diabetic control mice; D, diabetic mice

Gene and protein expression of PKC-α in the kidney. (a, c, f, h) Western blots showing the expression of renal cortical PKC-α (83 kDa) in the renal cortex of the respective control and Akita diabetic Sm22+NOX5− and Sm22+NOX5+ (a) or VEcad+NOX5− and VEcad+NOX5+ (f) transgenic mice and quantification of the expression levels (c, h). (b, g) Gene expression of Pkc-α (also known as Pkca) in control and Akita diabetic Sm22+NOX5− and Sm22+NOX5+ (b) or VEcad+NOX5− and VEcad+NOX5+ (g) transgenic mice (n=7–10/group). (d, e, i, j) Immunostaining of glomerular PKC-α in control and Akita diabetic Sm22+NOX5− and Sm22+NOX5+ (d) or VEcad+NOX5− and VEcad+NOX5+ (i) transgenic mice and quantification (e, j) (n=6 or 7/group). 18s RNA and β-actin (42 kDa) serve as housekeeping gene and protein, respectively. Scale bar, 20 μm in all photomicrographs. Data are shown as mean ± SEM. *p<0.05 vs respective control Sm22+NOX5− or VEcad+NOX5− mice; †p<0.05 vs respective control Sm22+NOX5+ or VEcad+NOX5+ mice; ‡p<0.05 vs respective diabetic Sm22+NOX5− or diabetic VEcad+NOX5− mice; ¶p=0.05 vs control Sm22+NOX5− mice. C, non-diabetic control mice; D, diabetic mice
Renal function (albuminuria and creatinine clearance)
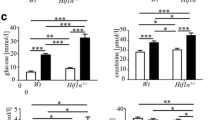
Increased creatinine clearance was seen in both diabetic groups of Akita mice when compared with their respective controls but expression of NOX5 in both VSMCs (Table 1) and endothelial cells (Table 2) had no significant effect on creatinine clearance. The effect of NOX5 expression per se in both renal cell populations did not increase albuminuria in the absence of diabetes. Albuminuria was significantly increased in both groups of diabetic mice when compared with their respective non-diabetic control groups (Table 1). A non-significant increase in urinary albumin/creatinine ratio (p = 0.057) was observed in diabetic Sm22+NOX5+ Akita mice when compared with diabetic Sm22+NOX5− mice (Table 1). In contrast, endothelial cell-specific NOX5 expression (VEcad+NOX5+) in the presence of diabetes resulted in a twofold increase in both 24 h albuminuria and albumin/creatinine ratio (Table 2) when compared with diabetic VEcad+NOX5− Akita mice.
Renal structure (mesangial expansion, glomerulosclerosis and extracellular matrix accumulation)
Diabetic Akita mice showed increased mesangial area, GSI (Fig. 8) and tubulointerstitial injury (ESM Fig. 1) when compared with non-diabetic control mice. NOX5 expression in VSMCs/mesangial cells further increased mesangial area and glomerulosclerosis in the presence of diabetes when compared with diabetic Sm22+NOX5− Akita mice (Fig. 8a–c). Findings in endothelial cell-specific NOX5-expressing Akita mice were not statistically significant with respect to increases in mesangial area (p = 0.06) and glomerulosclerosis (p = 0.055) (Fig. 8d–f) when compared with diabetic VEcad+NOX5− mice. Furthermore, the diabetes-induced increase in tubulointerstitial injury was unchanged by NOX5 expression (ESM Fig. 1).

Expression of NOX5 exacerbates glomerular injury in Akita diabetic mice. Periodic Acid–Schiff staining (a, d), mesangial area expansion (b, e) and GSI (c, f) in the respective control and Akita diabetic Sm22+NOX5− and Sm22+NOX5+ (a–c) or VEcad+NOX5− and VEcad+NOX5+ (d–f) transgenic mice (n=8–10/group). Scale bar, 20 μm in all photomicrographs. Data are shown as mean ± SEM. *p<0.05 vs respective control Sm22+NOX5− or VEcad+NOX5− mice; †p<0.05 vs respective control Sm22+NOX5+ or VEcad+NOX5+ mice; ‡p<0.05 vs diabetic Sm22+NOX5− mice; ¶p=0.055 vs diabetic VEcad+NOX5− mice. C, non-diabetic control mice; D, diabetic mice
Extracellular matrix gene and protein expression
Renal cortical gene expression of profibrotic markers, including collagen III, fibronectin and α-smooth muscle actin (α-SMA) (Fig. 9a,d), and protein expression of both glomerular and renal cortical collagen IV (Fig. 9b,c,e,f and ESM Fig. 2) in mice were not altered by expression of NOX5 in the absence of diabetes. However, the diabetes-induced increase in the gene expression of collagen III, fibronectin and α-SMA was further upregulated in both VSMC- and endothelial cell-specific NOX5 transgenic diabetic mice (Fig. 9a,d). Extracellular matrix (ECM) protein expression of glomerular collagen IV was further increased in diabetic Sm22+NOX5+ Akita mice when compared with diabetic Sm22+NOX5− Akita mice (Fig. 9b,c). In addition, endothelial cell-specific NOX5 expression caused a non-significant increase in glomerular collagen IV accumulation (p = 0.056) in the presence of diabetes, as observed when comparing VEcad+NOX5+ with VEcad+NOX5− diabetic mice (Fig. 9e,f).

Expression of NOX5 exacerbates renal fibrosis in Akita diabetic mice. Renal gene expression of collagen III, fibronectin and α-SMA (a, d), immunostaining of collagen IV (b, e) and its quantification (c, f) in the respective control and Akita diabetic Sm22+NOX5− and Sm22+NOX5+ (a–c) or VEcad+NOX5− and VEcad+NOX5+ (d–f) transgenic mice (n=8–10/group). Scale bar, 20 μm in all photomicrographs. Data are shown as mean ± SEM. *p<0.05 vs respective control Sm22+NOX5− or VEcad+NOX5− mice; †p<0.05 vs respective control Sm22+NOX5+ or VEcad+NOX5+ mice; ‡p<0.05 vs diabetic Sm22+NOX5− or VEcad+NOX5− mice and ¶p=0.056 vs diabetic VEcad+NOX5− mice. C, non-diabetic control mice; D, diabetic mice
HSP90 and TRPC-6
We examined the regulatory elements of NOX5 including HSP90 and TRPC-6 which modulate NOX5 function. Expression of either NOX5 alone in endothelial and VSMCs or diabetes alone had no effect on the protein expression of HSP90 or TRPC-6 in Akita mice (Figs 10a–d, 11a–d). Interestingly, expression of NOX5 in VSMCs/mesangial cells in the presence of diabetes significantly upregulated the expression of HSP90 (Fig. 10a,b) and TRPC-6 (Fig. 11a,b) when compared with diabetic Sm22+NOX5− Akita mice. Similar patterns were found with regards to HSP90 (p < 0.05, Fig. 10c, d) and TRPC-6 (p = 0.06, Fig. 11c,d) expression in endothelial cell-specific NOX5 transgenic diabetic Akita mice.

Protein expression of HSP90 in the kidney. (a, c) Western blots showing the expression of HSP90 (85 kDa) in the renal cortex of the respective control and Akita diabetic Sm22+NOX5− and Sm22+NOX5+ (a) or VEcad+NOX5− and VEcad+NOX5+ (c) transgenic mice and quantification of expression levels (b, d) (n=3 or 4/group). β-Actin (42 kDa) serves as housekeeping protein. Data are shown as mean ± SEM. †p<0.05 vs respective control Sm22+NOX5+ or VEcad+NOX5+ mice; ‡p<0.05 vs respective diabetic Sm22+NOX5− or VEcad+NOX5− mice

Protein expression of TRPC-6 in the kidney. (a, c) Western blots showing the expression of TRPC-6 (106 kDa) in the renal cortex of the respective control and Akita diabetic Sm22+NOX5− and Sm22+NOX5+ (a) or VEcad+NOX5− and VEcad+NOX5+ (c) transgenic mice and quantification of expression levels (b, d) (n=3/group). β-Actin (42 kDa) serves as housekeeping protein. Data are shown as mean ± SEM. †p<0.05 vs respective control Sm22+NOX5+ or VEcad+NOX5+ mice; ‡p<0.05 vs diabetic Sm22+NOX5− mice; ¶p=0.06 vs VEcad+NOX5− diabetic Akita mice
Discussion
Renal ROS have been shown to play a key role in the activation of proinflammatory cytokines, chemokines and profibrotic factors in DKD [6, 7, 11]. In this study we provide critical information on directly targeting the source of renal ROS in diabetes by specifically exploring the role of the prooxidant enzyme NOX5 in the pathogenesis of DKD. Since mesangial and endothelial cells are key components of glomeruli and their structural and functional alterations contribute to renal impairment in diabetes, we examined the effect of inducible human NOX5 expression either in VSMCs, which represent mesangial cells in the kidney, or in endothelial cells in the Akita mouse model of DKD. The key finding of this study is that NOX5 expression in these two cell populations in the diabetic Akita mouse leads to additive effects on top of hyperglycaemia with regards to accelerated glomerular injury, inflammation, fibrosis and albuminuria (Fig. 12).

Schematic representation of the contribution of NOX5 expression in renal injury in diabetes via activation of inflammatory elements (TLR-4, NF-κB and MCP-1) and PKC-α
We and others have previously demonstrated increased renal expression of NOX5 in individuals with diabetes [10, 11]. We demonstrated an upregulation of NOX5 in human mesangial cells exposed to glucose in vitro and that silencing of NOX5 attenuated glucose induced ROS in association with a decrease in a range of markers of inflammation and fibrosis [11]. Consistent with the findings of previous studies [6, 7, 11], diabetic Akita mice showed increased levels of ROS, including nitrotyrosine and DHE, in the kidney. NOX5 expression either in endothelial or in smooth muscle cells did not cause an increase in ROS in the absence of diabetes. Diabetes caused an increase in renal ROS and the expression of NOX5 in both renal cell populations increased ROS further; this ‘double-hit’ resulted in accelerated renal injury.
With respect to other NOX isoforms, we have previously shown that NOX1 does not play a role in DKD [6], deletion of NOX2 in diabetes is not a favourable target because of its role in phagocytosis with lethality in the setting of diabetes [14] and NOX4 plays a detrimental role in experimental DKD [5,6,7, 15]. In the current study, NOX5 expression did not modulate NOX2 expression but a downregulation of NOX4 was observed in the NOX5 transgenic mice in both diabetic and non-diabetic contexts. These findings are consistent with the further increase in renal ROS formation in NOX5 transgenic diabetic mice, most likely occurring as a result of concomitant expression of NOX5. This contrasts with a study in which expression of NOX5 in podocytes induced podocyte injury and albuminuria even in the absence of diabetes [10].
Oxidative stress occurs as a result of not only increased ROS formation but also depletion in antioxidant defence. NRF2 is an important element in antioxidant defence since it regulates the expression of antioxidant proteins that protect against oxidative damage triggered by injury and inflammation [16, 17]. A recent study, albeit in the diabetic heart, reported time-dependent effects of hyperglycaemia on NRF2 expression, as reflected by increased expression of NRF2 after 2 months but decreased expression of NRF2 after 5 months of STZ-induced diabetes in mice [18]. In line with this, we identified increased expression of renal NRF2 in diabetic mice after 10 weeks of diabetes and this was further increased by NOX5 expression. Notably, an upregulation of renal NRF2 was also found in response to expression of NOX5 even in the absence of diabetes. These data suggest that NOX5 is involved in the regulation of NRF2 and that upregulation of NRF2 may potentially play a compensatory role in response to the high levels of ROS accumulation mediated via NOX5 activity at least in the early stage of diabetes. This may also explain why NOX5 expression per se is not associated with increased ROS levels and renal injury in the absence of diabetes. Thus, only the presence of both diabetes and NOX5 expression leads to overwhelming ROS production resulting in significant end organ injury.
Inflammation is considered to be a key pathological feature of DKD [19, 20]. Proinflammatory chemokines such as MCP-1 play a key role in promoting macrophage infiltration and the transcription factor NF-κB is key in regulating the production of cytokines in the setting of diabetes [19, 21]. Indeed, diabetic Akita mice showed increased renal expression levels of MCP-1 and NF-κB, compared with non-diabetic mice, and levels were further increased by expression of both endothelial cell- and VSMC-specific NOX5. Interestingly, expression of NOX5 alone even in the absence of diabetes was able to induce the expression of these two proinflammatory molecules in the kidney. These findings are consistent with those of previous in vitro studies by our group which showed that knockdown of NOX5 in human mesangial cells attenuated the expression of high-glucose-induced increased expression of MCP-1 and NF-κB [11]. There is also increasing evidence of a role for toll-like receptors, particularly TLR-4, in macrophage infiltration as observed in DKD [22, 23]. Glucose per se upregulates TLR-4 in human renal cells and silencing attenuates glucose-induced increases in the expression of various inflammatory mediators including IL-6 and MCP-1 via inactivation of NF-κB [22]. We observed similar effects in relation to increased expression of TLR-4 in the kidney of diabetic Akita mice and further increases in the NOX5-expressing Akita mice. Our findings extend the results of a recent study which suggested that expression of NOX5 in podocytes of transgenic mice promotes renal inflammation via a TLR-4-dependent pathway albeit in an non-diabetic inflammatory model [24].
Activation of the PKC pathway, particularly with regard to the PKC-α and PKC-β isoforms, correlates with oxidative stress and inflammation and plays a key role in DKD [7, 8, 25]. In addition, PKC-α and PKC-β [26, 27] directly phosphorylate NOX5 and influence its enzymatic activity [28, 29]. Moreover, a downregulation of both PKC-α and PKC-β has been seen in NOX5-knockdown human mesangial cells [11]. In this study we observed upregulation of renal PKC-α in the presence of either NOX5 expression or diabetes alone and which was accelerated in NOX5-expressing diabetic mice. These findings suggest the presence of a self-perpetuating vicious cycle with a positive feedback loop among NOX5, PKC-α and chronic hyperglycaemia which likely exacerbates renal pathology in diabetes.
Diabetes leads to alterations in the glomerular filtration barrier including ultrastructural damage to the endothelial glycocalyx, endothelial cells and podocytes, leading to albuminuria [6, 7, 30]. We report here that diabetic Akita mice displayed increased albuminuria which was further increased in the context of concomitant NOX5 expression in endothelial cells, supporting the important role of the endothelial cell in the glomerular filtration barrier, a phenomenon not seen in non-diabetic mice. This effect was less pronounced when NOX5 was expressed in VSMCs/mesangial cells in diabetic Akita mice. These findings suggest that ROS derived from one cell type in the kidney, either endothelial cells or VSMCs, lead to local renal damage primarily in glomeruli but potentially also in the renal cortex, leading to functional and structural abnormalities including albuminuria and renal fibrosis.
Given the importance of mesangial and endothelial cells in the morphological and functional manifestations of DKD [31, 32], diabetic Akita mice showed increased mesangial expansion and glomerulosclerosis. These structural variables were further increased in diabetic mice expressing NOX5 in VSMCs/mesangial cells, with the further increase being statistically insignificant in diabetic mice expressing NOX5 in endothelial cells. Consistent with the mesangial expansion and glomerulosclerosis findings, the diabetes-induced increase in expression levels of ECM components, including renal collagens, fibronectin and α-SMA, was enhanced by NOX5 expression in both VSMCs/mesangial and endothelial cells of diabetic mice. Thus, this study provides evidence that NOX5 plays an important role in the evolution of structural changes in DKD via activation of ROS-mediated profibrotic/sclerotic and proinflammatory pathways in diabetes.
In addition to renal fibrosis and inflammation, the involvement of key intracellular signalling pathways implicated in the regulation of NOX5 activity, including TRPC-6 and HSP90, was examined [9, 33,34,35]. A correlation between NOX-derived ROS and the regulation of Ca2+ channels by TRPC-6 has been demonstrated in several studies, including one using a model of podocyte injury [36,37,38]. Moreover, unlike other NOX isoforms, NOX5 requires binding of intracellular Ca2+ to its EF-hand regulatory domains for the activation and catalytic production of superoxide [39]. Indeed, we observed that expression of human NOX5 in either of the renal cell types in the mouse kidney was associated with an upregulation of TRPC-6 only in the presence of diabetes. This is in line with our previous study showing that silencing of NOX5 in human mesangial cells attenuated high-glucose-induced increased expression of TRPC-6 [11]. In addition, it has been reported that HSP90 binds to the C-terminus of NOX5 and is necessary for enzyme stability and superoxide production [33, 35]. Furthermore, long-term inhibition of HSP90 was associated with reduced expression of NOX5 [34]. In this study we have demonstrated that diabetes alone or expression of NOX5 alone in both mesangial cells and endothelial cells had no direct effect on the expression of HSP90 in the kidney. However, expression of NOX5 in either cell type in the presence of diabetes resulted in increased expression of renal HSP90. Thus, the findings from this study suggest that TRPC-6 and HSP90 act downstream and upstream of NOX5 and that there is either a positive feedback loop or a bidirectional activation pathway between TRPC-6 and NOX5 as well as HSP90 and NOX5. Other signalling pathways linked to NOX5, such as Ca2+, may also be important, as previously demonstrated by us in NOX5-expressing mice [40].
In conclusion, identification of a key role for NOX5 in promoting renal injury in diabetes provides the rationale for examining NOX5 in models where this isoform is endogenously expressed, including humans and other animals such as rabbits. Our findings also provide the impetus to develop NOX5-specific inhibitors.
Data availability
All data that support the conclusions are available from the corresponding author on request.
Abbreviations
- DHE:
-
Dihydroethidium
- DKD:
-
Diabetic kidney disease
- ECM:
-
Extracellular matrix
- GSI:
-
Glomerulosclerosis index
- HSP90:
-
Heat shock protein 90
- MCP-1:
-
Monocyte chemoattractant protein-1
- NOX:
-
NADPH oxidase
- NRF2:
-
Nuclear factor erythroid 2-related factor 2
- PKC-α:
-
Protein kinase C-α
- ROS:
-
Reactive oxygen species
- αSMA:
-
α-Smooth muscle actin
- SM22:
-
Transgelin
- STZ:
-
Streptozotocin
- TLR-4:
-
Toll-like receptor-4
- TRPC-6:
-
Transient receptor potential cation channel subfamily C member 6
- VEcad:
-
Vascular endothelial cadherin
- VSMC:
-
Vascular smooth muscle cell
References
Molitch ME, DeFronzo RA, Franz MJ et al (2004) Nephropathy in diabetes. Diabetes Care 27(Suppl 1):S79–S83
Dimke H, Maezawa Y, Quaggin SE (2015) Crosstalk in glomerular injury and repair. Curr Opin Nephrol Hypertens 24(3):231–238. https://doi.org/10.1097/MNH.0000000000000117
Dimke H, Sparks MA, Thomson BR, Frische S, Coffman TM, Quaggin SE (2015) Tubulovascular cross-talk by vascular endothelial growth factor a maintains peritubular microvasculature in kidney. J Am Soc Nephrol 26(5):1027–1038. https://doi.org/10.1681/ASN.2014010060
Mason RM, Wahab NA (2003) Extracellular matrix metabolism in diabetic nephropathy. J Am Soc Nephrol 14(5):1358–1373. https://doi.org/10.1097/01.ASN.0000065640.77499.D7
Gorin Y, Block K, Hernandez J et al (2005) Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J Biol Chem 280(47):39616–39626. https://doi.org/10.1074/jbc.M502412200
Jha JC, Gray SP, Barit D et al (2014) Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J Am Soc Nephrol 25(6):1237–1254. https://doi.org/10.1681/ASN.2013070810
Jha JC, Thallas-Bonke V, Banal C et al (2016) Podocyte-specific Nox4 deletion affords renoprotection in a mouse model of diabetic nephropathy. Diabetologia 59(2):379–389. https://doi.org/10.1007/s00125-015-3796-0
Thallas-Bonke V, Jha JC, Gray SP et al (2014) Nox-4 deletion reduces oxidative stress and injury by PKC-α-associated mechanisms in diabetic nephropathy. Phys Rep 2(11):e12192. https://doi.org/10.14814/phy2.12192
Fulton DJ (2009) Nox5 and the regulation of cellular function. Antioxid Redox Signal 11(10):2443–2452. https://doi.org/10.1089/ars.2009.2587
Holterman CE, Thibodeau JF, Towaij C et al (2014) Nephropathy and elevated BP in mice with podocyte-specific NADPH oxidase 5 expression. J Am Soc Nephrol 25(4):784–797. https://doi.org/10.1681/ASN.2013040371
Jha JC, Banal C, Okabe J et al (2017) NADPH oxidase Nox5 accelerates renal injury in diabetic nephropathy. Diabetes 66(10):2691–2703. https://doi.org/10.2337/db16-1585
Watson AM, Li J, Schumacher C et al (2010) The endothelin receptor antagonist avosentan ameliorates nephropathy and atherosclerosis in diabetic apolipoprotein E knockout mice. Diabetologia 53(1):192–203. https://doi.org/10.1007/s00125-009-1540-3
Lassila M, Seah KK, Allen TJ et al (2004) Accelerated nephropathy in diabetic apolipoprotein e-knockout mouse: role of advanced glycation end products. J Am Soc Nephrol 15(8):2125–2138. https://doi.org/10.1097/01.ASN.0000133025.23732.46
Gray SP, Di Marco E, Okabe J et al (2013) NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 127(18):1888–1902. https://doi.org/10.1161/CIRCULATIONAHA.112.132159
Gray SP, Jha JC, Kennedy K et al (2017) Combined NOX1/4 inhibition with GKT137831 in mice provides dose-dependent reno- and atheroprotection even in established micro- and macrovascular disease. Diabetologia 60(5):927–937. https://doi.org/10.1007/s00125-017-4215-5
Cheng X, Siow RC, Mann GE (2011) Impaired redox signaling and antioxidant gene expression in endothelial cells in diabetes: a role for mitochondria and the nuclear factor-E2-related factor 2-Kelch-like ECH-associated protein 1 defense pathway. Antioxid Redox Signal 14(3):469–487. https://doi.org/10.1089/ars.2010.3283
He X, Kan H, Cai L, Ma Q (2009) Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. J Mol Cell Cardiol 46(1):47–58. https://doi.org/10.1016/j.yjmcc.2008.10.007
Tan Y, Ichikawa T, Li J et al (2011) Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes 60(2):625–633. https://doi.org/10.2337/db10-1164
Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Rollin BJ, Tesch GH (2006) Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int 69(1):73–80. https://doi.org/10.1038/sj.ki.5000014
Navarro-Gonzalez JF, Mora-Fernandez C (2008) The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol 19(3):433–442. https://doi.org/10.1681/ASN.2007091048
Jha JC, Banal C, Chow BS, Cooper ME, Jandeleit-Dahm K (2016) Diabetes and kidney disease: role of oxidative stress. Antioxid Redox Signal 25(12):657–684. https://doi.org/10.1089/ars.2016.6664
Lin M, Yiu WH, Wu HJ et al (2012) Toll-like receptor 4 promotes tubular inflammation in diabetic nephropathy. J Am Soc Nephrol 23(1):86–102. https://doi.org/10.1681/ASN.2010111210
Pulskens WP, Rampanelli E, Teske GJ et al (2010) TLR4 promotes fibrosis but attenuates tubular damage in progressive renal injury. J Am Soc Nephrol 21(8):1299–1308. https://doi.org/10.1681/ASN.2009070722
Holterman CE, Boisvert NC, Thibodeau JF et al (2018) Podocyte NADPH oxidase 5 promotes renal inflammation regulated by the Toll-like receptor pathway. Antioxid Redox Signal 30:1817–1830
Ren J, Wang Q, Morgan S et al (2014) Protein kinase C-δ (PKCδ) regulates proinflammatory chemokine expression through cytosolic interaction with the NF-κB subunit p65 in vascular smooth muscle cells. J Biol Chem 289(13):9013–9026. https://doi.org/10.1074/jbc.M113.515957
Chen F, Yu Y, Haigh S et al (2014) Regulation of NADPH oxidase 5 by protein kinase C isoforms. PLoS One 9(2):e88405. https://doi.org/10.1371/journal.pone.0088405
Pandey D, Fulton DJ (2011) Molecular regulation of NADPH oxidase 5 via the MAPK pathway. Am J Phys Heart Circ Phys 300(4):H1336–H1344. https://doi.org/10.1152/ajpheart.01163.2010
Jagnandan D, Church JE, Banfi B, Stuehr DJ, Marrero MB, Fulton DJ (2007) Novel mechanism of activation of NADPH oxidase 5. calcium sensitization via phosphorylation. J Biol Chem 282(9):6494–6507. https://doi.org/10.1074/jbc.M608966200
Serrander L, Jaquet V, Bedard K et al (2007) NOX5 is expressed at the plasma membrane and generates superoxide in response to protein kinase C activation. Biochimie 89(9):1159–1167. https://doi.org/10.1016/j.biochi.2007.05.004
Wolf G, Ziyadeh FN (1999) Molecular mechanisms of diabetic renal hypertrophy. Kidney Int 56(2):393–405. https://doi.org/10.1046/j.1523-1755.1999.00590.x
Steffes MW, Bilous RW, Sutherland DE, Mauer SM (1992) Cell and matrix components of the glomerular mesangium in type I diabetes. Diabetes 41(6):679–684. https://doi.org/10.2337/diab.41.6.679
Mauer SM, Steffes MW, Ellis EN, Sutherland DE, Brown DM, Goetz FC (1984) Structural-functional relationships in diabetic nephropathy. J Clin Invest 74(4):1143–1155. https://doi.org/10.1172/JCI111523
Chen F, Haigh S, Yu Y et al (2015) Nox5 stability and superoxide production is regulated by C-terminal binding of Hsp90 and CO-chaperones. Free Radic Biol Med 89:793–805. https://doi.org/10.1016/j.freeradbiomed.2015.09.019
Chen F, Pandey D, Chadli A, Catravas JD, Chen T, Fulton DJ (2011) Hsp90 regulates NADPH oxidase activity and is necessary for superoxide but not hydrogen peroxide production. Antioxid Redox Signal 14(11):2107–2119. https://doi.org/10.1089/ars.2010.3669
Chen F, Yu Y, Qian J et al (2012) Opposing actions of heat shock protein 90 and 70 regulate nicotinamide adenine dinucleotide phosphate oxidase stability and reactive oxygen species production. Arterioscler Thromb Vasc Biol 32(12):2989–2999. https://doi.org/10.1161/ATVBAHA.112.300361
Hool LC, Corry B (2007) Redox control of calcium channels: from mechanisms to therapeutic opportunities. Antioxid Redox Signal 9(4):409–435. https://doi.org/10.1089/ars.2006.1446
Moller CC, Wei C, Altintas MM et al (2007) Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J Am Soc Nephrol 18(1):29–36. https://doi.org/10.1681/ASN.2006091010
Wang Z, Wei X, Zhang Y et al (2009) NADPH oxidase-derived ROS contributes to upregulation of TRPC6 expression in puromycin aminonucleoside-induced podocyte injury. Cell Physiol Biochem 24(5-6):619–626. https://doi.org/10.1159/000257517
Tirone F, Radu L, Craescu CT, Cox JA (2010) Identification of the binding site for the regulatory calcium-binding domain in the catalytic domain of NOX5. Biochemistry 49(4):761–771. https://doi.org/10.1021/bi901846y
Montezano AC, De Lucca Camargo L, Persson P et al (2018) NADPH oxidase 5 is a pro-contractile Nox isoform and a point of cross-talk for calcium and redox signaling-implications in vascular function. J Am Heart Assoc 7:1–15
Acknowledgements
The authors thank E. Lastavec and M. Lyttle (Department of Diabetes, Central Clinical School, Monash University) for experimental animal handling and technical support and S. Urner (Institute for Clinical Diabetology, German Diabetes Centre, Dusseldorf, Germany) for editing and drafting of the manuscript.
Funding
This work was supported by the National Health & Medical Research Council of Australia, a JDRF Program/Project Grant. KAMJD and MEC are supported by NHMRC Senior Research and Senior Principal Research Fellowships, respectively. JCJ is supported by JDRF and an NHMRC- Early Career Fellowship.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study concept and design and the interpretation of the data and have approved the final version of the manuscript. JCJ collected research data, contributed to discussion and wrote, reviewed and edited the manuscript. AD and CEH collected research data and contributed to the manuscript. MEC, RMT, CRK and KAMJD contributed to analysis and interpretation of data and to drafting and editing the manuscript. JCJ and KAMJD are the guarantors of this work and, as such, had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding author
Ethics declarations
The authors declare that there is no duality of interest associated with this manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM
(PDF 459 kb)
Rights and permissions
About this article

Cite this article
Jha, J.C., Dai, A., Holterman, C.E. et al. Endothelial or vascular smooth muscle cell-specific expression of human NOX5 exacerbates renal inflammation, fibrosis and albuminuria in the Akita mouse. Diabetologia 62, 1712–1726 (2019). https://doi.org/10.1007/s00125-019-4924-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-019-4924-z