
Abstract
Necroptosis was initially identified as a backup cell death program when apoptosis is blocked. However, it is now recognized as a cellular defense mechanism against infections and is presumed to be a detrimental factor in several pathologies driven by cell death. Necroptosis is a prototypic form of regulated necrosis that depends on activation of the necrosome, which is a protein complex in which receptor interacting protein kinase (RIPK) 3 is activated. The RIP homotypic interaction motif (RHIM) is the core domain that regulates activation of the necrosome. To date, three RHIM-containing proteins have been reported to activate the kinase activity of RIPK3 within the necrosome: RIPK1, Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF), and DNA-dependent activator of interferon regulatory factors (DAI). Here, we review and discuss commonalities and differences of the increasing number of activators of the necrosome. Since the discovery that activation of mixed lineage kinase domain-like (MLKL) by RIPK3 kinase activity is crucial in necroptosis, interest has increased in monitoring and therapeutically targeting their activation. The availability of new phospho-specific antibodies, pharmacologic inhibitors, and transgenic models will allow us to further document the role of necroptosis in degenerative, inflammatory and infectious diseases.
Similar content being viewed by others

Introduction
Rudolf Virchow (1821–1902, Prussia), founder of the cell theory (Omnis cellula e cellula) and cellular pathology, referred to tissue injury as “parenchymatous inflammation” and introduced the idea that tissue injury is caused by pathological changes within the cells. In 1858, he introduced the notion of cell death as a potential basis for pathology, with ‘necrobiosis’ being a physiological process of spontaneous wearing out of living parts from the body and ‘necrosis’ an accidental process. The term ‘necrosis’ comes from the Greek word ‘nekros,’ which means ‘dead body.’ Virchow’s necrobiosis–necrosis dichotomy resembles to some extent the current apoptosis–necrosis classification [1]. Together with cellular and molecular insights into inflammation came a shift in our understanding of the molecular interplay between cell death and inflammation at the site of tissue injury. This emerging field of research is crucial for understanding organismal homeostasis and how its processes contribute to a growing list of inflammatory and degenerative pathologies. Although cell death during inflammation was initially considered a manifestation of tissue damage, it was later recognized as a mechanism for eliminating pathogens and regulating inflammation by exposing or releasing molecular patterns that attract and alter the functions of other cells [2]. More recently, it became clear that phagocytosis of apoptotic cells can also initiate anti-inflammatory and tissue-regenerative responses [3, 4].
The current notion that not only apoptotic but also necrotic cell death is molecularly controlled by defined signaling mechanisms has increased the interest in studying regulated necrosis in the context of development, homeostasis and inflammation. Although RIPK3 knockout mice and MLKL knockout mice do not show an overt phenotypic abnormality under non-challenged conditions [5–8], it became clear that RIPK3-mediated necroptosis is a highly controlled cell death program that is executed when negative regulators such as caspase-8, IAPs or even RIPK1 are absent [9–17]. This suggests that the default mode during development and homeostasis is strong inhibition of the necroptosis pathway. Regulated necrosis, like passive necrosis due to physico-chemical insult, is caused by loss of plasma membrane integrity leading to cellular rounding followed by swelling (oncosis). When the immune system recognizes cellular content exposed or released due to loss of membrane integrity, it initiates an inflammatory response. Regulated necrosis can be classified into several cell death modalities, such as necroptosis, parthanatos, ferroptosis, cyclophilin D-dependent necrosis, (n)etosis and pyroptosis [18]. Each type of regulated necrosis has particular biochemical features, yet it is not clear whether the common morphological features of these forms of cell death share or converge on common pathways. Other forms of cellular death are being identified, such as entosis [19], autosis [20, 21] and autoschizis [22].
Necroptosis, the best-characterized form of regulated necrosis, is mediated by the concerted action of receptor interacting protein kinase (RIPK) 3 and mixed lineage kinase domain-like (MLKL). In this review, we provide a snapshot of the activation of RIPK3 within the necrosome, typically by the three RHIM-containing proteins RIPK1, Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF) and DNA-dependent activator of interferon regulatory factors (DAI). We briefly discuss the pluripotent roles of RIPK1 and RIPK3 in gene regulation and cell death induction.
RIPK1-dependent necroptosis
The mechanism of RIPK1-dependent necroptosis has been discovered mostly from studying tumor necrosis factor (TNF) signaling under conditions favoring cell death. This is because the major function of TNFR1, like that of DR3 (TRAMP/APO-3), is to induce pro-survival and pro-inflammatory genes, in contrast to some other death receptor family members such as CD95 (FAS/APO-1), TRAILR1 (DR4) and TRAILR2 (APO-2/TRICK/DR5/KILLER). The following model is currently proposed for TNFR1 signaling: sensing of trimeric TNF by TNFR1 induces the assembly of a primary receptor-bound complex that triggers activation of signaling pathways leading to gene induction [23–25]. Subsequently, assembly of a secondary TNFR1-unbound cytosolic complex induces cell death. For FAS and TRAILR1/2, the opposite situation is observed [26]. This sequential signaling provides a backup response by the secondary cytosolic complex in case the default pathway activated by the receptor-associated complex fails to resolve the infectious or inflammatory condition. Typically, pathogens and (epi)genetic factors can interfere with gene activation or cell death induction. Thus, this sequential signaling probably evolved as a host defense against pathogens or conditions that might perturb either pathway.
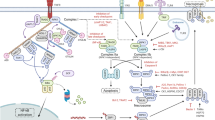
In this review, we will briefly describe cell death signaling downstream of TNFR1, with a focus on necrosome activation. A cytosolic cell death-inducing complex is formed upon stimulation of TNFR1 only in conditions that sensitize to cell death, for example when cellular inhibitor of apoptosis proteins (c-IAPs) are absent (IAP-antagonist treatment), TAK1 or translation is inhibited, or RIPK1 is deubiquitinated [18]. This cytosolic complex (often referred to as complex II), which is composed of at least RIPK1, the death-fold-containing proteins Fas-associated protein via death domain (FADD), CASP8, and cFLIP (Fig. 1), induces either apoptosis or necroptosis. The formation and/or activity of complex II is tightly regulated by inhibitor κB kinases (IKKs) through mechanisms that are either dependent or independent of NF-κB [27].

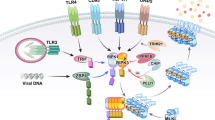
Canonical versus noncanonical necrosome activation by three distinct RHIM-containing host adaptors. RIPK1 is a key pro-necrotic kinase that responds to several DRs, TCR, IFN-γR and genotoxic stress by forming a RIPK1–RIPK3 complex through RHIM-dependent interactions (referred to as canonical necrosome). Necrostatin-1s (Nec-1s) blocks the cytotoxicity induced by the kinase activity of RIPK1. In TNFR1 signaling, cell death is efficiently induced only upon formation of a cytosolic cell death–inducing complex (complex II) when, for example, cellular inhibitor of apoptosis proteins (c-IAPs) are absent or depleted (IAP-antagonist treatment), TAK1 or translation is inhibited, or RIPK1 is deubiquitinated. In addition, caspases need to be inactivated or depleted, for example by pharmacological or viral inhibitors, to allow necroptosis to occur. TRIF and DAI are two other RHIM-containing adaptors that can activate RIPK3 via RHIM-dependent interactions in response to TLR3/-4 and MCMV, respectively (referred to as noncanonical necrosomes). RIPK3, upon activation by RIPK1, TRIF or DAI, phosphorylates itself and subsequently MLKL to stimulate oligomerization and translocation of MLKL to intracellular and plasma membranes. It is assumed that MLKL induces membrane rupture by binding directly to membrane phospholipids or indirectly with the help of calcium or sodium ion channels. Necrosulfonamide targets human MLKL and blocks necroptosis induction. DAI DNA-dependent activator of interferon regulatory factors, DR death receptor, FADD Fas-associated protein via a death domain, IFN interferon, MCMV murine cytomegalovirus, MLKL mixed lineage kinase domain-like, NSA necrosulfonamide, LPS lipopolysaccharide, RIPK receptor-interacting protein kinase, TCR T cell receptor, TLR Toll like receptor, TNF tumor necrosis factor, TRIF Toll-interleukin-1 receptor domain-containing adaptor inducing interferon-β
Necroptosis is typically initiated when caspases are insufficiently activated or their activity is blocked, for example by pharmacological or viral inhibitors. This concept was first proposed based on studies of DR signaling in fibrosarcoma cells [28, 29], and later confirmed in vivo by the rescue of the embryonal lethality of CASP8 or FADD deficiency by RIPK3 depletion [9, 14, 16]. CASP8 cleaves and inactivates RIPK1 [30], RIPK3 [31] and cylindromatosis (CYLD) [32]. This could at least partially explain the protective role of CASP8 against necroptosis [33, 34]. It is thought that CYLD is crucial for translocation of RIPK1 from receptor-bound complex I to the cytosolic death-inducing complex II by removing ubiquitin chains from RIPK1 [35, 36]. However, recently it became clear that complex II is also ubiquitylated, although the E3 ligases have not been identified yet [37–41]. While complex II is formed through interactions that depend on the death effector domain (DED) and the death domain (DD), RIPK1 and RIPK3 interact through RIP homotypic interaction motifs (RHIM) [6, 42–45]. It is assumed that this RHIM-dependent binding of RIPK1 and RIPK3 involves a conformational change that releases the RHIM domain [46]; the conformational change depends on posttranslational modifications, particularly the phosphorylation and ubiquitination status. A series of auto- and cross-phosphorylations between RIPK1 and RIPK3 result in the formation and activation of the canonical necrosome [43, 47], which appears as an amyloid-like structure of RHIM-dependent oligomerized RIPK3 [48]. The phosphorylation of human RIPK3 at Ser227 and mouse RIPK3 at Ser232 is crucial for recruitment of mixed lineage kinase domain-like (MLKL) [49–52]. Subsequent phosphorylation of MLKL at Thr357/Ser358 by human RIPK3 [50] or at Ser345/Ser347/Ser352/Thr349 by mouse RIPK3 [52] stimulates its oligomerization and translocation to intracellular and plasma membranes. The precise mechanism by which MLKL induces membrane rupture is controversial. Some reports implicate the influx of calcium or sodium through ion channels [53, 54] whereas others show direct binding to membrane phosphatidylinositol phosphates and loss of membrane integrity [55, 56].
In addition to TNF receptor signaling, other receptors induce necroptosis through RIPK1-dependent necrosome activation (Fig. 1; Table 1). These receptors include CD95L (FasL/APO-1L) [57], TRAIL (TNF-related apoptosis-inducing ligand or Apo2L) [57], TWEAK (TNF-like weak inducer of apoptosis) [58], and T cell receptor (TCR) [59]. Also, genotoxic stress [60, 61] and some anti-cancer drugs such as shikonin [62, 63] and obatoclax [64] have been shown to induce RIPK1-dependent necroptosis (Table 2). However, the assumption that many chemotherapeutics induce RIPK1/3-mediated necroptosis was recently challenged [65].
TRIF-dependent necroptosis
RIPK1 is the central RHIM-containing protein involved in the activation of RIPK3 during TNF-induced necroptosis, which leads to the formation of the so-called canonical necrosome complex. However, in response to some Toll-like receptors (TLRs), the RHIM-containing protein TRIF somehow activates RIPK3 independently of RIPK1 [44, 45], leading to assembly of the noncanonical necrosome complex [46]. Each member of the TLR family senses particular pathogen-associated molecular patterns [66]. When activated, TLRs recruit adaptors containing the Toll/IL-1R (TIR) domain and initiate NF-κB and IRF3/7 signaling that trigger the expression of cytokines, chemokines and interferons. All TLRs, with the exception of TLR3 and TLR4, mediate the signal through the adaptor myeloid differentiation primary-response gene 88 (MYD88). On the other hand, TLR3 and TLR4, after binding of the ligand (dsRNA and LPS, respectively), recruit the RHIM domain containing adaptor TRIF [67]. Both TLR3 and TLR4 directly induce apoptosis or, if caspase activity is compromised, necroptosis [42, 68, 69]. Several other TLRs, such as TLR2, TLR5 and TLR9, also induce cell death but through an endocrine or paracrine TNF-dependent mechanism [70]. TLR3/4-induced necroptosis is critically dependent on RHIM-mediated recruitment of TRIF to activate RIPK3 (Fig. 1) [69, 70]. Like RIPK1 and RIPK3, TRIF is a cleavage substrate of CASP8 that inhibits its ability to stimulate NF-κB-dependent cytokine expression [71]. Notably, RIPK1 seems to have a cell type specific function in TLR3/4-induced cell death. While fibroblast and endothelial cells undergo TLR3-dependent necroptosis independently of RIPK1, macrophages require RIPK1 to commit to TLR3/4-mediated necroptosis [70].
DAI-dependent necroptosis
In addition to RIPK1 and TRIF, a third RHIM-containing protein, DAI, has been reported to activate the necrosome (Fig. 1). The DAI pathway is typically activated in response to DNA viruses and leads to inhibition of viral replication [72]. Like TLR signaling, the intracellular DNA sensor activates the NF-κB and IRF3 pathways to promote the synthesis of cytokines and interferons, which is dependent on RHIM-mediated recruitment of RIPK1 [73]. In addition, in response to DNA viruses, DAI induces necroptosis through RHIM-mediated activation of RIPK3 in the noncanonical necrosome [74]. As a virus-encoded countermeasure, the murine cytomegalovirus (CMV) M45-encoded viral inhibitor of RIP activation (vIRA) acts as a RHIM competitor and blocks necroptosis, which explains the virus’s successful replication in the host. The potency of this cell autonomous host defense pathway is demonstrated by the remarkable attenuation of M45-deficient viruses in mice. Importantly, as in RIPK3-deficient mice, mCMV lacking M45 has the same pathogenesis in DAI-deficient mice, consistent with the notion of the existence of a DAI–RIPK3 complex as the natural target of M45 [74]. M45 encodes a ribonucleotide reductase (RNR) lacking enzymatic activity. Interestingly, many RNRs from herpesviruses also encode a RHIM [75]. This suggests that viral inhibitors that target the RIPKs via the RHIM represent a common viral evasion strategy.
The Janus faces of RIPK1
The ‘two faces’ of RIPK1 refers to its dual role. It has a cell death inhibitory role that is shown by the massive cell death observed in RIPK1-deficient models, whereas its necroptosis-inducing capacity is executed by its kinase activity. In the absence of RIPK1, massive apoptosis is observed in cells [34, 76], in postnatal death knockout mice [77], and in intestinal specific knockout mice [12, 13]. It was initially thought that this was due to the role of RIPK1 in mediating NF-κB activation, which results in the expression of survival genes such as Flip L [77]. In this respect, cFLIPL-CASP8 heterodimers have partial enzymatic activity, leading to incomplete cleavage of CASP8 [78, 79], and this consequently prevents apoptosis. Nevertheless, it is thought that CASP8 has some local activity within complex II resulting in cleavage of RIPKs and CYLD [80], which may contribute to the anti-necroptotic role of CASP8. However, mounting evidence questions the necessity or uniqueness of the role of NF-κB activation in controlling cell death. For example, NF-κB is still activated in response to TNF stimulation in the absence of RIPK1 in cultured MEF cells [81] and in intestinal organoids [13]. TAB2-deficient mice have a functional NF-κB pathway, yet they die from massive liver apoptosis like mice deficient in p65, IKKβ, TAK1 or NEMO [82]. Moreover, the rescue of mutant RIPK1 kinase-dead knockin mice from TNF-induced shock [10, 11, 83, 84] and from the lethal TNF-induced inflammation in Sharpin mutant mice [83] also calls into question the dominance of NF-κB activation (that occurs in a RIPK1 kinase independent way). This is underscored by the recent finding that IKKα and IKKβ control RIPK1-mediated cell death independently of NF-κB activation [27].
The dual role of RIPK1 in controlling cell death is also illustrated by the perinatal death of RIPK1 knockout mice due to the aberrant activation of caspase-8 and RIPK3; mice lacking all three enzymes survived to adulthood [10, 14, 85]. Indeed, in addition to its anti-apoptotic role, RIPK1 also prevents RIPK3-driven necroptosis promoted by IFN and the TLR-adapter TRIF [14]. Since RIPK1 is reported to be essential for RIPK3 activation and subsequent necroptosis induction by TNF, the identification of settings in which RIPK1 actively suppresses RIPK3 was surprising. Moreover, conditional depletion of RIPK1 leads to apoptosis in the intestine and necroptosis in the skin [12, 13]. This dynamic interplay and interdependence of these complex II components confers a crucial host-defense function to limit pathogen spread, especially when any one of these processes is disrupted [72, 86]. This may explain why this complex interrelationship exists and why ablation of specific elements (including RIPK1, FADD, caspase-8 and cFLIP) push the system to lethality [87]. In line with this reasoning, the tissues most affected by disruption of these gene products (intestine, lung, skin, endothelium, hematopoietic cells) represent crucial barriers to infection that are constantly engaged by pathogens [88]. Depending on the tissue, cell type and developmental stage, RIPK1 can certainly either activate or inhibit cell death.
The pleiotropic role of RIPK3
Whereas RIPK3 knockout mice are viable and fertile [5, 89], RIPK3 D161N kinase dead knockin mice die on embryonic day E10.5 due to massive levels of apoptosis in the embryo and yolk sac vasculature [11]. But this was not observed in RIPK3 D51A kinase dead knockin mice Mandal et al. [89]. The embryonic death of RIPK3 D161N kinase dead knockin mice was rescued by ablation of RIPK1 or caspase-8, indicating that RIPK3 can engage both RIPK1 and caspase-8 [11]. It remains unclear structurally why the D161N kinase-dead mutation in RIPK3 is proapoptotic, though it is likely that the kinase domain functionally “masks” the RHIM domain to prevent spurious activation [90]. In this scenario, the D161N alters the conformation of RIPK3 so that the RHIM domain is exposed for binding to RIPK1 to initiate apoptosis. This model predicts that the kinase and RHIM domains collaborate to control scaffolding of the necroptotic and apoptotic machineries. Some RIPK3 inhibitors were also found to induce apoptosis in a similar way through RHIM-dependent RIPK1 docking and subsequent FADD/CASP8-mediated apoptosis [70, 89]. Note that RIPK3 has also been reported to positively contribute to RIPK1-dependent apoptosis independently of its kinase activity but remarkably also of its RHIM domain [37]. While TNF signaling typically requires RIPK1 to activate RIPK3 in order to induce necroptosis, it has been noted that TNF can trigger RIPK3 activation even in the absence of RIPK1 if RIPK3 levels are high enough [91]. In the absence of RIPK1 and the presence of elevated levels of RIPK3, TNF can activate RIPK3 to induce cell death by both a caspase-8-dependent mechanism and a caspase-independent mechanism [37, 91]. Finally, similar to depletion of RIPK1 [34, 76], blocking TNF-induced necroptosis by suppressing RIPK3 or MLKL toggles the cell death response to apoptosis, albeit with different kinetics [92]. Collectively, these studies indicate that precise control of the complex II machinery is necessary to prevent a lethal imbalance of necroptotic or apoptotic pathways.
Concluding remarks
There has been a revival of interest in the close interconnection between cell death and inflammation originally recognized by Virchow. This interconnection is emphasized by some recent findings that classical cell death inducers such as caspase-8 and RIPK3 seem to act also upstream of inflammasome activation in a cell autonomous way [93–98]. However, the precise mechanisms of this interaction are unclear. RIPK1 as well as RIPK3 and other cytosolic TNFR complex II components have been implicated in regulating cell death and inflammation, though if these functions could be uncoupled is not clear. In addition, the potential signal transduction interplay between parenchymal cell necrosis and some forms of necrosis that occur in immune cells, such as pyroptosis and netosis, remains unknown. Considering the central role of RHIM domains in controlling the cell death induced by several stimuli, small molecules that disrupt RHIM signaling might also be therapeutically useful.
The number of genetic (Table 3) and pharmacological studies (Table 4) demonstrating an important role for RIPK1, RIPK3 or MLKL in murine experimental disease models is still increasing, highlighting the therapeutic potential of these necrosome members. In addition, the expression and activation of RIPK1, RIPK3 and MLKL is being increasingly explored in biopsies of patients with particular pathologies driven by cell death and inflammation (Table 5). The availability of new phospho-specific antibodies, pharmacologic inhibitors and transgenic models will allow us to document further the role of necroptosis in degenerative, inflammatory and infectious diseases. It is noteworthy that therapeutic targeting of only necroptosis might be insufficient in some complex pathologies, as exemplified by the additive protective effect of targeting different types of regulated necrosis [99–101]. This observed redundancy of necrosome proteins and interplay between different modalities of necrotic cell death in vivo is an intriguing topic for further research and will generate further insight into how the targeting of these molecules in some cases looks very effective.
Abbreviations
- cIAP:
-
Cellular inhibitor of apoptosis protein
- cFLIP:
-
Cellular FLICE-like inhibitory protein
- CypD:
-
Cyclophilin D
- CYLD:
-
Cylindromatosis
- DAI:
-
DNA-dependent activator of interferon regulatory factors
- DR:
-
Death receptor
- FADD:
-
Fas-associated protein via a death domain
- FasL:
-
Fas
- MLKL:
-
Mixed lineage kinase domain-like
- Nec-1:
-
Necrostatin-1
- NLR:
-
NOD-like receptor
- NLRP3:
-
NOD-like receptor family pyrin domain-containing protein 3
- PAMP:
-
Pathogen-associated molecular patterns
- PARP1:
-
Poly(ADP-ribose) polymerase 1
- RHIM:
-
RIP homotypic interaction motif
- RIG-I:
-
Retinoic acid-inducible gene-I
- RIPK:
-
Receptor-interacting protein kinase
- ROS:
-
Reactive oxygen species
- TAK1:
-
Transforming growth factor-β-activated kinase 1
- TLR:
-
Toll-like receptor
- TNFR:
-
Tumor necrosis factor receptor
- TRADD:
-
TNFR-associated death domain protein
- TRAF2:
-
TNFR-associated factor 2
- TRAIL-R:
-
TNF-related apoptosis-inducing ligand receptor
- TRIF:
-
Toll-interleukin-1 receptor domain-containing adaptor inducing interferon-β
- TWEAKR:
-
TNF-like weak inducer of apoptosis receptor
- VV:
-
Vaccinia virus
References
Virchow R (1860) Cellular pathology: as based upon physiological and pathological histology. Twenty lectures delivered in the pathological Institute of Berlin during the months of February, March and April, 1858
Wallach D, Kang TB, Kovalenko A (2014) Concepts of tissue injury and cell death in inflammation: a historical perspective. Nat Rev Immunol 14(1):51–59. doi:10.1038/nri3561
Felton JM, Lucas CD, Rossi AG, Dransfield I (2014) Eosinophils in the lung—modulating apoptosis and efferocytosis in airway inflammation. Front Immunol 5:302. doi:10.3389/fimmu.2014.00302
Saas P, Kaminski S, Perruche S (2013) Prospects of apoptotic cell-based therapies for transplantation and inflammatory diseases. Immunotherapy 5(10):1055–1073. doi:10.2217/imt.13.103
Newton K, Sun X, Dixit VM (2004) Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol 24(4):1464–1469
He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X (2009) Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137(6):1100–1111. doi:10.1016/j.cell.2009.05.021
Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, Ma J, Chen W, Zhang Y, Zhou X, Yang Z, Wu SQ, Chen L, Han J (2013) Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res 23(8):994–1006. doi:10.1038/cr.2013.91
Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, Young SN, Varghese LN, Tannahill GM, Hatchell EC, Majewski IJ, Okamoto T, Dobson RC, Hilton DJ, Babon JJ, Nicola NA, Strasser A, Silke J, Alexander WS (2013) The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39(3):443–453. doi:10.1016/j.immuni.2013.06.018
Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES (2011) RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471(7338):368–372. doi:10.1038/nature09857
Kaiser WJ, Daley-Bauer LP, Thapa RJ, Mandal P, Berger SB, Huang C, Sundararajan A, Guo H, Roback L, Speck SH, Bertin J, Gough PJ, Balachandran S, Mocarski ES (2014) RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc Natl Acad Sci USA 111(21):7753–7758. doi:10.1073/pnas.1401857111
Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, Roose-Girma M, Warming S, Dixit VM (2014) Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343(6177):1357–1360. doi:10.1126/science.1249361
Dannappel M, Vlantis K, Kumari S, Polykratis A, Kim C, Wachsmuth L, Eftychi C, Lin J, Corona T, Hermance N, Zelic M, Kirsch P, Basic M, Bleich A, Kelliher M, Pasparakis M (2014) RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 513(7516):90–94. doi:10.1038/nature13608
Takahashi N, Vereecke L, Bertrand MJ, Duprez L, Berger SB, Divert T, Goncalves A, Sze M, Gilbert B, Kourula S, Goossens V, Lefebvre S, Gunther C, Becker C, Bertin J, Gough PJ, Declercq W, van Loo G, Vandenabeele P (2014) RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature 513(7516):95–99. doi:10.1038/nature13706
Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong YN, Janke LJ, Kelliher MA, Kanneganti TD, Green DR (2014) RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157(5):1189–1202. doi:10.1016/j.cell.2014.04.018
Wong WW, Vince JE, Lalaoui N, Lawlor KE, Chau D, Bankovacki A, Anderton H, Metcalf D, O’Reilly L, Jost PJ, Murphy JM, Alexander WS, Strasser A, Vaux DL, Silke J (2014) cIAPs and XIAP regulate myelopoiesis through cytokine production in an RIPK1- and RIPK3-dependent manner. Blood 123(16):2562–2572. doi:10.1182/blood-2013-06-510743
Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR (2011) Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471(7338):363–367. doi:10.1038/nature09852
Lu JV, Weist BM, van Raam BJ, Marro BS, Nguyen LV, Srinivas P, Bell BD, Luhrs KA, Lane TE, Salvesen GS, Walsh CM (2011) Complementary roles of Fas-associated death domain (FADD) and receptor interacting protein kinase-3 (RIPK3) in T-cell homeostasis and antiviral immunity. Proc Natl Acad Sci USA 108(37):15312–15317. doi:10.1073/pnas.1102779108
Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P (2014) Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 15(2):135–147. doi:10.1038/nrm3737
Kroemer G, Perfettini JL (2014) Entosis, a key player in cancer cell competition. Cell Res 24(11):1280–1281. doi:10.1038/cr.2014.133
Liu Y, Levine B (2015) Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ 22(3):367–376. doi:10.1038/cdd.2014.143
Munoz-Pinedo C, Martin SJ (2014) Autosis: a new addition to the cell death tower of Babel. Cell Death Dis 5:e1319. doi:10.1038/cddis.2014.246
Jamison JM, Gilloteaux J, Taper HS, Calderon PB, Summers JL (2002) Autoschizis: a novel cell death. Biochem Pharmacol 63(10):1773–1783. doi:10.1016/S0006-2952(02)00904-8
Vandenabeele P, Declercq W, Beyaert R, Fiers W (1995) Two tumour necrosis factor receptors: structure and function. Trends Cell Biol 5(10):392–399. doi:10.1016/S0962-8924(00)89088-1
Micheau O, Tschopp J (2003) Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114(2):181–190
Schneider-Brachert W, Tchikov V, Neumeyer J, Jakob M, Winoto-Morbach S, Held-Feindt J, Heinrich M, Merkel O, Ehrenschwender M, Adam D, Mentlein R, Kabelitz D, Schutze S (2004) Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity 21(3):415–428. doi:10.1016/j.immuni.2004.08.017
Vanden Berghe T, Kaiser WJ, Bertrand MJM, Vandenabeele P (2015) Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mole Cell Oncol 2(4):e975093. doi:10.4161/23723556.2014.975093
Dondelinger Y, Jouan-Lanhouet S, Divert T, Theatre E, Bertin J, Gough PJ, Giansanti P, Heck AJ, Dejardin E, Vandenabeele P, Bertrand MJ (2015) NF-κB-independent role of IKKα/IKKβ in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol Cell 60(1):63–76. doi:10.1016/j.molcel.2015.07.032
Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, Vandenabeele P (1998) Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med 188(5):919–930
Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P (1998) Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med 187(9):1477–1485
Lin Y, Devin A, Rodriguez Y, Liu ZG (1999) Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev 13(19):2514–2526
Feng S, Yang Y, Mei Y, Ma L, Zhu DE, Hoti N, Castanares M, Wu M (2007) Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal 19(10):2056–2067. doi:10.1016/j.cellsig.2007.05.016
O’Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, Green DR, Ting AT (2011) Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol 13(12):1437–1442. doi:10.1038/ncb2362
Salvesen GS, Walsh CM (2014) Functions of caspase 8: the identified and the mysterious. Semin Immunol 26(3):246–252. doi:10.1016/j.smim.2014.03.005
Vanlangenakker N, Bertrand MJ, Bogaert P, Vandenabeele P, Vanden Berghe T (2011) TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis 2:e230. doi:10.1038/cddis.2011.111
O’Donnell MA, Legarda-Addison D, Skountzos P, Yeh WC, Ting AT (2007) Ubiquitination of RIP1 regulates an NF-κB-independent cell-death switch in TNF signaling. Curr Biol 17(5):418–424. doi:10.1016/j.cub.2007.01.027
Wright A, Reiley WW, Chang M, Jin W, Lee AJ, Zhang M, Sun SC (2007) Regulation of early wave of germ cell apoptosis and spermatogenesis by deubiquitinating enzyme CYLD. Dev Cell 13(5):705–716. doi:10.1016/j.devcel.2007.09.007
Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, Vandenabeele P, Bertrand MJ (2013) RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ 20(10):1381–1392. doi:10.1038/cdd.2013.94
Moquin DM, McQuade T, Chan FK (2013) CYLD deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS One 8(10):e76841. doi:10.1371/journal.pone.0076841
Onizawa M, Oshima S, Schulze-Topphoff U, Oses-Prieto JA, Lu T, Tavares R, Prodhomme T, Duong B, Whang MI, Advincula R, Agelidis A, Barrera J, Wu H, Burlingame A, Malynn BA, Zamvil SS, Ma A (2015) The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat Immunol 16(6):618–627. doi:10.1038/ni.3172
Biton S, Ashkenazi A (2011) NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-alpha feedforward signaling. Cell 145(1):92–103. doi:10.1016/j.cell.2011.02.023
de Almagro MC, Goncharov T, Newton K, Vucic D (2015) Cellular IAP proteins and LUBAC differentially regulate necrosome-associated RIP1 ubiquitination. Cell Death Dis 6:e1800. doi:10.1038/cddis.2015.158
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J (2009) RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325(5938):332–336. doi:10.1126/science.1172308
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK (2009) Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137(6):1112–1123. doi:10.1016/j.cell.2009.05.037
Guo H, Kaiser WJ, Mocarski ES (2015) Manipulation of apoptosis and necroptosis signaling by herpesviruses. Med Microbiol Immunol 204(3):439–448. doi:10.1007/s00430-015-0410-5
Mocarski ES, Guo H, Kaiser WJ (2015) Necroptosis: the Trojan horse in cell autonomous antiviral host defense. Virology 479–480:160–166. doi:10.1016/j.virol.2015.03.016
Chan FK, Luz NF, Moriwaki K (2015) Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev Immunol 33:79–106. doi:10.1146/annurev-immunol-032414-112248
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1(2):112–119. doi:10.1038/nchembio711
Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, Chan FK, Wu H (2012) The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150(2):339–350. doi:10.1016/j.cell.2012.06.019
Chen W, Zhou Z, Li L, Zhong CQ, Zheng X, Wu X, Zhang Y, Ma H, Huang D, Li W, Xia Z, Han J (2013) Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J Biol Chem 288(23):16247–16261. doi:10.1074/jbc.M112.435545
Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148(1–2):213–227. doi:10.1016/j.cell.2011.11.031
McQuade T, Cho Y, Chan FK (2013) Positive and negative phosphorylation regulates RIP1- and RIP3-induced programmed necrosis. Biochem J 456(3):409–415. doi:10.1042/BJ20130860
Xie T, Peng W, Yan C, Wu J, Gong X, Shi Y (2013) Structural insights into RIP3-mediated necroptotic signaling. Cell Rep 5(1):70–78. doi:10.1016/j.celrep.2013.08.044
Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG (2014) Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 16(1):55–65. doi:10.1038/ncb2883
Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Li W, Zheng X, Chen P, Han J (2014) Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res 24(1):105–121. doi:10.1038/cr.2013.171
Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, Bertin J, Gough PJ, Savvides S, Martinou JC, Bertrand MJ, Vandenabeele P (2014) MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 7(4):971–981. doi:10.1016/j.celrep.2014.04.026
Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X (2014) Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 54(1):133–146. doi:10.1016/j.molcel.2014.03.003
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1(6):489–495. doi:10.1038/82732
Wilson CA, Browning JL (2002) Death of HT29 adenocarcinoma cells induced by TNF family receptor activation is caspase-independent and displays features of both apoptosis and necrosis. Cell Death Differ 9(12):1321–1333. doi:10.1038/sj.cdd.4401107
Ch’en IL, Beisner DR, Degterev A, Lynch C, Yuan J, Hoffmann A, Hedrick SM (2008) Antigen-mediated T cell expansion regulated by parallel pathways of death. Proc Natl Acad Sci USA 105(45):17463–17468. doi:10.1073/pnas.0808043105
Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, MacFarlane M, Cain K, Meier P (2011) The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell 43(3):432–448. doi:10.1016/j.molcel.2011.06.006
Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Hacker G, Leverkus M (2011) cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell 43(3):449–463. doi:10.1016/j.molcel.2011.06.011
Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, Luo J, Hu X (2007) Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther 6(5):1641–1649. doi:10.1158/1535-7163.MCT-06-0511
Huang C, Luo Y, Zhao J, Yang F, Zhao H, Fan W, Ge P (2013) Shikonin kills glioma cells through necroptosis mediated by RIP-1. PLoS One 8(6):e66326. doi:10.1371/journal.pone.0066326
Basit F, Cristofanon S, Fulda S (2013) Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ 20(9):1161–1173. doi:10.1038/cdd.2013.45
Moriwaki K, Bertin J, Gough PJ, Orlowski GM, Chan FK (2015) Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis 6:e1636. doi:10.1038/cddis.2015.16
Gay NJ, Symmons MF, Gangloff M, Bryant CE (2014) Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol 14(8):546–558. doi:10.1038/nri3713
Kaiser WJ, Offermann MK (2005) Apoptosis induced by the Toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J Immunol 174(8):4942–4952
Kalai M, Van Loo G, Vanden Berghe T, Meeus A, Burm W, Saelens X, Vandenabeele P (2002) Tipping the balance between necrosis and apoptosis in human and murine cells treated with interferon and dsRNA. Cell Death Differ 9(9):981–994. doi:10.1038/sj.cdd.4401051
He S, Liang Y, Shao F, Wang X (2011) Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci USA 108(50):20054–20059. doi:10.1073/pnas.1116302108
Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES (2013) Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem 288(43):31268–31279. doi:10.1074/jbc.M113.462341
Rebsamen M, Meylan E, Curran J, Tschopp J (2008) The antiviral adaptor proteins Cardif and Trif are processed and inactivated by caspases. Cell Death Differ 15(11):1804–1811. doi:10.1038/cdd.2008.119
Upton JW, Chan FK (2014) Staying alive: cell death in antiviral immunity. Mol Cell 54(2):273–280. doi:10.1016/j.molcel.2014.01.027
Kaiser WJ, Upton JW, Mocarski ES (2008) Receptor-interacting protein homotypic interaction motif-dependent control of NF-κB activation via the DNA-dependent activator of IFN regulatory factors. J Immunol 181(9):6427–6434
Upton JW, Kaiser WJ, Mocarski ES (2012) DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11(3):290–297. doi:10.1016/j.chom.2012.01.016
Lembo D, Brune W (2009) Tinkering with a viral ribonucleotide reductase. Trends Biochem Sci 34(1):25–32. doi:10.1016/j.tibs.2008.09.008
Vanden Berghe T, Kalai M, van Loo G, Declercq W, Vandenabeele P (2003) Disruption of HSP90 function reverts tumor necrosis factor-induced necrosis to apoptosis. J Biol Chem 278(8):5622–5629. doi:10.1074/jbc.M208925200
Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P (1998) The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 8(3):297–303
Wachter T, Sprick M, Hausmann D, Kerstan A, McPherson K, Stassi G, Brocker EB, Walczak H, Leverkus M (2004) cFLIPL inhibits tumor necrosis factor-related apoptosis-inducing ligand-mediated NF-κB activation at the death-inducing signaling complex in human keratinocytes. J Biol Chem 279(51):52824–52834. doi:10.1074/jbc.M409554200
Scaffidi C, Schmitz I, Krammer PH, Peter ME (1999) The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem 274(3):1541–1548
Feoktistova M, Leverkus M (2015) Programmed necrosis and necroptosis signalling. FEBS J 282(1):19–31. doi:10.1111/febs.13120
Wong WW, Gentle IE, Nachbur U, Anderton H, Vaux DL, Silke J (2010) RIPK1 is not essential for TNFR1-induced activation of NF-κB. Cell Death Differ 17(3):482–487. doi:10.1038/cdd.2009.178
Sanjo H, Takeda K, Tsujimura T, Ninomiya-Tsuji J, Matsumoto K, Akira S (2003) TAB2 is essential for prevention of apoptosis in fetal liver but not for interleukin-1 signaling. Mol Cell Biol 23(4):1231–1238. doi:10.1128/mcb.23.4.1231-1238.2003
Berger SB, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, Capriotti C, Cook M, Finger J, Hughes-Earle A, Harris PA, Kaiser WJ, Mocarski ES, Bertin J, Gough PJ (2014) Cutting edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol 192(12):5476–5480. doi:10.4049/jimmunol.1400499
Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van TM, Lee TH, Chan FK, Pasparakis M, Kelliher MA (2014) Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol 193(4):1539–1543. doi:10.4049/jimmunol.1400590
Rickard JA, O’Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, Vince JE, Lawlor KE, Ninnis RL, Anderton H, Hall C, Spall SK, Phesse TJ, Abud HE, Cengia LH, Corbin J, Mifsud S, Di Rago L, Metcalf D, Ernst M, Dewson G, Roberts AW, Alexander WS, Murphy JM, Ekert PG, Masters SL, Vaux DL, Croker BA, Gerlic M, Silke J (2014) RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 157(5):1175–1188. doi:10.1016/j.cell.2014.04.019
Kaiser WJ, Upton JW, Mocarski ES (2013) Viral modulation of programmed necrosis. Curr Opin Virol 3(3):296–306. doi:10.1016/j.coviro.2013.05.019
Silke J, Rickard JA, Gerlic M (2015) The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol 16(7):689–697. doi:10.1038/ni.3206
Khan N, Lawlor KE, Murphy JM, Vince JE (2014) More to life than death: molecular determinants of necroptotic and non-necroptotic RIP3 kinase signaling. Curr Opin Immunol 26:76–89. doi:10.1016/j.coi.2013.10.017
Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, Ouellette M, King BW, Wisnoski D, Lakdawala AS, DeMartino MP, Casillas LN, Haile PA, Sehon CA, Marquis RW, Upton J, Daley-Bauer LP, Roback L, Ramia N, Dovey CM, Carette JE, Chan FK, Bertin J, Gough PJ, Mocarski ES, Kaiser WJ (2014) RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell 56(4):481–495. doi:10.1016/j.molcel.2014.10.021
Zhang J, Chan FK (2014) Cell biology. RIPK3 takes another deadly turn. Science 343(6177):1322–1323. doi:10.1126/science.1252526
Moujalled DM, Cook WD, Okamoto T, Murphy J, Lawlor KE, Vince JE, Vaux DL (2013) TNF can activate RIPK3 and cause programmed necrosis in the absence of RIPK1. Cell Death Dis 4:e465. doi:10.1038/cddis.2012.201
Remijsen Q, Goossens V, Grootjans S, Van den Haute C, Vanlangenakker N, Dondelinger Y, Roelandt R, Bruggeman I, Goncalves A, Bertrand MJ, Baekelandt V, Takahashi N, Berghe TV, Vandenabeele P (2014) Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis 5:e1004. doi:10.1038/cddis.2013.531
Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O’Reilly L, Mason K, Gross O, Ma S, Guarda G, Anderton H, Castillo R, Hacker G, Silke J, Tschopp J (2012) Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 36(2):215–227. doi:10.1016/j.immuni.2012.01.012
Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D (2013) Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 38(1):27–40. doi:10.1016/j.immuni.2012.09.015
Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, Mocarski ES, Pouliot K, Chan FK, Kelliher MA, Harris PA, Bertin J, Gough PJ, Shayakhmetov DM, Goguen JD, Fitzgerald KA, Silverman N, Lien E (2014) Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc Natl Acad Sci USA 111(20):7391–7396. doi:10.1073/pnas.1403477111
Man SM, Tourlomousis P, Hopkins L, Monie TP, Fitzgerald KA, Bryant CE (2013) Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1β production. J Immunol 191(10):5239–5246. doi:10.4049/jimmunol.1301581
Gurung P, Anand PK, Malireddi RK, Vande Walle L, Van Opdenbosch N, Dillon CP, Weinlich R, Green DR, Lamkanfi M, Kanneganti TD (2014) FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol 192(4):1835–1846. doi:10.4049/jimmunol.1302839
Philip NH, Dillon CP, Snyder AG, Fitzgerald P, Wynosky-Dolfi MA, Zwack EE, Hu B, Fitzgerald L, Mauldin EA, Copenhaver AM, Shin S, Wei L, Parker M, Zhang J, Oberst A, Green DR, Brodsky IE (2014) Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-κB and MAPK signaling. Proc Natl Acad Sci USA 111(20):7385–7390. doi:10.1073/pnas.1403252111
Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz PS, Weinlich R, Vanden Berghe T, Vandenabeele P, Pasparakis M, Bleich M, Weinberg JM, Reichel CA, Brasen JH, Kunzendorf U, Anders HJ, Stockwell BR, Green DR, Krautwald S (2014) Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci USA 111(47):16836–16841. doi:10.1073/pnas.1415518111
Linkermann A, Brasen JH, Darding M, Jin MK, Sanz AB, Heller JO, De Zen F, Weinlich R, Ortiz A, Walczak H, Weinberg JM, Green DR, Kunzendorf U, Krautwald S (2013) Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci USA 110(29):12024–12029. doi:10.1073/pnas.1305538110
Zhao H, Ning J, Lemaire A, Koumpa FS, Sun JJ, Fung A, Gu J, Yi B, Lu K, Ma D (2015) Necroptosis and parthanatos are involved in remote lung injury after receiving ischemic renal allografts in rats. Kidney Int 87(4):738–748. doi:10.1038/ki.2014.388
Trichonas G, Murakami Y, Thanos A, Morizane Y, Kayama M, Debouck CM, Hisatomi T, Miller JW, Vavvas DG (2010) Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc Natl Acad Sci USA 107(50):21695–21700. doi:10.1073/pnas.1009179107
Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, Declercq W, Libert C, Cauwels A, Vandenabeele P (2011) RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 35(6):908–918. doi:10.1016/j.immuni.2011.09.020
Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S (2012) Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol 13(10):954–962. doi:10.1038/ni.2397
Moulin M, Anderton H, Voss AK, Thomas T, Wong WW, Bankovacki A, Feltham R, Chau D, Cook WD, Silke J, Vaux DL (2012) IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J 31(7):1679–1691. doi:10.1038/emboj.2012.18
Liu S, Wang X, Li Y, Xu L, Yu X, Ge L, Li J, Zhu Y, He S (2014) Necroptosis mediates TNF-induced toxicity of hippocampal neurons. Biomed Res Int 2014:290182. doi:10.1155/2014/290182
Kitur K, Parker D, Nieto P, Ahn DS, Cohen TS, Chung S, Wachtel S, Bueno S, Prince A (2015) Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog 11(4):e1004820. doi:10.1371/journal.ppat.1004820
Godwin A, Sharma A, Yang WL, Wang Z, Nicastro J, Coppa GF, Wang P (2015) Receptor-interacting protein kinase 3 deficiency delays cutaneous wound healing. PLoS One 10(10):e0140514. doi:10.1371/journal.pone.0140514
Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE (2013) Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology 57(5):1773–1783. doi:10.1002/hep.26200
Meng L, Jin W, Wang X (2015) RIP3-mediated necrotic cell death accelerates systematic inflammation and mortality. Proc Natl Acad Sci USA 112(35):11007–11012. doi:10.1073/pnas.1514730112
Lin J, Li H, Yang M, Ren J, Huang Z, Han F, Huang J, Ma J, Zhang D, Zhang Z, Wu J, Huang D, Qiao M, Jin G, Wu Q, Huang Y, Du J, Han J (2013) A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep 3(1):200–210. doi:10.1016/j.celrep.2012.12.012
Cuda CM, Misharin AV, Khare S, Saber R, Tsai F, Archer AM, Homan PJ, Haines GK 3rd, Hutcheson J, Dorfleutner A, Budinger GR, Stehlik C, Perlman H (2015) Conditional deletion of caspase-8 in macrophages alters macrophage activation in a RIPK-dependent manner. Arthr Res Ther 17:291. doi:10.1186/s13075-015-0794-z
Murakami Y, Matsumoto H, Roh M, Suzuki J, Hisatomi T, Ikeda Y, Miller JW, Vavvas DG (2012) Receptor interacting protein kinase mediates necrotic cone but not rod cell death in a mouse model of inherited degeneration. Proc Natl Acad Sci USA 109(36):14598–14603. doi:10.1073/pnas.1206937109
Murakami Y, Matsumoto H, Roh M, Giani A, Kataoka K, Morizane Y, Kayama M, Thanos A, Nakatake S, Notomi S, Hisatomi T, Ikeda Y, Ishibashi T, Connor KM, Miller JW, Vavvas DG (2014) Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death Differ 21(2):270–277. doi:10.1038/cdd.2013.109
Luedde M, Lutz M, Carter N, Sosna J, Jacoby C, Vucur M, Gautheron J, Roderburg C, Borg N, Reisinger F, Hippe HJ, Linkermann A, Wolf MJ, Rose-John S, Lullmann-Rauch R, Adam D, Flogel U, Heikenwalder M, Luedde T, Frey N (2014) RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc Res 103(2):206–216. doi:10.1093/cvr/cvu146
Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, Kreggenwinkel K, Schneider AT, Bartneck M, Neumann UP, Canbay A, Reeves HL, Luedde M, Tacke F, Trautwein C, Heikenwalder M, Luedde T (2014) A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med 6(8):1062–1074. doi:10.15252/emmm.201403856
Petersen SL, Chen TT, Lawrence DA, Marsters SA, Gonzalvez F, Ashkenazi A (2015) TRAF2 is a biologically important necroptosis suppressor. Cell Death Differ 22(11):1846–1857. doi:10.1038/cdd.2015.35
Vitner EB, Salomon R, Farfel-Becker T, Meshcheriakova A, Ali M, Klein AD, Platt FM, Cox TM, Futerman AH (2014) RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat Med 20(2):204–208. doi:10.1038/nm.3449
Chen W, Wu J, Li L, Zhang Z, Ren J, Liang Y, Chen F, Yang C, Zhou Z, Su SS, Zheng X, Zhang Z, Zhong CQ, Wan H, Xiao M, Lin X, Feng XH, Han J (2015) Ppm1b negatively regulates necroptosis through dephosphorylating Rip3. Nat Cell Biol 17(4):434–444. doi:10.1038/ncb3120
Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z, Wang Y, Huang Z, Ren J, Liu S, Chen X, Han J (2015) A role for tubular necroptosis in cisplatin-induced AKI. J Am Soc Nephrol 26(11):2647–2658. doi:10.1681/ASN.2014080741
Dara L, Johnson H, Suda J, Win S, Gaarde W, Han D, Kaplowitz N (2015) Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology 62(6):1847–1857. doi:10.1002/hep.27939
Yang X, Chao X, Wang ZT, Ding WX (2015) The end of RIPK1-RIPK3-MLKL-mediated necroptosis in acetaminophen-induced hepatotoxicity? Hepatology. doi:10.1002/hep.28263
Zhao Y, Scott NA, Fynch S, Elkerbout L, Wong WW, Mason KD, Strasser A, Huang DC, Kay TW, Thomas HE (2015) Autoreactive T cells induce necrosis and not BCL-2-regulated or death receptor-mediated apoptosis or RIPK3-dependent necroptosis of transplanted islets in a mouse model of type 1 diabetes. Diabetologia 58(1):140
Rickard JA, Anderton H, Etemadi N, Nachbur U, Darding M, Peltzer N, Lalaoui N, Lawlor KE, Vanyai H, Hall C, Bankovacki A, Gangoda L, Wong WW, Corbin J, Huang C, Mocarski ES, Murphy JM, Alexander WS, Voss AK, Vaux DL, Kaiser WJ, Walczak H, Silke J (2014) TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. Elife 3. doi: 10.7554/eLife.03464
Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, Liu ZG (2012) Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci USA 109(14):5322–5327. doi:10.1073/pnas.1200012109
Wu X, Tian L, Li J, Zhang Y, Han V, Li Y, Xu X, Li H, Chen X, Chen J, Jin W, Xie Y, Han J, Zhong CQ (2012) Investigation of receptor interacting protein (RIP3)-dependent protein phosphorylation by quantitative phosphoproteomics. Mol Cell Proteomics 11(12):1640–1651. doi:10.1074/mcp.M112.019091
Zhong CQ, Li Y, Yang D, Zhang N, Xu X, Wu Y, Chen J, Han J (2014) Quantitative phosphoproteomic analysis of RIP3-dependent protein phosphorylation in the course of TNF-induced necroptosis. Proteomics 14(6):713–724. doi:10.1002/pmic.201300326
Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, Pierotti C, Garnier JM, Dobson RC, Webb AI, Tripaydonis A, Babon JJ, Mulcair MD, Scanlon MJ, Alexander WS, Wilks AF, Czabotar PE, Lessene G, Murphy JM, Silke J (2014) Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci USA 111(42):15072–15077. doi:10.1073/pnas.1408987111
Kearney CJ, Cullen SP, Clancy D, Martin SJ (2014) RIPK1 can function as an inhibitor rather than an initiator of RIPK3-dependent necroptosis. FEBS J 281(21):4921–4934. doi:10.1111/febs.13034
Li D, Xu T, Cao Y, Wang H, Li L, Chen S, Wang X, Shen Z (2015) A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proc Natl Acad Sci USA 112(16):5017–5022. doi:10.1073/pnas.1505244112
Yoon S, Bogdanov K, Kovalenko A, Wallach D (2015) Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ. doi:10.1038/cdd.2015.92
Schenk B, Fulda S (2015) Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death. Oncogene. doi:10.1038/onc.2015.35
Fauster A, Rebsamen M, Huber KV, Bigenzahn JW, Stukalov A, Lardeau CH, Scorzoni S, Bruckner M, Gridling M, Parapatics K, Colinge J, Bennett KL, Kubicek S, Krautwald S, Linkermann A, Superti-Furga G (2015) A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell Death Dis 6:e1767. doi:10.1038/cddis.2015.130
Najjar M, Suebsuwong C, Ray SS, Thapa RJ, Maki JL, Nogusa S, Shah S, Saleh D, Gough PJ, Bertin J, Yuan J, Balachandran S, Cuny GD, Degterev A (2015) Structure guided design of potent and selective ponatinib-based hybrid inhibitors for RIPK1. Cell Rep 10(11):1850–1860. doi:10.1016/j.celrep.2015.02.052
Xu M, Cai C, Sun X, Chen W, Li Q, Zhou H (2015) Clnk plays a role in TNF-alpha-induced cell death in murine fibrosarcoma cell line L929. Biochem Biophys Res Commun 463(3):275–279. doi:10.1016/j.bbrc.2015.05.046
Preyat N, Rossi M, Kers J, Chen L, Bertin J, Gough PJ, Le Moine A, Rongvaux A, Van Gool F, Leo O (2015) Intracellular nicotinamide adenine dinucleotide promotes TNF-induced necroptosis in a sirtuin-dependent manner. Cell Death Differ. doi:10.1038/cdd.2015.60
Karch J, Kanisicak O, Brody MJ, Sargent MA, Michael DM, Molkentin JD (2015) Necroptosis interfaces with MOMP and the MPTP in mediating cell death. PLoS One 10(6):e0130520. doi:10.1371/journal.pone.0130520
Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP, Oberst A, Quarato G, Low J, Cripps JG, Chen T, Green DR (2015) Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. doi:10.1038/cdd.2015.70
Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, Hall C, Kaur Spall S, Anderton H, Masters SL, Rashidi M, Wicks IP, Alexander WS, Mitsuuchi Y, Benetatos CA, Condon SM, Wong WW, Silke J, Vaux DL, Vince JE (2015) RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun 6:6282. doi:10.1038/ncomms7282
Kim SJ, Li J (2013) Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis 4:e716. doi:10.1038/cddis.2013.238
Kang S, Fernandes-Alnemri T, Rogers C, Mayes L, Wang Y, Dillon C, Roback L, Kaiser W, Oberst A, Sagara J, Fitzgerald KA, Green DR, Zhang J, Mocarski ES, Alnemri ES (2015) Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat Commun 6:7515. doi:10.1038/ncomms8515
Kang YJ, Bang BR, Han KH, Hong L, Shim EJ, Ma J, Lerner RA, Otsuka M (2015) Regulation of NKT cell-mediated immune responses to tumours and liver inflammation by mitochondrial PGAM5-Drp1 signalling. Nat Commun 6:8371. doi:10.1038/ncomms9371
Moujalled DM, Cook WD, Murphy JM, Vaux DL (2014) Necroptosis induced by RIPK3 requires MLKL but not Drp1. Cell Death Dis 5:e1086. doi:10.1038/cddis.2014.18
Cook WD, Moujalled DM, Ralph TJ, Lock P, Young SN, Murphy JM, Vaux DL (2014) RIPK1- and RIPK3-induced cell death mode is determined by target availability. Cell Death Differ 21(10):1600–1612. doi:10.1038/cdd.2014.70
Wu XN, Yang ZH, Wang XK, Zhang Y, Wan H, Song Y, Chen X, Shao J, Han J (2014) Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis. Cell Death Differ 21(11):1709–1720. doi:10.1038/cdd.2014.77
Orozco S, Yatim N, Werner MR, Tran H, Gunja SY, Tait SW, Albert ML, Green DR, Oberst A (2014) RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ 21(10):1511–1521. doi:10.1038/cdd.2014.76
Qiu X, Klausen C, Cheng JC, Leung PC (2015) CD40 ligand induces RIP1-dependent, necroptosis-like cell death in low-grade serous but not serous borderline ovarian tumor cells. Cell Death Dis 6:e1864. doi:10.1038/cddis.2015.229
Steinwascher S, Nugues AL, Schoeneberger H, Fulda S (2015) Identification of a novel synergistic induction of cell death by Smac mimetic and HDAC inhibitors in acute myeloid leukemia cells. Cancer Lett 366(1):32–43. doi:10.1016/j.canlet.2015.05.020
Miki Y, Akimoto J, Moritake K, Hironaka C, Fujiwara Y (2015) Photodynamic therapy using talaporfin sodium induces concentration-dependent programmed necroptosis in human glioblastoma T98G cells. Lasers Med Sci 30(6):1739–1745. doi:10.1007/s10103-015-1783-9
Koo MJ, Rooney KT, Choi ME, Ryter SW, Choi AM, Moon JS (2015) Impaired oxidative phosphorylation regulates necroptosis in human lung epithelial cells. Biochem Biophys Res Commun 464(3):875–880. doi:10.1016/j.bbrc.2015.07.054
Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR, Hashimoto S, Ryter SW, Choi AM (2014) Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest 124(9):3987–4003. doi:10.1172/JCI74985
Fu Z, Deng B, Liao Y, Shan L, Yin F, Wang Z, Zeng H, Zuo D, Hua Y, Cai Z (2013) The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer 13:580. doi:10.1186/1471-2407-13-580
Dunai ZA, Imre G, Barna G, Korcsmaros T, Petak I, Bauer PI, Mihalik R (2012) Staurosporine induces necroptotic cell death under caspase-compromised conditions in U937 cells. PloS One 7(7). doi:10.1371/journal.pone.0041945
Chromik J, Safferthal C, Serve H, Fulda S (2014) Smac mimetic primes apoptosis-resistant acute myeloid leukaemia cells for cytarabine-induced cell death by triggering necroptosis. Cancer Lett 344(1):101–109. doi:10.1016/j.canlet.2013.10.018
Saveljeva S, Mc Laughlin SL, Vandenabeele P, Samali A, Bertrand MJ (2015) Endoplasmic reticulum stress induces ligand-independent TNFR1-mediated necroptosis in L929 cells. Cell Death Disease 6. doi:10.1038/cddis.2014.548
Melo-Lima S, Celeste Lopes M, Mollinedo F (2014) Necroptosis is associated with low procaspase-8 and active RIPK1 and -3 in human glioma cells. Oncoscience 1(10):649–664
Ratovitski EA (2015) Phospho-ΔNp63α-responsive microRNAs contribute to the regulation of necroptosis in squamous cell carcinoma upon cisplatin exposure. FEBS Lett 589(12):1352–1358. doi:10.1016/j.febslet.2015.04.020
Koo GB, Morgan MJ, Lee DG, Kim WJ, Yoon JH, Koo JS, Kim SI, Kim SJ, Son MK, Hong SS, Levy JM, Pollyea DA, Jordan CT, Yan P, Frankhouser D, Nicolet D, Maharry K, Marcucci G, Choi KS, Cho H, Thorburn A, Kim YS (2015) Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res 25(6):707–725. doi:10.1038/cr.2015.56
Philipp S, Sosna J, Plenge J, Kalthoff H, Adam D (2015) Homoharringtonine, a clinically approved anti-leukemia drug, sensitizes tumor cells for TRAIL-induced necroptosis. Cell Commun Signal 13:25. doi:10.1186/s12964-015-0103-0
Blohberger J, Kunz L, Einwang D, Berg U, Berg D, Ojeda SR, Dissen GA, Frohlich T, Arnold GJ, Soreq H, Lara H, Mayerhofer A (2015) Readthrough acetylcholinesterase (AChE-R) and regulated necrosis: pharmacological targets for the regulation of ovarian functions? Cell Death Dis 6:e1685. doi:10.1038/cddis.2015.51
Desai J, Vr SK, Mulay SR, Konrad L, Romoli S, Schauer C, Herrmann M, Bilyy R, Muller S, Popper B, Nakazawa D, Weidenbusch M, Thomasova D, Krautwald S, Linkermann A, Anders HJ (2015) Neutrophil extracellular trap formation can involve RIPK1-RIPK3-MLKL signalling. Eur J Immunol. doi:10.1002/eji.201545605
Amini P, Stojkov D, Wang X, Wicki S, Kaufmann T, Wong WW, Simon H-UU, Yousefi S (2015) NET formation can occur independently of RIPK3 and MLKL signaling. Eur J Immunol. doi:10.1002/eji.201545615
Suzuki T, Kikuguchi C, Sharma S, Sasaki T, Tokumasu M, Adachi S, Natsume T, Kanegae Y, Yamamoto T (2015) CNOT3 suppression promotes necroptosis by stabilizing mRNAs for cell death-inducing proteins. Sci Rep 5:14779. doi:10.1038/srep14779
Yu X, Deng Q, Li W, Xiao L, Luo X, Liu X, Yang L, Peng S, Ding Z, Feng T, Zhou J, Fan J, Bode AM, Dong Z, Liu J, Cao Y (2015) Neoalbaconol induces cell death through necroptosis by regulating RIPK-dependent autocrine TNFα and ROS production. Oncotarget 6(4):1995–2008
He W, Wang Q, Srinivasan B, Xu J, Padilla MT, Li Z, Wang X, Liu Y, Gou X, Shen HM, Xing C, Lin Y (2014) A JNK-mediated autophagy pathway that triggers c-IAP degradation and necroptosis for anticancer chemotherapy. Oncogene 33(23):3004–3013. doi:10.1038/onc.2013.256
Huang CY, Kuo WT, Huang YC, Lee TC, Yu LC (2013) Resistance to hypoxia-induced necroptosis is conferred by glycolytic pyruvate scavenging of mitochondrial superoxide in colorectal cancer cells. Cell Death Dis 4:e622. doi:10.1038/cddis.2013.149
Tian W, Xu D, Han W, He H, Cai H, Chen H, Zhou M, Chen J, Deng YC (2013) Cyclophilin D modulates cell death transition from early apoptosis to programmed necrosis induced by honokiol. Int J Oncol 42(5):1654–1663. doi:10.3892/ijo.2013.1863
Wang XQ, Jiang W, Yan YQ, Gong T, Han JH, Tian ZG, Zhou RB (2014) RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat Immunol 15(12):1126–1133. doi:10.1038/ni.3015
Omoto S, Guo H, Talekar GR, Roback L, Kaiser WJ, Mocarski ES (2015) Suppression of RIP3-dependent necroptosis by human cytomegalovirus. J Biol Chem 290(18):11635–11648. doi:10.1074/jbc.M115.646042
Wang X, Li Y, Liu S, Yu X, Li L, Shi C, He W, Li J, Xu L, Hu Z, Yu L, Yang Z, Chen Q, Ge L, Zhang Z, Zhou B, Jiang X, Chen S, He S (2014) Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci USA 111(43):15438–15443. doi:10.1073/pnas.1412767111
Yu X, Li Y, Chen Q, Su C, Zhang Z, Yang C, Hu Z, Hou J, Zhou J, Gong L, Jiang X, Zheng C, He S (2015) Herpes simplex virus type 1 (HSV-1) and HSV-2 mediate species specific modulations of programmed necrosis through the viral ribonucleotide reductase large subunit R1. J Virol. doi:10.1128/JVI.02446-15
Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, Kaiser WJ, Mocarski ES (2015) Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 17(2):243–251. doi:10.1016/j.chom.2015.01.003
Huang Z, Wu SQQ, Liang Y, Zhou X, Chen W, Li L, Wu J, Zhuang Q, Ca Chen, Li J, Zhong CQQ, Xia W, Zhou R, Zheng C, Han J (2015) RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe 17(2):229–242. doi:10.1016/j.chom.2015.01.002
Wagner RN, Reed JC, Chanda SK (2015) HIV-1 protease cleaves the serine-threonine kinases RIPK1 and RIPK2. Retrovirology 12:74. doi:10.1186/s12977-015-0200-6
Lim SY, Davidson SM, Mocanu MM, Yellon DM, Smith CC (2007) The cardioprotective effect of necrostatin requires the cyclophilin-D component of the mitochondrial permeability transition pore. Cardiovasc Drugs Ther 21(6):467–469. doi:10.1007/s10557-007-6067-6
You Z, Savitz SI, Yang J, Degterev A, Yuan J, Cuny GD, Moskowitz MA, Whalen MJ (2008) Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab 28(9):1564–1573. doi:10.1038/jcbfm.2008.44
Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, Cuny GD, Yuan J, Savitz SI (2010) Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res 88(7):1569–1576. doi:10.1002/jnr.22314
Wang Y, Wang H, Tao Y, Zhang S, Wang J, Feng X (2014) Necroptosis inhibitor necrostatin-1 promotes cell protection and physiological function in traumatic spinal cord injury. Neuroscience 266:91–101. doi:10.1016/j.neuroscience.2014.02.007
Zhu Y, Cui H, Gan H, Xia Y, Wang L, Wang Y, Sun Y (2015) Necroptosis mediated by receptor interaction protein kinase 1 and 3 aggravates chronic kidney injury of subtotal nephrectomised rats. Biochem Biophys Res Commun 461(4):575–581. doi:10.1016/j.bbrc.2015.03.164
Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP, Ye J, Zhang X, Chang A, Vakifahmetoglu-Norberg H, Geng J, Py B, Zhou W, Amin P, Berlink Lima J, Qi C, Yu Q, Trapp B, Yuan J (2015) Activation of necroptosis in multiple sclerosis. Cell Rep 10(11):1836–1849. doi:10.1016/j.celrep.2015.02.051
Re DB, Le Verche V, Yu C, Amoroso MW, Politi KA, Phani S, Ikiz B, Hoffmann L, Koolen M, Nagata T, Papadimitriou D, Nagy P, Mitsumoto H, Kariya S, Wichterle H, Henderson CE, Przedborski S (2014) Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 81(5):1001–1008. doi:10.1016/j.neuron.2014.01.011
Li JX, Feng JM, Wang Y, Li XH, Chen XX, Su Y, Shen YY, Chen Y, Xiong B, Yang CH, Ding J, Miao ZH (2014) The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis 5:e1278. doi:10.1038/cddis.2014.241
Kim SK, Kim WJ, Yoon JH, Ji JH, Morgan MJ, Cho H, Kim YC, Kim YS (2015) Upregulated RIP3 expression potentiates MLKL phosphorylation-mediated programmed necrosis in toxic epidermal necrolysis. J Invest Dermatol 135(8):2021–2030. doi:10.1038/jid.2015.90
He L, Peng K, Liu Y, Xiong J, Zhu FF (2013) Low expression of mixed lineage kinase domain-like protein is associated with poor prognosis in ovarian cancer patients. Onco Targets Ther 6:1539–1543. doi:10.2147/OTT.S52805
Pierdomenico M, Negroni A, Stronati L, Vitali R, Prete E, Bertin J, Gough PJ, Aloi M, Cucchiara S (2014) Necroptosis is active in children with inflammatory bowel disease and contributes to heighten intestinal inflammation. Am J Gastroenterol 109(2):279–287. doi:10.1038/ajg.2013.403
Geserick P, Wang J, Schilling R, Horn S, Harris PA, Bertin J, Gough PJ, Feoktistova M, Leverkus M (2015) Absence of RIPK3 predicts necroptosis resistance in malignant melanoma. Cell Death Dis 6:e1884. doi:10.1038/cddis.2015.240
Gaiha GD, McKim KJ, Woods M, Pertel T, Rohrbach J, Barteneva N, Chin CR, Liu D, Soghoian DZ, Cesa K, Wilton S, Waring MT, Chicoine A, Doering T, Wherry EJ, Kaufmann DE, Lichterfeld M, Brass AL, Walker BD (2014) Dysfunctional HIV-specific CD8+ T cell proliferation is associated with increased caspase-8 activity and mediated by necroptosis. Immunity 41(6):1001–1012. doi:10.1016/j.immuni.2014.12.011
Nugues AL, El Bouazzati H, Hetuin D, Berthon C, Loyens A, Bertrand E, Jouy N, Idziorek T, Quesnel B (2014) RIP3 is downregulated in human myeloid leukemia cells and modulates apoptosis and caspase-mediated p65/RelA cleavage. Cell Death Dis 5:e1384. doi:10.1038/cddis.2014.347
Acknowledgments
We thank Amin Bredan for excellent editorial help. TV is Assistant Professor at Ghent University. His research is supported by the Methusalem Grant of PV. Research in Vandenabeele’s unit is further supported by Belgian Grants (Interuniversity Attraction Poles, IAP 7/32), Flemish Grants (Research Foundation Flanders: FWO G.0875.11, FWO G.0973.11, FWO G.0A45.12N, FWO G.0787.13N, FWO G.0C31.14N, FWO G0E04.16N, Methusalem Grants—BOF09/01M00709 and BOF16/MET_V/007), Grants from Ghent University (MRP, GROUP-ID consortium), Foundation against Cancer, F94) and VIB.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Vanden Berghe, T., Hassannia, B. & Vandenabeele, P. An outline of necrosome triggers. Cell. Mol. Life Sci. 73, 2137–2152 (2016). https://doi.org/10.1007/s00018-016-2189-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2189-y