
Abstract
Ventricular chamber morphogenesis, characterized by trabeculae formation, is crucial for cardiac function and embryonic viability and depends on cellular interactions between the endocardium and myocardium. The ang1-Tie2 pathway is required for normal heart chamber development, but its molecular effectors are not well defined. Here we show that loss of Tie2 in endocardial cells (EC) results in mid-gestation lethality due to heart defects including hyperplastic myocardium trabeculation (fewer but thicker trabeculae), thinner compact myocardium and simplified endocardial network. The thicker trabeculae phenotype results from enhanced proliferation of trabecular cardiomyocytes. Meanwhile, endocardial loss of Tie2 reduces proliferation and production of ECs and impairs endocardial sprouting, which provides necessary support for trabecular assembly and extension, thus resulting in a dramatically simplified trabecular meshwork. These findings reveal two seemingly apposing functions of Tie2 in myocardium trabeculation: ensuring normal trabeculation by supporting EC proliferation and sprouting while preventing hypertrabeculation by inhibiting trabecular cardiomyocyte proliferation.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Congenital heart diseases are some of the most common human birth defects. In many congenital heart diseases, genetic defects lead to impaired embryonic heart development or growth. A key development process in cardiac ontogeny is ventricular chamber formation. One of the first signs of chamber development is the formation of trabeculae [1]. Lack of trabeculation frequently causes embryonic lethality in mice and excess trabeculation causes cardiomyopathy and heart failure in humans [2]. A number of signaling molecules from myocardium, endocardium and cardiac extracellular matrix (ECM) have been implicated in trabeculation and an intimate communication between endocardium and myocardium promotes cardiomyocyte (CM) proliferation and differentiation leading to trabeculae formation [2]. The Notch signaling pathway has been the most intensely investigated in this process with Bmp10, Nrg1 and EphrinB2 identified as downstream targets of Notch signaling during trabeculation [3]. However, the molecular and cellular mechanisms controlling trabeculation are still poorly understood.
The Tie (tyrosine kinase with immunoglobulin-like and EGF homology) receptors, Tie1 and Tek/Tie2, are type 1 transmembrane protein receptor tyrosine kinases (RTKs) [4]. Along with the vascular endothelial growth factor (VEGF) receptors, these are the only known endothelial cell (EC)-specific RTKs. Although deficiency in Tie1 results in cardiovascular demise in utero [5,6,7], Tie1 is dispensable for cardiac trabeculation. Mutations in Tie2 have been described to cause ventricular septal defects and cutaneomucosal venous malformations in humans [8, 9]. Previous reports have suggested a role for Tie2 signaling in trabeculation because Tie2-null embryos have no trabeculae [5, 10] and deficiency of Ang1; the primary agonist for Tie2 results in a marked reduction in cardiac trabeculation [11, 12]. However, the molecular basis for these phenotypes has remained elusive. In addition, Tie2-deficient mouse embryos with abnormal trabeculation also had lethal vascular defects raising the question of whether the cardiac defects observed were primary or secondary to more global vascular compromise. Here, we show that loss of endocardial Tie2 results in embryonic heart failure that is characterized by impaired endocardial growth and hyperplastic trabeculation.
2 Mouse Models for Endocardial Specific Gene Deletion
In order to allow temporal and tissue-specific gene inactivation, we have developed a conditional allele of the mouse Tie2 gene by introducing Cre recombinase recognition sites (loxP) into the Tie2 locus. This strategy involved introducing a loxP site within the first intron, and another loxP site just upstream of the minimal promoter region. To specifically determine the role of Tie2 in cardiac trabeculation, we utilized this floxed Tie2 allele and an endocardial specific Cre transgenic line previously generated in our lab (Nfatc1 Cre) [13]. We first analyzed Cre activity by X-gal staining of R26 fslz;Nfatc1 Cre heart sections showing that Cre expression was restricted to the endocardium from E9.0 but not in the ECs of the peripheral vasculature (Fig. 52.1).

Nfatc1 Cre mediates loxP excision exclusively in the endocardium. X-gal staining of R26 fslz;Nfatc1 Cre heart sections at E12.5 shows that Nfatc1 Cre-mediated recombination is restricted to the embryonic atrial and ventricular endocardium but not in the endothelial cells (ECs) of the peripheral vasculature. RA right atrium, RV right ventricle, LA left atrium, LV left ventricle, IVS, interventricular septum
3 Tie2 Is Essential for Ventricular Chamber Development
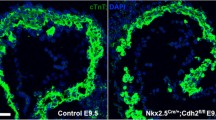
Mice with a floxed Tie2 allele were crossed with the Nfatc1 Cre line. We refer to Nfatc1 Cre/+;Tie2 f/f mice as Tie2 conditional knockout (Tie2-cko). Similar to the global deletion of Tie2, no endocardial Tie2-cko animals survived to birth. Tie2-cko embryos were grossly indistinguishable from their littermates until E12.5 at which point most began to show dorsal edema that increased until demise at E13.0–E13.5. Histological analysis of their hearts confirmed abnormal trabeculation, thin compact myocardium and ventricular septal defects compared with littermate controls (Tie2 +/f and Tie2 f/f). Dual-fluorescence staining of heart sections with antibodies for troponin T (myocardial marker) and endomucin (which identifies ECs) (Fig. 52.2) revealed fewer but thicker trabeculae in Tie2-cko hearts. In addition, the endomucin-positive endocardial network and sproutings were abnormal.

Endocardial specific deletion of Tie2 results in abnormal trabeculation. (a, b) Gross images of control (a) and Tie2-cko (b) embryos at E12.5 showing pericardial effusions (arrow) and edema (arrowhead) in the mutant embryos. (c, d) Control and Tie2-cko ventricular sections at E12.5 were stained with troponin T (myocardial marker, red) and endomucin (endocadial marker, green) antibodies. Compared to the control, the mutant heart had fewer but thicker trabeculae (arrows), thinner compact myocardium and simplified endocardial network
To determine if the thicker trabeculae in Tie2-cko hearts were due to enhanced proliferation rate in myocardium, we assessed MCs proliferation rate by BrdU pulse labeling. There is no significant difference in the percentage of BrdU-positive cardiomyocytes in the compact zone between the control and Tie2-cko hearts at E11.5 and E12.5. However, in the trabecular zone the percentage of BrdU-positive MCs in the Tie2-cko was significantly higher than that in the control, 36.6% vs. 24.6% at E11.5 and 32.4 vs. 11.8% at E12.5. Co-immunostaining for endomucin and Erg (EC-specific ETS transcription factor) revealed that the reduced endocardial sproutings in Tie2-cko mice was accompanied by a decreased number of ECs; the total endocardial EC number was determined by counting cells positive for Erg (Fig. 52.3). BrdU analysis also revealed that EC proliferation in the Tie2-cko endocardium was reduced by nearly 50% compared with control mice at E10.5 and E11.5. Thus, endocardial attenuation of Tie2 leads to impaired endocardial growth and hyperplastic trabeculation.

Endocardial attenuation of Tie2 results in decreased EC number and simplified endocardial network. Dual immunostaining of control and Tie2-cko heart sections (left ventricle) for Erg (red) and endomucin (green) at E11.5 reveals a significant decrease in Erg-positive endocardial cell number in the mutant ventricles (b) compared to control ventricles (a) following endocardial loss of Tie2. DAPI, blue
In summary, we propose a model of trabeculation regulation by Endocardial Tie2 (Fig. 52.4). In wild-type mice, at E9.0, the first stage or initiation of trabeculation is characterized by endocardial sprout formation and touchdown coincident with myocardial delamination. Next endocardial sproutings progress laterally beneath the myocardial lamina resulting in isolated clusters of myocardial cells within a “bubble” of cardiac ECM, covered by endocardium. By E9.25 the endocardial sprouting-enveloped myocardial lamina assemble to isolated short trabecular clusters (stage 2: assembly), which is followed by trabecular extension (stage 3) at E9.5, leading to finger-like long trabecular structures. In Tie2-cko mice, only limited endocardial sproutings reached the outer myocardial layer. Although the myocardial wall in mutant ventricles was able to thicken and form multicellular sheet-like protrusions (myocardial lamina), assembly and extension of trabeculae were impaired, thus leading to thicker but reduced trabeculae.

Proposed model of trabeculation regulation by endocardial Tie2. In wild-type mice, myocardial trabeculation develops as a three-step model: stage 1 at E9.0, initiation of trabeculation, stage 2 at E9.25, trabecular assembly and stage 3 at E9.5, trabecular extension. In Tie2-cko mice, only limited endocardial sproutings reached the outer layer myocardium. Although the myocardial wall in mutant ventricles was able to thicken to a multicellular layer and form sheet-like protrusions (myocardial lamina), assembly and extension of trabeculae were impaired, thus leading to thicker but reduced trabeculae. CJ cardiac jelly, CM cardiomyocytes, End, endocardium
4 Future Directions and Clinical Implications
Despite the absolute requirement for functional Tie2 signaling in normal cardiovascular development as well as its essential role in the progression of adult heart and vascular diseases, there is a paucity of information delineating the mechanism(s) of Tie2 activation or the subsequent signaling events that modulate heart ontogeny. This work reveals a critical role for the endocardium in the orchestration of ventricular trabeculation which is dependent on endocardial RTK Tie2 signaling. Tie2 not only plays a primary autocrine function in support of EC proliferation and angiogenic sprouting and touchdown but also is instrumental in the generation of paracrine signaling that results in inhibition of trabecular CM proliferation that prevents hypertrabeculation. Further investigation on this project will focus on delineating downstream targets of Tie2 activation in both endocardium (cell autonomous effect) and myocardium (paracrine effect) during ventricular trabeculation. These new insights will be important in further defining mechanisms of disease pathology related to ventricular chamber morphogenesis and function and will ultimately be critical in elucidating the etiology of congenital heart diseases.
References
Sedmera D, Pexieder T, Vuillemin M, Thompson RP, Anderson RH. Developmental patterning of the myocardium. Anat Rec. 2000;258(4):319–37.
Zhang W, Chen H, Qu X, Chang CP, Shou W. Molecular mechanism of ventricular trabeculation/compaction and the pathogenesis of the left ventricular noncompaction cardiomyopathy (LVNC). Am J Med Genet C Semin Med Genet. 2013;163C(3):144–56.
Captur G, et al. Morphogenesis of myocardial trabeculae in the mouse embryo. J Anat. 2016;229(2):314–25.
Augustin HG, et al. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol. 2009;10(3):165–77.
Sato TN, et al. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature. 1995;376(6535):70–4.
Puri MC, et al. The receptor tyrosine kinase TIE is required for integrity and survival of vascular endothelial cells. EMBO J. 1995;14(23):5884–91.
Qu X, Tompkins K, Batts LE, Puri M, Baldwin S. Abnormal embryonic lymphatic vessel development in Tie1 hypomorphic mice. Development. 2010;137(8):1285–95.
Szot JO, Cuny H, Blue GM, Humphreys DT, Ip E, Harrison K, Sholler GF, Giannoulatou E, Leo P, Duncan EL, Sparrow DB, Ho JWK, Graham RM, Pachter N, Chapman G, Winlaw DS, Dunwoodie SL. A screening approach to identify clinically actionable variants causing congenital heart disease in exome data. Circ Genom Precis Med. 2018;11(3):e001978.
Wouters V, Limaye N, Uebelhoer M, Irrthum A, Boon LM, Mulliken JB, et al. Hereditary cutaneomucosal venous malformations are caused by TIE2 mutations with widely variable hyper-phosphorylating effects. Eur J Hum Genet. 2010;18:414–20.
Dumont DJ, et al. Dominant-negative and targeted null mutations in the endothelial receptor tyrosine kinase, tek, reveal a critical role in vasculogenesis of the embryo. Genes Dev. 1994;8(16):1897–909.
Suri C, et al. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87(7):1171–80.
Jeansson M, et al. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J Clin Invest. 2011;121(6):2278–89.
Wu B, et al. Endocardial cells form the coronary arteries by angiogenesis through myocardial-endocardial VEGF signaling. Cell. 2012;151(5):1083–96.
Acknowledgments
This work was supported by grants from NHLB/NIH: RL1HL0952551 (H.S.B.).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this paper

Cite this paper
Qu, X., Baldwin, H.S. (2020). The Endocardium as a Master Regulator of Ventricular Trabeculation. In: Nakanishi, T., Baldwin, H., Fineman, J., Yamagishi, H. (eds) Molecular Mechanism of Congenital Heart Disease and Pulmonary Hypertension. Springer, Singapore. https://doi.org/10.1007/978-981-15-1185-1_52
Download citation
DOI: https://doi.org/10.1007/978-981-15-1185-1_52
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-1184-4
Online ISBN: 978-981-15-1185-1
eBook Packages: MedicineMedicine (R0)