
Abstract
Aims/hypothesis
Sodium–glucose cotransporter (SGLT) 2 inhibitors constitute a new class of glucose-lowering drugs, but they increase glucagon secretion, which may counteract their glucose-lowering effect. Previous studies using static incubation of isolated human islets or the glucagon-secreting cell line α-TC1 suggested that this results from direct inhibition of alpha cell SGLT1/2-activity. The aim of this study was to test whether the effects of SGLT2 on glucagon secretion demonstrated in vitro could be reproduced in a more physiological setting.
Methods
We explored the effect of SGLT2 activity on glucagon secretion using isolated perfused rat pancreas, a physiological model for glucagon secretion. Furthermore, we investigated Slc5a2 (the gene encoding SGLT2) expression in rat islets as well as in mouse and human islets and in mouse and human alpha, beta and delta cells to test for potential inter-species variations. SGLT2 protein content was also investigated in mouse, rat and human islets.
Results
Glucagon output decreased three- to fivefold within minutes of shifting from low (3.5 mmol/l) to high (10 mmol/l) glucose (4.0 ± 0.5 pmol/15 min vs 1.3 ± 0.3 pmol/15 min, p < 0.05). The output was unaffected by inhibition of SGLT1/2 with dapagliflozin or phloridzin or by addition of the SGLT1/2 substrate α-methylglucopyranoside, whether at low or high glucose concentrations (p = 0.29–0.99). Insulin and somatostatin secretion (potential paracrine regulators) was also unaffected. Slc5a2 expression and SGLT2 protein were marginal or below detection limit in rat, mouse and human islets and in mouse and human alpha, beta and delta cells.
Conclusions/interpretation
Our combined data show that increased plasma glucagon during SGLT2 inhibitor treatment is unlikely to result from direct inhibition of SGLT2 in alpha cells, but instead may occur downstream of their blood glucose-lowering effects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.

Introduction
Sodium–glucose cotransporter (SGLT) 2 inhibitors represent the newest addition to the armamentarium for treatment of type 2 diabetes [1]. SGLT2 inhibitor treatment lowers blood glucose but at the same time is associated with increased hepatic glucose production and increased glucagon secretion [2,3,4], which appears to counteract the glucose-lowering effects. The mechanism whereby SGLT2 inhibitors stimulate glucagon secretion is not well understood but has, based on static incubations of isolated human islets, been proposed to involve a direct effect on the alpha cell [5]. Thus, the aim of this study was to test whether the effects of SGLT2 on glucagon secretion demonstrated in vitro could be reproduced in a more physiological setting. As glucagon secretion is tightly regulated by blood glucose, which is lowered by SGLT2 inhibitors, we chose the isolated perfused rat pancreas as experimental model [6, 7], where perfusate glucose is kept constant. We studied the effects of the SLGT2 inhibitor dapagliflozin, the SGLT1/2 inhibitor phloridzin and the metabolically inert SGLT-specific substrate methyl-α-d-glucopyranoside (α-MGP) on glucagon secretion in this model. We also measured the secretion of insulin and somatostatin, both known to powerfully influence glucagon secretion. In addition, we performed a thorough expression analysis of SGLT1 and SGLT2, as well as glucose transporters 1–5 for comparison, in mouse, rat and human islets and in alpha, beta and delta cells from mice and humans; the latter using two different human populations. Finally, we investigated SGLT2 protein expression by western blot and immunohistochemistry.
Methods
Ethical considerations
Animal studies in Copenhagen were approved by the Danish Animal Experiments Inspectorate (2013-15-2934-00833) and the local ethics committee and were performed in accordance with the guidelines of Danish legislation governing animal experimentation (1987) and the National Institutes of Health (publication number 85-23). Animal studies in Cambridge, UK (isolation of mouse pancreatic tissue) were approved by the University of Cambridge Animal Welfare and Ethical Review Body, conformed to the Animals (Scientific Procedures) Act 1986 Amendment Regulations (SI 2012/3039) and were performed under the UK Home Office Project License 70/7824.
Human islets were isolated by the European Consortium for Islet Transplantation (ECIT) under local approval in Milan, Italy, and were received in fully anonymous form. Human kidney cDNA was derived from kidneys surgically removed due to renal cell carcinoma. Kidney tissue was obtained from Sahlgrenska Hospital (Gothenburg, Sweden) in accordance with relevant ethical guidelines and patient consent. Human kidney and pancreas tissue used for immunohistochemistry were anonymous archival tissue.
Animals
Male Wistar rats were obtained from Janvier (Saint Berthevin Cedex, France) and kept at animal facilities at the Faculty of Health and Medical Sciences, University of Copenhagen. Rats were housed two per cage, had ad libitum access to standard chow and water and were kept on a 12 h light–dark cycle. Animals were acclimatised for at least 1 week.
Chickens (Bowans Brown hens) were also kept at the animal facilities at the Faculty of Health and Medical Sciences, University of Copenhagen and housed with ad libitum access to standard chow and drinking water for at least 2 weeks before experiments.
Mice for expression analysis were bred in-house at the University of Cambridge, UK (Central Biomedical Services [CBS]) and had been bred for at least 8 generations on a C57BL/6 background. Kidney and whole islets were isolated from female Glu-Cre-ERT2 × GCaMP3 mice (29 weeks old). For RNA sequencing analysis, delta cells were isolated from SstCre × EYFP mice (4 females, 2 males; 18–39 weeks old). Alpha and beta cells were isolated from Glu-Venus mice (2 females, 2 males; 22–29 weeks old). Further information about the strains has been detailed previously [8].
Isolated perfused rat pancreas procedure
Non-fasted rats were anaesthetised with Hypnorm/midazolam (0.3 ml/100 g body weight, per ml: Hypnorm: 0.08 mg fentanyl, 2.5 mg fluanisone, 0.45 mg methyl parahydroxybenzoate, 0.05 mg propyl parahydroxybenzoate; midazolam: 1.25 mg; Matrix Pharmaceuticals, Hellerup, Denmark) and the pancreas was isolated by removing the stomach, spleen and small and large intestine (except for the part of duodenum connected to pancreas) and by tying off the renal stalks. The abdominal aorta was tied off just below the diaphragm and, immediately, a catheter was inserted retrogradely into the abdominal aorta just distally to the renal arteries so that the aorta now exclusively supplied the pancreas (through both the coeliac and the superior mesenteric arteries). The pancreas was perfused by use of a UP100 Universal Perfusion System from Hugo Sachs (Harvard Apparatus, March-Hugstetten, Germany) (5.0 ml/min, 37°C) with a Krebs–Ringer bicarbonate buffer gassed with 95% O2, 5% CO2 [9]. A catheter was inserted into the portal vein for collection of venous effluent. After the start of the perfusion, the rat was killed by diaphragm perforation. The preparation was allowed to stabilise for 25 min and then 1 min samples were collected, transferred to ice and stored at −20°C. Glucose concentration was 3.5 and 10 mmol/l as indicated in Results. The following reagents were administered: the SGLT1/2-specific and metabolically inert substrate α-MGP (20 mmol/l); the SGLT1/2 inhibitor phloridzin dihydrate (100 μmol/l); the SGLT2 inhibitor dapagliflozin (0.5 μmol/l) and l-arginine (10 mmol/l, as positive control). Dapagliflozin was from Seleckchem.com (cat. no. S1548, Munich, Germany). The remaining reagents were from Sigma-Aldrich, Brøndby, Denmark (α-MGP; cat. no. M9376, phloridzin; 274313 and l-arginine; A92600). Pre-analysis inclusion criteria were quick insertion of catheter in aorta, stable perfusion output and perfusion pressure. Post-analysis inclusion criteria were stable hormone output before addition of test compounds as well as responsiveness to the positive control, l-arginine. Perfusion pressure and flow were closely monitored and remained stable during the perfusion.
Ussing chamber experiments
Experiments were carried out using fresh colonic tissue collected from Bowans Brown hens, which had been used for other purposes. Hens (n = 6) were killed by cervical dislocation and immediately after the colon was isolated, opened along the mesenteric border and cleaned by rinsing in cold Krebs–Ringer buffer. Tissue was placed in Krebs Ringer buffer (room temperature) and the mucosa was isolated by a careful scrape within 10 min after collection and placed in Ussing chambers (exposed mucosal area ~0 .6cm2) with oxygenated (95% O2 and 5% CO2) Krebs–Ringer buffer (37°C). The preparation was left for ~35 min to stabilise and then clamped at a potential difference of 0 mV from 10 min before the start until the end of the experiment. Short circuit currents (IScc), potential difference (mV) and resistance (Ω) were recorded every 2 s and shown in real-time by use of Clampdata software (Clamp version 2.14, Mussler Scientific Instruments, Aachen, Germany). Glucose, dapagliflozin or phloridzin was added to the luminal side of the mucosal layer (final concentration 100 mmol/l, 55 μmol/l and 100 μmol/l, respectively, corresponding to ~50 × IC50 on human SGLT1 [10] in case of dapagliflozin). l-Lysine (10 mmol/l) was included as a positive control. Inclusion criteria were stable resistance throughout the experiment, stable IScc before addition of test compounds, and responsiveness to both d-glucose and l-arginine.
Immunohistochemistry
Formalin-fixed tissue from rat, mouse and human kidney and pancreas (n = 3 for all tissue types) was embedded in paraffin. Five micrometre dewaxed tissue sections were pre-treated by boiling for 15 min in a microwave oven in EGTA buffer at pH 9 (antigen retrieval). Sections were then pre-incubated in PBS with 10% (wt/vol.) BSA for 10 min and incubated with the primary antibody (rabbit anti-human, -rat and -horse SGLT2 antibody; cat. no. LS-A2810; LSBio Seattle, Seattle, WA, USA) diluted 1:1000 in PBS with 10% (wt/vol.) BSA overnight at 4°C. Next day, the tissues were incubated with biotin goat anti-rabbit IgG 1:200 (Vector Labs, Burlingame, CA, USA), ABC (PK4000; Vector labs) and DAB, lightly counterstained with haematoxylin and coverslipped with Pertex mounting medium. In initial validation and optimisation experiments, the primary SGLT2 antibody was tested diluted 1:100, 1.1000 and 1:10000 times on human kidney tissue and with and without microwave antigen retrieval treatment both at pH 6 and 9. The optimal dilution was found to be 1:1000. The antibody yielded positive immunostaining in kidney only after microwave pretreatment at pH 9. The antibody was tested in different tissues, with human and rat kidney as the positive controls (known to be localised on apical membrane of proximal tubules). No SGLT2 immunoreactive stain was found in other parts of the kidney or in the exocrine pancreas (negative control).
The SGLT2 antibody used is a commercially available antibody and the immunisation fragment was not available for pre-absorption experiments. The sections were viewed under a Leitz orthoplan microscope (Wetzlar, Germany) and images were taken with an AxioCam IcC5 camera (Zeiss, Jena, Germany) mounted onto the microscope.
Western blot
Rat islets, human donor islets and rat kidney tissue were lysed on ice with NP-40 lysis buffer containing protease inhibitor cocktail (Life Technologies, Nærum, Denmark, cat. no. FNN0021) and stored at −20°C. Total protein concentration was measured by Bradford protein assay kit (cat. no. 5000001, Bio Rad Laboratories, Copenhagen, Denmark). Ten micrograms of protein lysate/sample was loaded onto gels, separated by 4–20% SDS-PAGE and blotted on a PDVF membrane. To avoid stripping membranes when staining for multiple proteins, membranes were cut according to the desired molecular mass range. Membranes were then stained separately with rabbit anti-human, -rat and -horse SGLT2 antibody (cat. no. LS-A2810-50, LSBio) or anti-α-tubulin antibody (loading control; cat. no. T6074; Sigma-Aldrich, Denmark), both diluted to a concentration of 0.5 μg/ml. Blots were developed with the chemiluminescence detection system using Super Signal (Life Technologies, cat no. 34075). Light emission was captured using an Alpha imager system (Alpha-Innotech, Broager, Denmark).
Gene expression procedures
Pancreatic islets from non-diabetic 1-week-old rats (n = 5) were isolated as described previously [11]. Rat and donated human islets (anthropometric data supplied in electronic supplementary material [ESM] Table 1) were cultured for 72 h at 37°C (95% O2, 5% CO2) in RPMI-glutamax medium supplemented with 10% (vol./vol.) FBS, 1% (vol./vol.) P/S (cat. no. P4333, Sigma-Aldrich, Denmark) and 5.6 mmol/l glucose. Total RNA was extracted by use of Nucleo-Spin kit (Macheray-Nagel, Bethlehem, PA, USA). Quality and quantity of the extracted RNA were assessed using a NanoDrop-1000 (Thermo Scientific). Five-hundred nanograms of total RNA were used for cDNA synthesis with the iScript-cDNA Kit (BioRad). Real-time reverse-transcriptase quantitative PCR (RT-qPCR) was performed on 12 ng cDNA with SYBR Green PCR mastermix (Life Technologies, cat. no. 4309155) using specific primers (ESM Table 2) and run in a real-time PCR machine (Applied Biosystems, Naerum, Denmark). Gene expression levels were normalised to the housekeeping mRNA Hprt1 (encoding hypoxanthine-guanine phosphoribosyltransferase-1) through the −∆Ct method. Methods are described in detail elsewhere [12]. As positive control, kidney tissue from rats and human donors were subjected to the same procedure.
For whole islet studies, mouse islets were isolated as described previously [8] and RNA from homogenised tissues was extracted using TRI reagent (Sigma-Aldrich, Gillingham, UK) or RNeasy Plus Micro Kit (Qiagen, Manchester, UK). Genomic DNA was removed via treatment with DNAse1 (Thermo Fisher Scientific, Winsford, UK) or through the RNeasy Plus gDNA eliminator spin columns. Purified RNA was reverse transcribed using Superscript II kit (Thermo Fisher Scientific). RT-qPCR was performed with a QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems). The PCR reaction mix consisted of first-strand cDNA template, 6-carboxyfluorescein/quencher probe/primer mixes (Thermo Fisher Scientific) and PCR Master Mix (Thermo Fisher Scientific) and was amplified for 40 cycles. Expression of selected targets was normalised to that of the housekeeping mRNA Actb, and expression levels calculated as above. Primer information is provided in ESM Table 2. RNA sequencing was performed as described previously [8] on FACS separated transgenic mice expressing fluorescent marker proteins in either the delta cells (Sst-EYFP strain) or the alpha cells (Glu-Venus strain). Beta cells were isolated based on their size, using forward and side scatter characteristics distinguishing them from other nonfluorescent islet cells in Glu-Venus mice. Methods are described in detail elsewhere [8].
Single-cell RNA sequencing of human pancreatic islet cells
Single-cell expression levels of SGLT1 (also known as SLC5A1), SGLT2 (SLC5A2) and GLUT1–5 (SLC2A1–5) in human alpha, beta and delta cells were determined by RNA-sequencing-based analysis by two different independent groups of investigators, using their own methodology and populations.
Population 1
In population 1, expression was determined by reanalysing published human islet single-cell sequencing data, from donors with type 2 diabetes and non-diabetic donors (n = 4 with diabetes, n = 6 without diabetes) [13]. FastQ files were downloaded from ArrayExpress (accession E-MTAB-5061). Data were analysed with bcbio-nextgen (https://github.com/chapmanb/bcbio-nextgen), using the hisat2 algorithm [14] to align reads to human genome version hg38 and the Salmon algorithm [15] for quantification of gene counts. Only cells that passed the quality control in the original study [13] were maintained for further analysis: non-diabetic donors/diabetic donors: alpha cells 443/443; beta cells 171/99 and delta cells 59/55. Cell type classification from the original study was maintained. Log2 transformed counts per million (CPM) were plotted for individual cells.
Population 2
Publicly available RNA sequencing data from two different population sets (GSE85241, GSE81608) [16, 17] were used for this analysis. Only non-diabetic individuals were included in the analyses, with a combined sample size of 26. Reads per kilobase million (RPKM) values for SGLT1, SGLT2 and GLUT1–5 from the respective data bases were uploaded to the Jupyter Notebook (http://jupyter.org/, accessed 1207–2018). Data were then log2 transformed, and means were calculated for SGLT1, SGLT2 and GLUT1–5 in alpha, beta and delta cells, respectively. Further details about the donors, isolation of cells and RNA sequencing methods can be found in the original studies [16, 17].
Biochemical measurements
The in-house glucagon RIA employed a C-terminal directed antibody (code no. 4305 [18]). Insulin and somatostatin (total: SST-14 and SST-28) concentrations were determined by in-house RIA (code no. 2006-3 and 1758-5 [19, 20]). Sensitivities of the assays were around 1 pmol/l, allowing detection of secretory outputs as low as 5 fmol/min.
Data presentation and statistical analysis
Data are presented as means ± SEM. For perfusion data, averaged basal and response outputs were calculated by taking an average of output values over the entire stimulus administration (response, 15 min) and the period leading immediately up to stimulus administration (15 min, baseline). Output (fmol/min) was the product of peptide concentration (pmol/l) and flow rate (ml/min). Averaged IScc at baseline, and after glucose and dapagliflozin, was calculated over at least 50 consecutive recordings within the terminal part of the respective periods. Statistical significance was assessed by one-way ANOVA for repeated measurements followed by Bonferroni multiple comparison test using GraphPad Prism software (version 6, La Jolla, CA, USA). Graphs were constructed in GraphPad and edited in Adobe Illustrator (San Jose, CA). p < 0.05 was considered significant.
Results
Glucagon secretion
Glucagon secretion was tightly regulated by the glucose concentration in the perfusion buffer, decreasing to 20–30% within minutes of changing the concentration from 3.5 mmol/l to 10 mmol/l (p < 0.05); dapagliflozin had no effect regardless of glucose concentration (15 min output at 3.5 mmol/l glucose: baseline 4.0 ± 0.5 pmol vs dapagliflozin 3.6 ± 0.4 pmol, p = 0.53; at 10 mmol/l glucose: baseline 1.3 ± 0.3 pmol vs dapagliflozin 1.4 ± 0.4 pmol, p = 0.95, n = 6; Fig. 1a–c). Changing the order of glucose concentration, starting with 10 mmol/l and shifting to 3.5 mmol/l, gave similar results—no effects of dapagliflozin on glucagon secretion (data shown in ESM Fig. 1). Phloridzin also had no effect on glucagon secretion at 3.5 or 10 mmol/l glucose (Fig. 1d–f). Addition of the SGLT-specific substrate α-MGP (20 mmol/l) had no significant effect on glucagon secretion at 3.5 or 10 mmol/l glucose (15 min output at 3.5 mmol/l glucose: baseline 2.9 ± 0.5 pmol vs α-MGP 2.6 ± 0.5 pmol, p = 0.48 and at 10 mmol/l glucose: baseline 0.5 ± 0.01 pmol vs α-MGP 0.5 ± 0.1 pmol, p = 0.99, n = 6; Fig. 1g–i). In all experiments, glucagon output was significantly lower at high glucose, and l-arginine (10 mmol/l, positive control) markedly stimulated secretion (Fig. 1a, d, g).

Effects of SGLT activity on glucagon secretion in perfused pancreases isolated from male Wistar rats. (a) Glucagon output at 3.5 and 10 mmol/l glucose in the absence or presence of dapagliflozin. (b) Averaged total glucagon output at 3.5 and 10 mmol/l glucose exposure during dapagliflozin administration (15 min) or at baseline (15 min immediately before stimulation). (c) Glucagon outputs from individual rat pancreases. (d, e, f) As for (a, b, c) respectively, but with administration of phloridzin (an SGLT1/2 inhibitor) rather than dapagliflozin. (g, h, i) As for (a, b, c) respectively, but with administration of α-MGP (an SGLT-specific substrate) rather than dapagliflozin. l-Arginine, a known glucagon secretagogue, was added at the end of all experiments to control for responsiveness (positive control). Data are shown as mean (SEM); n = 6 for all groups. In (b, c, e, f, h, i) outputs from separate experiments are shown as dots. *p < 0.05 (one-way ANOVA for repeated measurements followed by Bonferroni post hoc test). At 3.5 mmol/l glucose: p = 0.53 for baseline vs dapagliflozin (a–c); p = 0.29 for baseline vs phloridzin (d–f); and p = 0.48 for baseline vs α-MGP (g–i). At 10 mmol/l glucose: p = 0.95 for baseline vs dapagliflozin (a–c); p = 0.71 for baseline vs phloridzin (d–f); and p = 0.99 for baseline vs α-MGP (g–i). BL, baseline; Dapa, dapagliflozin; Glu., glucose; Phlor, phloridzin
Insulin and somatostatin secretion
Insulin and somatostatin secretion increased after shifting the glucose concentration in the perfusion buffer from 3.5 mmol/l to 10 mmol/l. The presence of neither dapagliflozin nor phloridzin affected the secretion (p values for insulin/somatostatin in response to test substance compared with baseline: for dapagliflozin p = 0.99/0.36 at 3.5 mmol/l glucose and p = 0.69/0.29 at 10 mmol/l glucose; for phloridzin p = 0.85/0.64 at 3.5 mmol/l glucose and p = 0.43/0.98 at 10 mmol/l glucose, n = 6 in all cases) (Fig. 2a, b and d, e). Infusions of α-MGP were similarly ineffective (p values for insulin/somatostatin compared with baseline: p = 0.73/0.65 at 3.5 mmol/l glucose and p = 0.53/0.42 at 10 mmol/l glucose, n = 6) (Fig. 2b, e).

Effects of SGLT1/2 activity on insulin and somatostatin secretion in perfused pancreases isolated from male Wistar rats. (a–c) Insulin output at 3.5 and 10 mmol/l glucose, as indicated, and in the absence or presence of dapagliflozin (a), phloridzin (b) or α-MGP (c). (d–f) Somatostatin output at 3.5 and 10 mmol/l glucose, as indicated, in the absence or presence of dapagliflozin (d), phloridzin (e) or α-MGP (f). l-Arginine, a known insulin and somatostatin secretagogue, was added at the end of all experiments to control for responsiveness (positive control). Data are shown as mean (SEM); n = 6. Insulin secretion at 3.5 mmol/l glucose: p = 0.99 for baseline vs dapagliflozin (a); p = 0.85 for baseline vs phloridzin (b) and p = 0.72 for baseline vs α-MGP (c). Insulin secretion at 10 mmol/l glucose: p = 0.69 for baseline vs dapagliflozin (a); p = 0.64 for baseline vs phloridzin (b); and p = 0.53 for baseline vs α-MGP (c). Somatostatin secretion at 3.5 mmol/ glucose: p = 0.36 for baseline vs dapagliflozin (d); p = 0.43 for baseline vs phloridzin (e); and p = 0.65 for baseline vs α-MGP (f). Somatostatin secretion at 10 mmol/l glucose: p = 0.29 for baseline vs dapagliflozin (d); p = 0.98 for baseline vs phloridzin (e); and p = 0.42 for baseline vs α-MGP (f). Glu., glucose
Effects of dapagliflozin on glucose-sensitive currents
The bioactivity of the dapagliflozin and phloridzin used on SGLT1-mediated glucose absorption was investigated using the Ussing chamber technique, by recording the SGLT1-sensitive IScc in real time. Glucose administration (100 mmol/l) to the luminal side of the chicken mucosa doubled the IScc (from ~40 μA/cm2), but this response was abolished by the SGLT1/2 inhibitor phloridzin (100 μmol/l, positive control) (Fig. 3a, b). Importantly, dapagliflozin also eliminated the glucose-sensitive current when applied in a concentration well above (55 μmol/l) the IC50 on human SGLT1 (1.3 μmol/l [10]) (Fig. 3c, d).

Effects of dapagliflozin on colonic glucose-sensitive currents in chicken mucosa (reflecting SGLT1 activity). (a, c) Representative experimental traces at baseline or in response to luminal glucose, phloridzin (a, positive control) or dapagliflozin (c), followed by luminal l-Lys to control for responsiveness (positive control). (b, d) Collected responses (mean [SEM]) to the stimuli shown in (a) and (c), respectively. n = 1 (a, c) or n = 4 (b, d). *p < 0.05 (one-way ANOVA for repeated measurements followed by Bonferroni post hoc test). Lum., luminal
Expression of glucose transporters in rat, human and mouse islets
Expression of Slc5a2 (the gene encoding SGLT2) was investigated in rat as well as in human and mouse pancreatic islets to test for potential species differences. In all tested species, expression was below the detection limit (Ct = 35). In apparent contradiction to these results, high SLC5A2 expression levels in human islets were detected in our initial analysis (ESM Fig. 2), using primers annealing to the N-terminal part of the gene. However, we later discovered that this expression most likely reflects amplification of two expressed unrelated genes (AC026471.6 and C16orf58) that overlap with the gene encoding SGLT2 and are expressed in islets (ESM Fig. 3). Expression levels of the gene encoding SGLT1 (Slc5a1) were low in mouse, rat and human islets in comparison with levels of the gene encoding GLUT1 (Slc2a1) and in particular GLUT2 (Slc2a2) but were still detectable (Fig. 4a–d). Consistently, Sglt2 expression, as well as Sglt3a (encoded by Slc5a4a) and Sglt3b (encoded by Slc5a4b), was below detection limit in sorted alpha, beta and delta islet cells from the mouse, whereas genes encoding GLUT1 and GLUT2 were expressed at varying levels depending on the cell type (Fig. 4c). In the mouse, low levels of Sglt1 were detected in beta and delta cells, but not in alpha cells. As positive control, we used the same respective primer sets to amplify expression of SGLT1 and SGLT2 genes in mouse, rat and human kidney tissue. In all cases, high expression was detected (Fig. 4a, b, d).

Expression of glucose transporters in rat, human and mouse pancreatic islets. Relative expression levels of SGLT1, SGLT2, SGLT3a, SGLT3b and GLUT1–5 in rat, human and mouse islets are presented, as indicated. (a) Rat islets and rat kidney tissue (positive control). (b) Human islets and human kidney (positive control). (c) Fractionated expression in alpha, beta and delta cells from mice. (d) Mouse islets and mouse kidney (positive control). Expression was assessed by qPCR (a, b, d) or by RNA sequencing (c). Data are presented as mean (SEM); n = 5 (a, islets), n = 3 (a, kidney); n = 6 (b, islets), n = 3 (b, kidney); n = 3 (c, d). Horizontal dotted lines show a break in the y-axis
In addition to the qPCR-based expression analysis on human islets, we investigated the expression of glucose transporters in dispersed islet cells, using publicly available RNA sequencing data. Population 1 consisted of tissue from both non-diabetic donors and donors with type 2 diabetes, whereas population 2 only included tissue from non-diabetic donors. In both populations, SGLT2 expression was practically absent in all alpha, beta and delta cells and its expression was not altered in cells from donors with type 2 diabetes (Fig. 5a, b). SGLT1 was expressed in alpha cells in both human populations and its expression was upregulated approximately 1.5-fold in donors with vs without type 2 diabetes (p < 0.001, Fig. 5a). GLUT1 was highly expressed in all three cell types in both populations, whereas GLUT2 and GLUT3 were mostly expressed by beta cells (Fig. 5a, b). Expression of GLUT1–5 was not statistically different between donors with vs without type 2 diabetes. Consistent with the low expression level, SGLT2 protein was not detected in rat and human islet lysates or in pancreatic islets from mice, rats or humans using immunohistochemistry as detection method. SGLT2 was, however, detected in kidney tissue (positive control) by both western blot and immunohistochemistry (Figs. 5c and 6; uncropped western blots are shown in ESM Fig. 4).

Expression of glucose transporters in human alpha, beta and delta cells and SGLT2 protein in rat and human islets. (a) Single-cell expression of genes encoding SGLT1, SGLT2 and GLUT1–5 in human alpha, beta and delta cells from both non-diabetic donors and donors with type 2 diabetes (population 1). (b) Combined single-cell expression of genes encoding SGLT1, SGLT2 and GLUT1–5 from two different populations, all non-diabetic donors (population 2). Gene expression was investigated by RNA sequencing. (c) Protein level of SGLT2 in protein lysates from human and rat islets and rat kidney tissue lysates (positive control). After blotting, the membrane was cut as indicated by the dashed line and protein stainings were performed separately. Uncropped data are provided in ESM Fig. 4. Data are presented as mean (SEM); n = 10 (a, 4 donors that had T2D, 6 that did not), n = 26 (b; all non-diabetic donors), n = 3 (c, rat kidney, rat islets) or n = 5 (c, human islets). Dots in (a) show expression in respective single cells (non-diabetic donors/diabetic donors: alpha cells 443/443; beta cells 171/99 and delta cells 59/55). For SGLT1 and 2 number of positive cells (RPKM >0) are indicated above respective bars, as a fraction of the total. ***p < 0.001 (one-way ANOVA followed by Bonferroni post hoc test). ND, non-diabetic; T2D, type 2 diabetic; NC, no sample control

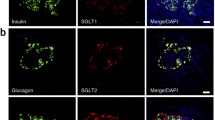
Representative images for immunohistochemical staining against SGLT2 (brown color) in rat, mouse and human pancreatic islets (islets are indicated by arrows) and kidney (positive control). Representative images are shown from n = 3 in all cases. Scale bar, 100 μm (applies to all images)
Discussion
The use of SGLT2 inhibitors is associated with increased plasma glucagon concentrations. This may limit their therapeutic effectiveness as glucagon is a powerful stimulus for hepatic glucose production, which increases during SGLT2 inhibitor therapy [2, 3, 21]. Based on studies performed on human islets or the glucagon-secreting cell line α-TC1, it was suggested that the hyperglucagonaemia might be a direct effect of the inhibitors on the pancreatic alpha cell [5, 22], leading to reduced glucose uptake by the alpha cell. Indeed, a mathematical model based on the study by Bonner et al [5] supported the notion that SGLT2 activity in the alpha cell leads to small depolarisations and subsequent inactivation of voltage-gated ion channels, resulting in reduced glucagon secretion [23]. In continuation hereof, attenuation of SGLT2 activity by SGLT2 inhibitors was predicted to partly relieve the suppressive actions of glucose on glucagon secretion [23]. However, this model assumes that SGLT2 is expressed by the alpha cell and contrasts to the findings in our study.
We used the physiologically relevant isolated perfused rat pancreas model [7] and found that SGLT2 activity had no effect on glucagon secretion whether transporter activity was inhibited by dapagliflozin or phloridzin or whether it was activated by the SGLT-specific substrate α-MGP. Of note, the dapagliflozin concentration used (0.5 μmol/l) matches peak plasma concentration in humans after intake of therapeutic doses (5–10 mg/day) [24] and, in all experiments, secretion was tightly regulated by the glucose concentration so glucagon secretion was high at low glucose. We have shown previously that α-MGP-mediated activation of SGLT1 in the perfused rat small intestine stimulates glucagon-like peptide-1 secretion [25], supporting the use of α-MGP as a suitable experimental tool. Besides being directly regulated by glucose, glucagon secretion is also thought to be regulated by paracrine effects of insulin and/or somatostatin [26,27,28]. However, we found that neither insulin nor somatostatin secretion was affected by acute dapagliflozin, phloridzin or α-MGP administration. Our data, therefore, do not support a recent report linking intra-islet cross talk to the activation of SGLT2 in delta cells downstream of insulin [29], although it is possible that SGLT2 recruitment requires the higher insulin concentrations used in static islet incubation studies, or exhibits species differences.
Our results are also in contrast to the findings of Bonner et al and Solini et al [5, 22]. However, although these two studies both report a stimulatory effect on glucagon secretion, they are inconsistent with respect to the suggested mechanisms of action. Thus, one [5] showed expression of SGLT2 in human islets and concluded that the stimulatory effect of dapagliflozin on glucagon secretion was attributable to a direct effect of SGLT2 inhibition at the level of the alpha cell, whereas in the other [22] SGLT2 expression was nearly undetectable in human pancreatic islets, mouse islets and the α-TC1 cell line. Instead, the authors found that dapagliflozin acutely resulted in upregulation of SGLT1 expression, which they concluded leads to increased glucose-stimulated glucagon secretion through an SGLT1-dependent mechanism [22]. Similar conclusions have been reached by another group [30]. The inconsistency between our study and the two previous studies is difficult to explain, but the lack of responses to dapagliflozin is unlikely to be dose-related, as the concentration we used was almost 500 times higher than the IC50 for SGLT2. The inconsistency is also unlikely to reflect inactivity of the dapagliflozin or phloridzin used in these in vitro experiments, since both compounds eliminated glucose-sensitive currents across intestinal mucosal preparations in Ussing chambers. Instead, the inconsistencies between our studies may be related to differences between the use of the intact perfused pancreas and static incubations of isolated islets from which secreted peptides and co-secreted molecules (both metabolites and active molecules like somatostatin) accumulate in the medium and may influence further secretion via feedback, whereas with perfusion secreted molecules are instantly removed, mimicking the situation in vivo. Supporting this, in a study with perifusion of mouse islets, both insulin and glucagon secretion was similar between global SGLT2 knockdown mice and wild-type littermates [31]. In another study, employing an experimental setup similar to ours, dapagliflozin suppressed glucagon secretion from rat pancreases perfused with 2.5 mmol/l glucose. However, these studies were carried out in rat models of type 1 diabetes, and the relevance of this observation for normal islets or islets in type 2 diabetes warrants further investigation [32]. To investigate whether the inconsistencies between the two studies reporting acute stimulatory effects on glucagon secretion [5, 22] and our present findings could result from inter-species differences, we investigated SGLT1 and SGLT2 expression in rat, mouse and human islets, as well as in mouse and human alpha, beta and delta cells. For comparison, expression of GLUT1–5 was also investigated. In rat, human and mouse islets, SGLT2 expression was below the detection limit and so was its expression in fractionated mouse alpha, beta and delta cells. In human islet cells from both non-diabetic and diabetic donors, expression was below the detection limit in >98% of the investigated alpha cells, and expression levels were low in the few positive cells. Moreover, SGLT2 protein was not detected by western blot or immunohistochemistry in rat, mouse and human pancreatic extracts/islets, while clear signals were evident in the control tissue (kidney). Our results agree with those of Solini et al [22], showing no detectable SLGT2 in mouse and human islets, but disagree with those of Bonner et al [5]. However, in the latter study, expression levels were only provided as relative levels (comparing levels in different groups of individuals), making it impossible to assess whether expression was high or close to the detection limit. Furthermore, the data cannot easily be reproduced as the primer sets employed are not listed and comparisons with the primary glucose transporters GLUT1 and GLUT2 are also lacking, again making the interpretation of these results difficult.
In addition, the signal detected by Bonner et al may reflect non-protein-coding SGLT2 transcripts as there are two additional overlapping genes (AC026471.6 and C16orf58) at the SGLT2 locus that may contribute to the signal, depending on the annealing sites of the primers. Indeed, we found C16orf58 to be highly expressed in all islet cell types (ESM Fig. 3). Furthermore, there may be expression of several non-protein-coding SGLT2 transcript variants. Indeed, our RNA-sequencing-based analysis (on population 1) identified seven putatively expressed SGLT2 transcript variants in islet cells (based on ENSEMBL build hg38). Of these, only one codes for full-length protein-coding SGLT2, and its expression level was generally low or undetectable in all cells. In an initial round of qPCR in human islets, we detected very high expression levels of SGLT2 (ESM Fig. 2) but later found by sequence analysis that the first primer set annealed to a sequence overlapping with eight different SGLT2 transcript variants that did not code for full-length protein-coding SGLT2 protein. Consistently, subsequent sequencing of the amplicon confirmed that irrelevant transcripts of non-coding SGLT2 variants had been amplified. Our results suggest that the lack of SGLT2 activity modulation on glucagon secretion measured using the isolated perfused rat pancreas is unlikely to be a species-dependent phenomenon. Rather, the increased circulating glucagon levels observed in patients treated with SGLT2 inhibitors may result from the glucose-lowering effect of the drugs, which in itself would be expected to increase glucagon secretion. In support of this, dapagliflozin did not increase plasma glucagon concentrations measurably in mice, rats or humans under clamped isoglycaemic conditions [30, 32, 33], although in another study in humans, dapagliflozin, but not placebo, increased plasma glucagon concentrations [2]. At any rate, the clinically relevant question is whether endogenous glucose production (EGP) caused by SGLT2 inhibitors is mediated downstream of increased glucagon secretion or by other mechanisms. In this context, it should be considered that the reported increases in plasma glucagon are marginal (0.5–1.5 pmol/l) [2, 3, 21] and perhaps too small to cause a quantitatively relevant increase in EGP. In support of this, a recent study in humans concluded that increased EGP after SGLT2 inhibition resulted from the glucose-lowering actions rather than from direct effects on glucagon and insulin secretion [33].
Data availability
Data are available upon request to Jens J. Holst.
Abbreviations
- CPM:
-
Counts per million
- EGP:
-
Endogenous glucose production
- IScc :
-
Short circuit current
- α-MGP:
-
Methyl-α-d-glucopyranoside
- RPKM:
-
Reads per kilobase million
- SGLT:
-
Sodium–glucose cotransporter
References
Cuypers J, Mathieu C, Benhalima K (2013) SGLT2-inhibitors: a novel class for the treatment of type 2 diabetes introduction of SGLT2-inhibitors in clinical practice. Acta Clin Belg 68(4):287–293. https://doi.org/10.2143/ACB.3349
Merovci A, Solis-Herrera C, Daniele G et al (2014) Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest 124(2):509–514. https://doi.org/10.1172/JCI70704
Ferrannini E, Muscelli E, Frascerra S et al (2014) Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest 124(2):499–508. https://doi.org/10.1172/JCI72227
Martinez R, Al-Jobori H, Ali AM et al (2018) Endogenous glucose production and hormonal changes in response to canagliflozin and liraglutide combination therapy. Diabetes 67(6):1182–1189. https://doi.org/10.2337/db17-1278
Bonner C, Kerr-Conte J, Gmyr V et al (2015) Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat Med 21(5):512–517. https://doi.org/10.1038/nm.3828
Kuhre RE, Wewer Albrechtsen NJ, Larsen O et al (2018) Bile acids are important direct and indirect regulators of the secretion of appetite- and metabolism-regulating hormones from the gut and pancreas. Mol Metab 11:84–95. https://doi.org/10.1016/j.molmet.2018.03.007
Christiansen CB, Svendsen B, Holst JJ (2015) The VGF-derived neuropeptide TLQP-21 shows no impact on hormone secretion in the isolated perfused rat pancreas. Horm Metab Res 47(7):537–543. https://doi.org/10.1055/s-0034-1395615
Adriaenssens AE, Svendsen B, Lam BY et al (2016) Transcriptomic profiling of pancreatic alpha, beta and delta cell populations identifies delta cells as a principal target for ghrelin in mouse islets. Diabetologia 59(10):2156–2165. https://doi.org/10.1007/s00125-016-4033-1
Kuhre RE, Frost CR, Svendsen B, Holst JJ (2014) Molecular mechanisms of glucose-stimulated GLP-1 secretion from perfused rat small intestine. Diabetes 64:370
Meng W, Ellsworth BA, Nirschl AA et al (2008) Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. J Med Chem 51(5):1145–1149. https://doi.org/10.1021/jm701272q
Carter JD, Dula SB, Corbin KL, Wu R, Nunemaker CS (2009) A practical guide to rodent islet isolation and assessment. Biol Proced Online 11(1):3–31. https://doi.org/10.1007/s12575-009-9021-0
Ghiasi SM, Krogh N, Tyrberg B, Mandrup-Poulsen T (2018) The no-go and nonsense-mediated RNA decay pathways are regulated by inflammatory cytokines in insulin-producing cells and human islets and determine β-cell insulin biosynthesis and survival. Diabetes 67(10):2019–2037. https://doi.org/10.2337/db18-0073
Segerstolpe A, Palasantza A, Eliasson P et al (2016) Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab 24(4):593–607. https://doi.org/10.1016/j.cmet.2016.08.020
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12(4):357–360. https://doi.org/10.1038/nmeth.3317
Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C (2017) Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14(4):417–419. https://doi.org/10.1038/nmeth.4197
Xin Y, Kim J, Okamoto H et al (2016) RNA sequencing of single human islet cells reveals type 2 diabetes genes. Cell Metab 24(4):608–615. https://doi.org/10.1016/j.cmet.2016.08.018
Muraro MJ, Dharmadhikari G, Grun D et al (2016) A single-cell transcriptome atlas of the human pancreas. Cell Syst 3:385–394.e383
Orskov C, Jeppesen J, Madsbad S, Holst JJ (1991) Proglucagon products in plasma of noninsulin-dependent diabetics and nondiabetic controls in the fasting state and after oral glucose and intravenous arginine. J Clin Invest 87(2):415–423. https://doi.org/10.1172/JCI115012
Brand CL, Jorgensen PN, Knigge U et al (1995) Role of glucagon in maintenance of euglycemia in fed and fasted rats. Am J Phys 269:E469–E477
Holst JJ, Bersani M (1991) Assays for peptide products of somatostatin gene expression. Methods Neurosci 5:3–22
Martinez F, Rizza R, Romero J (1994) High-fructose feeding elicits insulin resistance, hyperinsulinism, and hypertension in normal mongrel dogs. Hypertension 23(4):456–463. https://doi.org/10.1161/01.HYP.23.4.456
Solini A, Sebastiani G, Nigi L, Santini E, Rossi C, Dotta F (2017) Dapagliflozin modulates glucagon secretion in an SGLT2-independent manner in murine alpha cells. Diabetes Metab 43(6):512–520. https://doi.org/10.1016/j.diabet.2017.04.002
Pedersen MG, Ahlstedt I, El Hachmane MF, Gopel SO (2016) Dapagliflozin stimulates glucagon secretion at high glucose: experiments and mathematical simulations of human A-cells. Sci Rep 6(1):31214. https://doi.org/10.1038/srep31214
Kasichayanula S, Liu X, Lacreta F, Griffen SC, Boulton DW (2014) Clinical pharmacokinetics and pharmacodynamics of dapagliflozin, a selective inhibitor of sodium-glucose co-transporter type 2. Clin Pharmacokinet 53(1):17–27. https://doi.org/10.1007/s40262-013-0104-3
Kuhre RE, Bechmann LE, Wewer Albrechtsen NJ, Hartmann B, Holst JJ (2015) Glucose stimulates neurotensin secretion from the rat small intestine by mechanisms involving SGLT1 and GLUT2, leading to cell depolarization and calcium influx. Am J Phys Endocrinol Metab 308(12):E1123–E1130. https://doi.org/10.1152/ajpendo.00012.2015
de Heer J, Rasmussen C, Coy DH, Holst JJ (2008) Glucagon-like peptide-1, but not glucose-dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas. Diabetologia 51(12):2263–2270. https://doi.org/10.1007/s00125-008-1149-y
Cooperberg BA, Cryer PE (2010) Insulin reciprocally regulates glucagon secretion in humans. Diabetes 59(11):2936–2940. https://doi.org/10.2337/db10-0728
Cejvan K, Coy DH, Efendic S (2003) Intra-islet somatostatin regulates glucagon release via type 2 somatostatin receptors in rats. Diabetes 52(5):1176–1181. https://doi.org/10.2337/diabetes.52.5.1176
Vergari E, Knudsen JG, Ramracheya R et al (2019) Insulin inhibits glucagon release by SGLT2-induced stimulation of somatostatin secretion. Nat Commun 10(1):139. https://doi.org/10.1038/s41467-018-08193-8
Suga T, Kikuchi O, Kobayashi M et al (2018) SGLT1 in pancreatic alpha cells regulates glucagon secretion in mice, possibly explaining the distinct effects of SGLT2 inhibitors on plasma glucagon levels. Mol Metab 19:1–12
Jurczak MJ, Lee HY, Birkenfeld AL et al (2011) SGLT2 deletion improves glucose homeostasis and preserves pancreatic β-cell function. Diabetes 60(3):890–898. https://doi.org/10.2337/db10-1328
Wang MY, Yu X, Lee Y et al (2017) Dapagliflozin suppresses glucagon signaling in rodent models of diabetes. Proc Natl Acad Sci U S A 114(25):6611–6616. https://doi.org/10.1073/pnas.1705845114
Lundkvist P, Pereira MJ, Kamble PG et al (2019) Glucagon levels during short-term SGLT2 inhibition are largely regulated by glucose changes in patients with type 2 diabetes. J Clin Endocrinol Metab 104(1):193–201. https://doi.org/10.1210/jc.2018-00969
Acknowledgements
We thank M. Lal (AstraZeneca, Gothenburg, Sweden) for providing the human kidney tissue.
Funding
REK was supported by a postdoctoral scholarship from the Lundbeck foundation (Lundbeckfonden, R264-2017-3492). LC was supported by a postdoctoral scholarship from the AstraZeneca postdoc programme. The isolated perfused rat pancreas studies, immunohistochemistry, qPCR on human and rat islets and western blotting were supported by a grant from Lundbeck foundation to REK (Lundbeckfonden, R289-2018-1026) an unrestricted grant to JJH from the Novo Nordisk Center for Basic Metabolic Research (Novo Nordisk Foundation, Denmark), a separate grant from the Novo Nordisk Foundation (grant no. NNF15OC0016574), a grant to JJH from the European Research Council (grant no. 695069) and the European Union’s Seventh Framework Programme for Research, Technological Development, and Demonstration Activities (grant No. 266408). Gene expression analysis on mouse tissue was supported by grants to FR and FMG from Wellcome (106262/Z/14/Z and 106263/Z/14/Z) and the MRC (MRC_MC_UU_12012/3). The study sponsors had no involvement in the conception of study, study design, interpretation of data or writing of the manuscript.
Author information
Authors and Affiliations
Contributions
REK and JJH conceived the study. REK and DBA designed perfusion experiments and performed isolated perfused rat pancreas experiments and assayed for glucagon, insulin and somatostatin concentrations. REK designed and performed Ussing chamber experiments and constructed figures. REK, DBA and JJH interpreted pancreas perfusion data. SMG and TM-P designed and performed qPCR expression and western blot analyses on rat and human islets/kidney and produced and interpreted data. AA and BT carried out the glucose transporter analysis on sorted human alpha, beta and delta cells from population 1. LC performed the qPCR analyses on human kidney. NW provided intellectual content and interpreted data. NJWA did the expression analysis of glucose transporters, population 2. CØ designed, performed and interpreted the immunohistochemistry on mouse, rat and human pancreas and kidney. AEA, FMG and FR did all work related to the expression of SGLT2 and other glucose transporters in mouse pancreatic cells and produced and interpreted data. REK drafted the manuscript. REK, SMG, AEA, DBA, LC, AA, NJWA, CØ, TM-P, FMG, FR, NW, BT and JJH critically revised and edited the manuscript. All authors approved the final version of the manuscript. JJH is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding author
Ethics declarations
AA, LC and BT are employed by AstraZeneca. AstraZeneca had no involvement in the conception of study, design and execution of experiments, interpretation of data or writing of the manuscript. All other authors declare that there is no duality of interest associated with their contribution to this manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM
(PDF 3947 kb)
Rights and permissions
About this article

Cite this article
Kuhre, R.E., Ghiasi, S.M., Adriaenssens, A.E. et al. No direct effect of SGLT2 activity on glucagon secretion. Diabetologia 62, 1011–1023 (2019). https://doi.org/10.1007/s00125-019-4849-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-019-4849-6