
Abstract
Objective
Saikosaponin c (SSc), a compound purified from the traditional Chinese herb of Radix Bupleuri was previously identified to exhibit anti-HBV replication activity. However, the mechanism through which SSc acts against HBV remains unknown. In this study, we investigated the mechanism of SSc mediated anti-HBV activity.
Methods
HepG2.2.15 cells were cultured at 37 ℃ in the presence of 1–40 μg/mL of SSc or DMSO as a control. The expression profile of HBV markers, cytokines, HNF1α and HNF4α were investigated by real-time quantitative PCR, Elisa, Western blot and Dot blotting. Knockdown of HNF1α or HNF4α in HepG2.2.15 cells was mediated by two small siRNAs specifically targeting HNF1α or HNF4α.
Results
We found that SSc stimulates IL-6 expression, leading to attenuated HNF1α and HNF4α expression, which further mediates suppression of HBV pgRNA synthesis. Knockdown of HNF1α or HNF4α in HepG2.2.15 cells by RNA interference abrogates SSc’s anti-HBV role. Moreover, SSc is effective to both wild-type and drug-resistant HBV mutants.
Conclusion
SSc inhibits pgRNA synthesis by targeting HNF1α and HNF4α. These results indicate that SSc acts as a promising compound for modulating pgRNA transcription in the therapeutic strategies against HBV infection.
Similar content being viewed by others


Introduction
As a leading cause of liver cirrhosis and hepatocellular carcinoma (HCC), hepatitis B virus (HBV) chronic infection is one of the major public health problems worldwide. Currently, interferon and nucleoside analogs (NAs) such as lamivudine, telbivudine and entecavir have been widely used in anti-HBV therapy. However, none of them are perfect. NAs target viral polymerase, which can suppress HBV viral replication, but long term treatment may result in development of drug-resistant mutants. Therapy with IFN-α or PEG-IFN-α has the advantages of finite treatment duration, absence of drug resistance, and higher rates of hepatitis B surface antigen (HBsAg) and hepatitis B e antigen (HBeAg) seroconversion, but the disadvantage of great side effects is also obvious [1]. Therefore, screening or development of new drugs with potent anti-HBV activity, low toxicity and novel modes of actions is undoubtedly essential to combat the chronic HBV infection.
In the life cycle of HBV, the viral genome is transcribed to produce the 3.5-, 2.4-, 2.1- and 0.7 kb viral mRNAs, among which, the 3.5-kb pgRNA encodes both the polymerase and core protein and also acts as the template for HBV DNA replication intermediates synthesis inside nucleocapsid [2]. Previous work showed that the synthesis of HBV mRNA was governed by liver enriched transcription factors such as hepatocyte nuclear factor 4α (HNF4α), farnesoid X receptor (FXRα), peroxisome proliferator-activated receptors (PPARs), retinoid X receptor α (RXRα) and the CCAAT/enhancer binding protein (C/EBP). Among these transcription factors, HNF4α has been shown playing a central role in HBV transcription, especially for the generation of pgRNA [3,4,5].
HNF4α is a member of nuclear receptor superfamily that controls as much as 60% of liver genes expression. Since its discovery in the early 90’s, HNF4α has been well characterized and widely associated with a diverse group of cellular functions including carbohydrate, lipid, amino acid metabolism, differentiation, inflammation, morphogenesis and immune response [6]. In addition to these multiple known functions, HNF4α has been evidenced contribute to many pathologies development, including inflammatory bowel disease [7], cancer [8], and chronic HBV infection [9].
In liver biopsy specimens collected from chronic HBV infected patients,there is a positive correlation between HNF4α expression and HBV replication [10]. Samples with higher HNF4α expression contains higher levels of HBV DNA. Moreover, overexpression of HNF4α in Huh7 cells increases pgRNA synthesis 14-fold [11], while knockdown of HNF4α by specific short hairpin RNAs decreases pgRNA expression by more than 70% in both HepG2 cells and mice liver [12], suggesting that HNF4α may function as a promising drug target for anti-persistent HBV infection.
Chinese herb Radix Bupleuri represents one of the most important herbal drugs in Asia. It has been widely used to treat fever, malaria, influenza and hepatitis diseases in Eurasia, North Africa and many Asian countries [13]. In recent decades, many valuable biological activities of Radix Bupleuri have been identified, such as anti-tumor, anti-inflammatory, anti-depressant, immunomodulatory, hepatoprotective and antiviral [14, 15]. With the development of modern pharmacology, over 100 compounds have been isolated from Radix Bupleuri, among them saikosaponin a (SSa), saikosaponin b2 (SSb2), saikosaponin c (SSc) and saikosaponin d (SSd) are considered to be the major bioactive constituents [16]. Although the three sakosaponins of SSa, SSc and SSd share the same basal structure, their biological activities are not always the same. For example, both SSa and SSd have been well documented for their anti-inflammatory activities [17], whereas little has been reported about SSc’s inflammation regulatory role. In addition, among the four major bioactive components of Radix Bupleuri, SSc is the only saikosaponin that has been identified exhibiting anti-HBV replication activity [18, 19]. Interestingly, unlike SSd who stimulates mitochondrial apoptosis in hepatocytes to exhibit hepatotoxicity [20], SSc exerts anti-apoptotic effects on both primary human umbilical vein endothelial cells (HUVECs) and HepG2 cells [18, 21]. Therefore, SSc appears to be a promising therapeutic candidate for chronic HBV infection. Despite great progress, the mechanism through which SSc acts to inhibit HBV replication remains unknown.
In this study, we investigated the anti-HBV activity of SSc in a stably expressed HBV cell line, HepG2.2.15. We provide evidence that SSc attenuates pgRNA synthesis by stimulating IL-6 expression, which further attenuates HNF1α and HNF4α expression.
Materials and methods
Compounds
SSc was from J&K Scientific (Catalogue No: 143658). Dimethyl sulfoxide (Catalogue No: D2650) and PEG8000 (Catalogue No: 89510) were from Sigma.
Cell culture
The human hepatoma cell lines HepG2 and HepG2.2.15 were cultured in tissue culture dishes at 37 ℃, 5% CO2 in DMEM medium containing 10% FBS (Gibco), 100 unit/mL penicillin streptomycin (Hyclone) and 1% glutamine.
Cytotoxicity assay
The effect of SSc on HepG2 and HepG2.2.15 cell growth was determined by using cell counting kit-8 assay (TransGen Biotech, FC101-02) in 96-well microtiter plates with a total volume of 200 μL. Cells at a density of 8000 cells/well were treated with SSc (2–40 mg/mL) or 1% DMSO (v/v) for 72 h and replaced with drug-free medium. Then, the cell counting kit-8 assay was carried out using the manufacturer’s instructions.
PEG-precipitation and quantification of HBV DNA
Core-associated HBV DNA was extracted as described with minor modification [22]. Briefly, PEG8000 buffer (16% PEG8000 and 70 mM NaCl) was added to the cellular lysate or supernatant of HepG2.2.25 cells at a volume ratio of 1–1. The mixture was shaken slowly overnight at 4 ℃, and then centrifuged at 12,000g for 30 min at 4 ℃ (Centrifuge 5424R, Eppendorf, Germany). The PEG-precipitated pellet was digested with DNase I (Takara, 2212) and proceeded for DNA extraction. HBV DNA was then isolated using the Qiamp DNA blood mini kit (Qiagen, 51106) according to the manufacturer’s instructions. The primers used to detect HBV DNA were as follows: F: 5′-ATG GTG GAT TCG CAC TCC T -3′ and R: 5′-GAT TTC TTC TTC TAG GGG ACC TG-3′. A total of 20 μL PCR mixture containing 1 μL of forward primer (10 μM), 1 μL of reverse primer (10 μM), 10 μL of SYBR PCR mix (Takara, RR420A), 0.4 μL of ROX and 7.6 μL of HBV DNA was prepared for real-time PCR assay. The thermocycling conditions were: hot start at 95 ℃ for 5 min, and 40 cycles of 95 ℃ for 30 s, 60 ℃ for 20 s, 72 ℃ for 30 s, followed by 1 cycle of 72 ℃ for 60 s.
Reverse transcription and quantification of HBV pgRNA, HNF1α and HNF4α
The EasyPure Viral RNA Kit (TransGen Biotech, ER201-01) was used for RNA extraction. Isolated HBV RNA was reverse transcribed using PrimeScript™RT Master Mix (Takara, RR036A) with random primers and reverse transcription was carried out as the manufacturer instructed. The quantification of HBV DNA was performed with the ABI 7500 real-time PCR system. Primers to detect HBV 3.5 kb-pgRNA was described previously [22]. Primers used to detect HBV 3.5 kb RNA were as follows: F: 5′-AYA GAC CAT CAA ATG CCC-3′, and R: 5′-ATT CTC AGA CCG TAG CAC ACG ACA C-3′. The primers used to detect HBV total RNA were as follows: F: 5′-TCA CCA GCA CCA TGC AAC-3′, and R: 5′-AAG CCA CCC AAG GCA CAG-3′. Primers used to detect HNF4α and HNF1α were as follows: F-4a: 5′-GAG TGG GCC AAG TAC A-3′, R-4a: 5′-GGC TTT GAG GTA GGC ATA-3′, F-1a: 5′-TGT GCG CTA TGG ACA G-3′, R-1a: 5′-GTG TTG GTG AAC GTA GGA-3′. Primers specific to GAPDH (internal control) were as follows: F: 5′-GAT TCC ACC CAT GGC AAA TTC CA-3′, R: 5′-TGG TGA TGG GAT CAT TGA TGA-3′. The real-time PCR reaction mixture (20 μL) contained 0.8 μL forward primer (10 μM), 0.8 μL reverse primer (10 μM), 10 μL SYBR PCR mix (takara RR420A), 0.4 μL ROX and 8 μL cDNA. The thermocycling conditions were: hot start at 95 ℃ for 5 min, and 40 cycles of 95 ℃ for 30 s, 60 ℃ for 20 s, 72 ℃ for 30 s, followed by 1 cycle of 72 ℃ for 60 s.
Quantification of HBsAg and HBeAg
The levels of HBsAg and HBeAg in the culture medium were measured by a commercial enzyme linked immunosorbent kit (HBsAg kit: Abnova, KA0286; HBeAg kit: Abnova, KA0290) respectively. All samples were examined in triplicate and results were presented as fold changes to the negative controls after normalization with luciferase activities.
HBV cccDNA extraction and quantification
HBV cccDNA purification and quantification were carried out as described previously with slight modification [23]. In detail, total DNA extracted from either HepG2.2.15 cells or HepAD38 cells were treated for 5 h at 37 °C with 10 U of plasmid-safe ATP dependent DNase (PSAD, Epicentre, E3110K) to digest single-strand DNA and linear double-strand DNA. PSAD digestion was carried out using a 50 μL reaction volume containing 5 μL of 10× PSAD buffer, 2 μL of 25 mM ATP (Takara, 4041), 1 μL of PSAD, 37 μL of de-ionized water and 5 μL (400 ng/μL) of total DNA. Digested samples were further purified by a QIAamp DNA blood mini kit (Qiagen, 51104) and resuspended in 20 μL of steriled water. To quantify HBV cccDNA, specific primers targeting the HBV DNA gap region and a fluorescence hybridization probe were used. The sequence of primers and probes are as follows: HBV cccDNA-f: 5′-CGT CTG TGC CTT CTC ATC TGC-3′ (nt 1550-1570), HBV cccDNA-r: 5′-GCA CAG CTT GGA GGC TTG AA-3′ (nt 1882-1863), HBV cccDNA-probe: 5′-FAM-CTG TAG GCA TAA ATT GGT CTG CGA A-BHQ1-3′. HBV cccDNA was amplified using ABI 7500 real-time PCR system with the following amplification profile: an initial denaturation cycle of 10 min at 95 °C, followed by 40 cycles of denaturation for 30 s at 95 °C, annealing for 60 s at 63 °C and a final incubation of 10 min at 95 °C. Plasmid pHBV-1.3mer was used to generate the standard curve for quantification.
Western blot
Whole cell extracts were prepared in ice-cold RIPA buffer (Beyotime, P0013C), followed by Bradford protein determination using Bio-Rad protein assay Kit (Bio-Rad, 500-0002). Proteins were then separated by 12% SDS-PAGE, transferred to PVDF membranes (Amersham Biosciences, USA) and hybridized to anti-HNF1α (CST, 89670S) anti- HNF4α (CST, 3113S), anti-H3 (ProtienTech, 17168.I-AP), anti-GAPDH (CST, 3683S), anti-HBcAg (Abcam, GR24077-2) antibodies. After incubation with horseradish peroxidase-conjugated secondary antibodies (ProtienTech, SA00001), immunoblots were visualized by chemilluminescence (Beyotime, P0018A).
Gene cloning
The lamivudine-resistant (L180M plus M204V), telbivudine-resistant (M204V), or entecavir-resistant (L180M plus M204V plus S202G) were constructed by altering its wild-type HBV-1.3mer plasmid through substitution of fragment of the HBV polymerase gene with mutant one. In detail, fragments containing the above mutations were amplified by PCR, purified by a gel extraction kit, digested by two restriction endonucleases (HindIII and XhoI). The HBV-1.3mer plasmid was digested by the two restriction endonucleases (HindIII and XhoI). After ligation, all constructs were verified by DNA sequencing. For L180M amplification, four primers were used. NYF: 5′-CTG TTC TGA CTA CTG CCT CTC-3′, L180 M-R1: 5′-GTA AAC TGA GCC ATG AGA AAC G-3′ paired to generate frament1; L180M-F1: 5′-CTC ATG GCT CAG TTT ACT AGT GC-3′, NY-R: 5′-CAA GCT CTA ATA CGA CTC ACT-3′ paired to generate fragment 2. The fragment 1 and fragment 2 were purified by a gel extraction kit respectively. To get fragments with L180 M mutation, a third round of PCR was then performed with the fragment 1 and fragment 2 as the template and the NY-F and NY-R as primers. Using sequencing confirmed L180M mutants as the template and primers specific to M204V or S202GM204V, the lamivudine-resistant (L180M plus M204V), telbivudine-resistant (M204V), or entecavir-resistant (L180M plus M204V plus S202G) mutants were then constructed respectively. For M204V amplification, four primers including the above NYF, NFR and two other primers were used. M204V-R1: 5′-GTA AAC TGA GCC ATG AGA AAC G-3′, M204 V-F1: 5′-CTC ATG GCT CAG TTT ACT AGT GC′. For S202GM204V amplification, four primers were used as follows: NYF, NFR, S202G M204V-F: 5′-CTT TCG GTT ATG TGG ATG ATG TGG TAT TG-3′, S202GM204 V-R: 5′-ATC ATC CAC ATA ACC GAA AGC CAA ACA GTG-3′.
HNF4α and HNF1α knockdown
Two small interfering RNAs specifically targeting HNF1α (s529260), HNF4α (s6697) were purchased from Thermo Fisher Scientific. The knockdown effects of both siRNAs were measured by western blot assays. A nonspecific siRNA targeting the replicase 1A region of the severe acute respiratory syndrome (SARS) coronavirus genome (siSARS: 5′- GCA CUU GUC UAC CUU GAU GdTdT-3′), which had no homology to HBV or the human genome, was used as the negative control in this part of the study.
Results
SSc was noncytotoxic to either HepG2.2.15 or HepG2 cells
We first examined cytotoxicity of SSc by CCK-8 assay. In this experiment, SSc did not show any toxicity to either HepG2.2.15 or HepG2 cells (Fig. 1), consistent with previous study that SSc was non-toxic to HepG2.2.15 cells up to 40 μg/mL [18].

Effect of SSc on cell viability. a HepG2.2.15 cells were incubated with various concentrations of SSc or 1% DMSO for 48 h and cell survival rate was measured with CCK8 reagent. b Cell viability of HepG2 cells was measured using CCK8 reagent. Data were obtained from the average of six independent studies and cell survival rate was calculated by comparing data with that of the DMSO control
SSc suppressed 3.5-kb pgRNA synthesis and HBcAg expression
To fully evaluate the anti-HBV activity of SSc, we determined HBV DNA replication, 3.5-kb pgRNA synthesis and antigens (HBsAg, HBeAg) expression in HepG2.2.15 cells. As Fig. 2a showed, incubation of Hep2.2.15 cells with 20 μg/mL of SSc for 48 h decreased intracellular HBV DNA by 51% and supernatant HBV DNA by 72%. To confirm our results, HBsAg and HBeAg were examined by a commercially available Elisa kit. As Fig. 2b showed, in response to SSc treatment, HBeAg expression decreased by about 40%, while no HBsAg decrease was observed. Interestingly, we found that SSc treatment suppresses HBV 3.5-kb pgRNA synthesis in a dose dependent manner (Fig. 2c), which was not mentioned in any previous works before. In addition to 3.5-kb pgRNA, HBV cccDNA was also examined to investigate which step in the HBV life cycle was affected by SSc. As showed in Fig. 2d, although the intracellular pgRNA decreased with SSc treatment, the HBV cccDNA remained stable. It seems that the transcription from cccDNA to mRNA is interrupted by SSc. To further determine the function of SSc, we quantified HBV total mRNA and HBcAg expression in HepG2.2.15 cells. As showed in Fig. 2e and f, the addition of SSc resulted in decrease of both HBV total RNA and HBcAg.

SSc inhibits HBV replication and transcription. a HepG2.2.15 cells were treated with 20 μg/mL of SSc or 1% DMSO (v/v) for 48 h. HBV DNA were purified and quantified by real-time PCR assay. b Expression of HBsAg and HBeAg in response to SSc treatment was measured by a commercially available Elisa kit as described in Materials and methods. c Evaluation of pgRNA synthesis in response to various concentrations of SSc by real-time PCR assay with DMSO as a negative control. d HBV cccDNA purified in SSc incubated cells was quantified by real-time PCR as described in “Materials and methods”. e HepG2.2.15 cells incubated with 0, 10 and 20 μg/mL of SSc for 48 h were lysed and HBcAg production was analyzed by western blot. f Total HBV RNA extracted from SSc treated HepG2.2.15 cells was quantified by real-time PCR assay
SSc suppressed 3.5-kb pgRNA synthesis by downregulating HNF1α and HNF4α expression
Previous work showed that hepatocyte enriched transcription factors HNF1α and HNF4α determined pgRNA synthesis in a concerted action in hepatocytes [12, 24]. To evaluate whether HNF1α or HNF4α plays a role in SSc mediated pgRNA suppression, we compared expression of HNF1α and HNF4α in HepG2 and HepG2.2.15 cells. As Fig. 3a showed, HNF1α and HNF4α expression increased tremendously in HBV producing HepG2.2.15 cells. To confirm our results, expression of HNF1α and HNF4α in HepG2 cells transfected with either pEGFP or pHBV plasmid was analyzed by western blot. Compared with the control, HNF1α and HNF4α expression increased significantly in pHBV transfected HepG2 cells, testifying the strong link between these two molecules and HBV infection (Fig. 3b). We then examined HNF1α and HNF4α expression in HepG2.2.15 cells in the presence of 0–40 μg/mL of SSc. Compared with the control, SSc treatment significantly inhibited HNF1α and HNF4α expression (Fig. 3c and d). As previous work suggested, HNF4α can bind to enhancer regions of HBV and play a critical role in regulating HBV transcription [12, 25]. We knocked down HNF1α or HNF4α with respective small siRNAs and compared pgRNA expression in the presence or absence of SSc. As presented, anti-HBV activity of SSc was greatly attenuated when either HNF1α or HNF4α was knocked down (Fig. 3e–g), demonstrating that both HNF1α and HNF4α play important roles in SSc mediated pgRNA transcription.

SSc downregulates HBV pgRNA synthesis through HNF1α and HNF4α. a Expression of HNF1α and HNF4α in HepG2 and HepG2.2.15 cells was examined by western blot. b HepG2 cells transfected with pEGFP or pHBV1.3 plasmid was lysed and expression of HNF1α and HNF4α was detected by western blot. c and d HNF1α and HNF4α expression in response to SSc treatment was examined by real-time PCR and western blot assay. e–g Knockdown of HNF1α or HNF4α by specific siRNAs significantly suppressed anti-HBV activity of SSc
SSc downregulates HNF1α and HNF4α by stimulating IL-6 expression
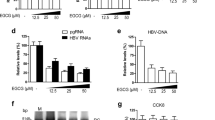
Extracellular signals such as cytokines can affect HBV transcription by dysregulating liver-enriched transcription factors. Previously, cytokines including IL-6, TGF-β, IL-1β, TNF-α and IFN-γ had been found control HNF1α or HNF4α expression through different signal pathways [24, 26]. To identify cytokines involved in SSc mediated anti-HBV signal pathway, HepG2.2.15 cells were treated with SSc for 48 h and mRNA was extracted for QPCR assay. As Fig. 4a and b showed, addition of SSc significantly stimulates IL-6 expression. Instead, other cytokines were not affected. Because IL-6 can activate the jun N-terminal kinase (JNK) and JNK phosphorylation controls HNF4α expression [24], we tested JNK phosphorylation in SSc incubated cells and found that SSc significantly enhances JNK phosphorylation (Fig. 4c). For detailed analysis of IL-6’s effects on HNF1α, HNF4α and pgRNA, we treated HepG2.2.15 cells with recombinant IL-6 (rIL-6, Thermo Fisher Scientific, PHC0066). As Fig. 4d and e showed, expression of HNF1α, HNF4α and pgRNA was significantly decreased in response to SSc treatment, consistent with previous studies that IL-6 targets nuclear cccDNA minichromosome and inhibits cccDNA transcription [27].

IL-6 downregulates expression of HNF1α and HNF4α in HepG2.2.15 cells. a Expression levels of IL-6, TGF-β, IL-1β, TNF-α and IFN-γ in response to SSc was examined by real-time PCR assay. b IL-6 expression was confirmed by dot botting with HBsAg as an internal control. c JNK phosphorylation in SSc incubated HepG2.2.15 cells were analyzed by western blot. d and e Transcription of HBV pgRNA and total RNA in response to recombinant IL-6 (rIL-6) was analyzed by real-time PCR. f Expression of HNF1α and HNF4α in SSc incubated HepG2.2.15 cells was measured by western blot. DMSO was used as a negative control
SSc’s activity against drug-resistant HBV mutants
To measure SSc’s activity against drug-resistant HBV mutants, we constructed three plasmid harboring HBV genomic DNA with respective mutations (lamivudine-resistant mutant of L180M plus M204V, telbivudine-resistant mutant of M204V, entecavir-resistant mutant of L180M plus M204V plus S202G) and examined anti-HBV activity of SSc to these three mutants. As showed in Fig. 5, SSc exhibited significant anti-HBV activity against all of the three nucleoside-resistant mutants. The inhibition efficacy of SSc to the three drug-resistant HBV mutants were similar to that in the wild-type one, demonstrating that SSc was effective against drug-resistant HBV mutants.

SSc was effective against drug-resistant HBV. HepG2 cells were transfected with wild-type HBV or HBV mutants resistant to lamivudine (LAM), telbivudine (TbL), or entecavir (ETV). The cells were then treated with 1% DMSO or SSc (10–20 μg/mL) followed by measurement of the cellular HBV DNA by real-time PCR
Discussion
Although the advent of nucleoside analogs and IFN-α makes significant progress in the management of HBV infection, for chronic HBV carriers that requires long-term antiviral treatment, the challenge to clinical therapy is substantial. There is an urgent need to search for novel drugs to address issues of availability, drug resistance and virus rebound. Natural products, especially Chinese herbal medicines (CHM) are important sources of new drugs. Over the last 20 years, CHM were widely used in the treatment of HBV carriers and some of the herbs including Radix Astragali, Radix Salviae miltiorrhizae and Radix Bupleuri, have been proved to be effective in terms of seroreversion of HBsAg, HBeAg and HBV DNA [28]. Recently, a number of natural compounds from CHM that block HBV replication have been identified, including epigallocatechin-3-gallate [29], curcumin [30], wogonin [31], ellagic acid [32], artemisinin and artesunate [33], chrysophanol 8-O-β-d-glucoside [34], SSc [18], asiaticoside [35], helioxanthin [36], and oxymatrine [37]. In this study we investigated the anti-HBV activity of SSc, one of the saikosaponins extracted from the medicinal plant Radix Bupleuri. As presented, SSc significantly suppressed pgRNA synthesis and showed little signs of toxicity in HepG2.2.15 cells, consistent with a previous study, which found that SSc inhibit apoptosis [21]. This has promoted us to search for potential intracellular targets of SSc.
Control of HBV at the level of transcription is regulated by precise recruitment of host factor with cccDNA. As demonstrated in our study, SSc significantly suppressed HNF4α expression but has no effect on PPARα, FXRα, RXRα, C/EBP or cccDNA (Supplementary Fig. 1 and Fig. 2d), suggesting that SSc regulates pgRNA transcription through HNF4α. As a global regulator of gene expression, HNF4α has been generally involved in inflammation processes occurring in liver. Extracellular pro-inflammatory cytokines including IL-6 and TNF-α have been proved inhibiting pgRNA transcription by dysregulating HNF1α and HNF4α expression. We questioned whether SSc could regulate extracellular cytokines expression in HBV replicating cells. Our data showed that IL-6 expression was upregulated in SSc incubated HepG2.2.15 cells. However, no signs of IL-6 promotion was observed in HepG2 cells without HBV infection (supplementary Fig. 2). It seems that SSc stimulated IL-6 expression in liver cells is specifically related to HBV infection. Consistent with our findings that SSc act as a pro-inflammatory molecular, Kou-Gi Shyu et al. found that SSc bore the properties to induce endothelial cells migration and the activation of mitogen-activated protein kinase (MAPK) and extracellular matrix-regulated kinase (ERK) [38]. In contrast, other saikosaponins such as SSa and SSd have been proved possess anti-inflammatory activities [17]. For example, SSa exerts anti-inflammatory activities by inhibiting the expression of pro-inflammatory cytokines and regulating inflammation-associated signal pathways, such as MAPK pathway and nuclear factor-κB (NF-κB) pathway [39]. In addition to inflammation, the contrary role of different saikosaponins in other signaling pathway was also be frequently observed. Kyo et al. found that while SSb2 and SSd stimulated prostaglandin E2 release from peritoneal macrophages and C6 rat glioma cells, SSa and SSc were inhibitory [40, 41]. As observed, saikosaponins of SSa, SSc and SSd share the same basal structure, but their side chain groups are different. The structure of SSa and SSd contains the same di-saccharide of glucose-fucose. However, SSc contains a tri-saccharide of glucose–glucose-rhamnose [17]. We speculate that the differentially presented functions of saikosapoins may be attributed to their different side chains, which specifically determines target binding, protein interaction or metabolism.
In addition to our findings, there are still a lot interesting points worth further study. Previous studies in cancer treatment suggested that saikosaponins (SSa, SSb, SSc, SSd) be potent adjuvant that are able to override primary drug resistance [42,43,44], improve sensitivity to chemotherapy and radiotherapy [45,46,47], and enhance liver-targeting effects of anticancer drugs [48]. For chronic HBV therapy, various drug combination therapies have been tried. Combination therapy offers several advantages over single drug therapy and has been found to be necessary in chronic HBV infection. For example, more rapid achievement of an undetectable HBV DNA level or increase the rates of seroconversion of hepatitis B e antigen [49]. It is seems that the selection of adjuvant is very important for chronic HBV combination therapy [50]. As we observed, SSc showed no cytotoxicity and was effective to both wild-type and drug-resistant HBV strain. These features make it a potent choice for combination with drugs currently used in clinical therapy.
Despite many advantages of SSc have been found, some issues about SSc’s anti-HBV activity are still unsolved. For example, while studies from Tajiri H et al. showed that the prescription of ‘Xiao-Chai-Hu-Tang’ promoted clearance of HBeAg in children with chronic HBV infection, the toxicity of Radix Bupleuri were also be observed [51, 52]. Although SSc has been proved with effective anti-HBV activity and little toxicity, the conclusion summarized here was based on in vitro experiments. To investigate the effects and mechanisms of SSc’s activity against HBV, related studies in vivo or in animals will be carried out in the near future.
References
Perrillo R. Benefits and risks of interferon therapy for hepatitis B. Hepatology. 2009;49:S103–11.
Yang X, Li H, Sun H, Fan H, Hu Y, Liu M, et al. Hepatitis B virus-encoded microrna controls viral replication. J Virol. 2017;91:e01919-16.
Mouzannar K, Fusil F, Lacombe B, Ollivier A, Menard C, Lotteau V, et al. Farnesoid X receptor-alpha is a proviral host factor for hepatitis B virus that is inhibited by ligands in vitro and in vivo. FASEB J. 2019;33:2472–83.
Mitra B, Thapa RJ, Guo H, Block TM. Host functions used by hepatitis B virus to complete its life cycle: implications for developing host-targeting agents to treat chronic hepatitis B. Antiviral Res. 2018;158:185–98.
Kim DH, Kang HS, Kim KH. Roles of hepatocyte nuclear factors in hepatitis B virus infection. World J Gastroenterol. 2016;22:7017–29.
Ko HL, Zhuo Z, Ren EC. Hnf4alpha combinatorial isoform heterodimers activate distinct gene targets that differ from their corresponding homodimers. Cell Rep. 2019;26:2549–57.
Meddens CA, Harakalova M, van den Dungen NA, Foroughi Asl H, Hijma HJ, Cuppen EP, et al. Systematic analysis of chromatin interactions at disease associated loci links novel candidate genes to inflammatory bowel disease. Genome Biol. 2016;17:247.
Walesky C, Apte U. Role of hepatocyte nuclear factor 4alpha (HNF4alpha) in cell proliferation and cancer. Gene Expr. 2015;16:101–8.
Chen EQ, Sun H, Feng P, Gong DY, Liu C, Bai L, et al. Study of the expression levels of hepatocyte nuclear factor 4 alpha and 3 beta in patients with different outcome of HBV infection. Virol J. 2012;9:23.
Long Y, Chen E, Liu C, Huang F, Zhou T, He F, et al. The correlation of hepatocyte nuclear factor 4 alpha and 3 beta with hepatitis B virus replication in the liver of chronic hepatitis B patients. J Viral Hepat. 2009;16:537–46.
Quasdorff M, Protzer U. Control of hepatitis B virus at the level of transcription. J Viral Hepat. 2010;17:527–36.
He F, Chen EQ, Liu L, Zhou TY, Liu C, Cheng X, et al. Inhibition of hepatitis B virus replication by hepatocyte nuclear factor 4-alpha specific short hairpin rna. Liver Int. 2012;32:742–51.
Zhao Y, Wang YJ, Zhao RZ, Xiang FJ. Vinegar amount in the process affected the components of vinegar-baked Radix Bupleuri and its hepatoprotective effect. BMC Complement Altern Med. 2016;16:346.
Liu G, Tian Y, Li G, Xu L, Song R, Zhang Z. Metabolism of saikosaponin a in rats: diverse oxidations on the aglycone moiety in liver and intestine in addition to hydrolysis of glycosidic bonds. Drug Metab Dispos. 2013;41:622–33.
Lin LT, Chung CY, Hsu WC, Chang SP, Hung TC, Shields J, et al. Saikosaponin b2 is a naturally occurring terpenoid that efficiently inhibits hepatitis C virus entry. J Hepatol. 2015;62:541–8.
Yang F, Dong X, Yin X, Wang W, You L, Ni J. Radix Bupleuri: a review of traditional uses, botany, phytochemistry, pharmacology, and toxicology. Biomed Res Int. 2017;2017:7597596.
Yuan B, Yang R, Ma Y, Zhou S, Zhang X, Liu Y. A systematic review of the active saikosaponins and extracts isolated from Radix Bupleuri and their applications. Pharm Biol. 2017;55:620–35.
Chiang LC, Ng LT, Liu LT, Shieh DE, Lin CC. Cytotoxicity and anti-hepatitis B virus activities of saikosaponins from bupleurum species. Planta Med. 2003;69:705–9.
Wohlfarth C, Efferth T. Natural products as promising drug candidates for the treatment of hepatitis B and C. Acta Pharmacol Sin. 2009;30:25–30.
Chen L, Zhang F, Kong D, Zhu X, Chen W, Wang A, et al. Saikosaponin D disrupts platelet-derived growth factor-beta receptor/p38 pathway leading to mitochondrial apoptosis in human LO2 hepatocyte cells: a potential mechanism of hepatotoxicity. Chem Biol Interact. 2013;206:76–82.
Lee TH, Chang J, Kim BM. Saikosaponin C inhibits lipopolysaccharide-induced apoptosis by suppressing caspase-3 activation and subsequent degradation of focal adhesion kinase in human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2014;445:615–21.
Wang J, Shen T, Huang X, Kumar GR, Chen X, Zeng Z, et al. Serum hepatitis B virus rna is encapsidated pregenome RNA that may be associated with persistence of viral infection and rebound. J Hepatol. 2016;65:700–10.
Hindson CM, Chevillet JR, Briggs HA, Gallichotte EN, Ruf IK, Hindson BJ, et al. Absolute quantification by droplet digital pcr versus analog real-time pcr. Nat Methods. 2013;10:1003–5.
Quasdorff M, Hosel M, Odenthal M, Zedler U, Bohne F, Gripon P, et al. A concerted action of HNF4alpha and HNF1alpha links hepatitis B virus replication to hepatocyte differentiation. Cell Microbiol. 2008;10:1478–90.
Moolla N, Kew M, Arbuthnot P. Regulatory elements of hepatitis B virus transcription. J Viral Hepat. 2002;9:323–31.
Xiang WQ, Feng WF, Ke W, Sun Z, Chen Z, Liu W. Hepatitis B virus x protein stimulates il-6 expression in hepatocytes via a myd88-dependent pathway. J Hepatol. 2011;54:26–33.
Palumbo GA, Scisciani C, Pediconi N, Lupacchini L, Alfalate D, Guerrieri F, et al. IL-6 inhibits HBV transcription by targeting the epigenetic control of the nuclear cccdna minichromosome. PLoS One. 2015;10:e0142599.
Yang Y, Jiang HY, Shi Y, He JL, Su S, Chen Z. Chinese herbal medicine for carriers of the hepatitis B virus: an updated systematic review and meta-analysis. Pharmazie. 2014;69:723–30.
Lai YH, Sun CP, Huang HC, Chen JC, Liu HK, Huang C. Epigallocatechin gallate inhibits hepatitis B virus infection in human liver chimeric mice. BMC Complement Altern Med. 2018;18:248.
Wei ZQ, Zhang YH, Ke CZ, Chen HX, Ren P, He YL, et al. Curcumin inhibits hepatitis B virus infection by down-regulating cccdna-bound histone acetylation. World J Gastroenterol. 2017;23:6252–60.
Liu Y, Yao W, Si L, Hou J, Wang J, Xu Z, et al. Chinese herbal extract su-duxing had potent inhibitory effects on both wild-type and entecavir-resistant hepatitis B virus (HBV) in vitro and effectively suppressed HBV replication in mouse model. Antiviral Res. 2018;155:39–47.
Kang EH, Kown TY, Oh GT, Park WF, Park SI, Park SK, et al. The flavonoid ellagic acid from a medicinal herb inhibits host immune tolerance induced by the hepatitis B virus-e antigen. Antiviral Res. 2006;72:100–6.
Romero MR, Efferth T, Serrano MA, Castano B, Macias RI, Briz O, et al. Effect of artemisinin/artesunate as inhibitors of hepatitis B virus production in an “in vitro” replicative system. Antiviral Res. 2005;68:75–83.
Li Z, Li LJ, Sun Y, Li J. Identification of natural compounds with anti-hepatitis B virus activity from Rheum palmatum L ethanol extract. Chemotherapy. 2007;53:320–6.
Huang Q, Zhang S, Huang R, Wei L, Chen Y, Lv S, et al. Isolation and identification of an anti-hepatitis B virus compound from hydrocotyle sibthorpioides lam. J Ethnopharmacol. 2013;150:568–75.
Tseng YP, Kuo YH, Hu CP, Jeng KS, Janmanchi D, Lin CH, et al. The role of helioxanthin in inhibiting human hepatitis b viral replication and gene expression by interfering with the host transcriptional machinery of viral promoters. Antiviral Res. 2008;77:206–14.
Wang YP, Zhao W, Xue R, Zhou ZX, Liu F, Han YX, et al. Oxymatrine inhibits hepatitis B infection with an advantage of overcoming drug-resistance. Antiviral Res. 2011;89:227–31.
Shyu KG, Tsai SC, Wang BW, Liu YC, Lee CC. Saikosaponin c induces endothelial cells growth, migration and capillary tube formation. Life Sci. 2004;76:813–26.
Zhou C, Liu W, He W, Wang H, Chen Q, Song H. Saikosaponin a inhibits RANKL-induced osteoclastogenesis by suppressing Nf-kappaB and MAPK pathways. Int Immunopharmacol. 2015;25:49–54.
Kyo R, Nakahata N, Kodama Y, Nakai Y, Kubo M, Ohizumi Y. Antagonism of saikosaponin-induced prostaglandin E2 release by baicalein in C6 rat glioma cells. Biol Pharm Bull. 1999;22:1385–7.
Shin JE, Kim HJ, Kim KR, Lee SK, Park J, Kim H, et al. Type I saikosaponins a and d inhibit osteoclastogenesis in bone marrow-derived macrophages and osteolytic activity of metastatic breast cancer cells. Evid Based Complement Alternat Med. 2015;2015:582437.
Wang Q, Zheng XL, Yang L, Shi F, Gao LB, Zhong YJ, et al. Reactive oxygen species-mediated apoptosis contributes to chemosensitization effect of saikosaponins on cisplatin-induced cytotoxicity in cancer cells. J Exp Clin Cancer Res. 2010;29:159.
Ye RP, Chen ZD. Saikosaponin A, an active glycoside from Radix Bupleuri, reverses p-glycoprotein-mediated multidrug resistance in MCF-7/ADR cells and HEPG2/ADM cells. Xenobiotica. 2017;47:176–84.
Li C, Guan X, Xue H, Wang P, Wang M, Gai X. Reversal of P-glycoprotein-mediated multidrug resistance is induced by saikosaponin D in breast cancer MCF-7/adriamycin cells. Pathol Res Pract. 2017;213:848–53.
Wong VK, Zhang MM, Zhou H, Lam KY, Chan PL, Law CK, et al. Saikosaponin-d enhances the anticancer potency of TNF-alpha via overcoming its undesirable response of activating NF-kappa B signalling in cancer cells. Evid Based Complement Alternat Med. 2013;2013:745295.
Wang BF, Dai ZJ, Wang XJ, Bai MH, Lin S, Ma HB, et al. Saikosaponin-d increases the radiosensitivity of smmc-7721 hepatocellular carcinoma cells by adjusting the g0/g1 and g2/m checkpoints of the cell cycle. BMC Complement Altern Med. 2013;13:263.
Ma H, Yokoyama S, Saiki I, Hayakawa Y. Chemosensitizing effect of saikosaponin B on B16F10 melanoma cells. Nutr Cancer. 2017;69:505–11.
Feng L, Liu L, Zhao Y, Zhao R. Saikosaponins A, C and D enhance liver-targeting effects of anticancer drugs by modulating drug transporters. Oncotarget. 2017;8:110092–102.
Liaw YF, Sheen IS, Lee CM, Akarca US, Papatheodoridis GV, Suet-Hing Wong F, et al. Tenofovir disoproxil fumarate (TDF), emtricitabine/TDF, and entecavir in patients with decompensated chronic hepatitis B liver disease. Hepatology. 2011;53:62–72.
Su TH, Liu CJ. Combination therapy for chronic hepatitis B: current updates and perspectives. Gut Liver. 2017;11:590–603.
Tajiri H, Kozaiwa K, Ozaki Y, Miki K, Shimuzu K, Okada S. Effect of sho-saiko-to(xiao-chai-hu-tang) on HBeAG clearance in children with chronic hepatitis b virus infection and with sustained liver disease. Am J Chin Med. 1991;19:121–9.
Gao X, Liang M, Fang Y, Zhao F, Tian J, Zhang X, et al. Deciphering the differential effective and toxic responses of Bupleuri Radix following the induction of chronic unpredictable mild stress and in healthy rats based on serum metabolic profiles. Front Pharmacol. 2017;8:995.
Acknowledgements
This work is supported by the National Science Foundation of China (21502026), China Postdoctoral Science Foundation Funded Project (2017M620369), Shenzhen Science and Technology Innovation Committee (KQJSCX20170329152357439, JCYJ20160427152241973) and the Sanming Project of Medicine in Shenzhen (SZSM20151205).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Responsible Editor: John Di Battista.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pan, Y., Ke, Z., Ye, H. et al. Saikosaponin C exerts anti-HBV effects by attenuating HNF1α and HNF4α expression to suppress HBV pgRNA synthesis. Inflamm. Res. 68, 1025–1034 (2019). https://doi.org/10.1007/s00011-019-01284-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-019-01284-2