
Abstract
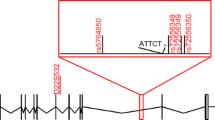
Machado-Joseph disease (MJD) is the most common autosomal dominant spinocerebellar ataxia reported worldwide, but it shows marked geographic differences in prevalence. The study of ancestral origins and spreading routes of MJD mutational events has contributed to explain such differences. During human evolution, at least two independent de novo MJD expansions occurred in distinct haplotype backgrounds: TTACAC and GTGGCA (named Joseph and Machado lineages). The most ancient Joseph lineage, probably of Asian origin, has been introduced recently in Europe, where founder effects are responsible for the high MJD prevalence, as occurs in the Portuguese/Azorean island of Flores and Northeastern mainland. The Machado lineage is geographically more restricted, with most known families in Portugal (island of São Miguel and along the Tagus valley). The hypothesis of other mutational origins has been raised, namely to explain the disease among Australian aborigines; however, a comprehensive haplotype study suggested the introduction of the Joseph lineage in that community via Asia. Also, additional SNP-based haplotypes (TTAGAC, TTGGAC and GTGCCA) were observed in other MJD families, but phylogenetic analysis with more polymorphic flanking markers did not point to independent mutational events, reinforcing the hypothesis of a very low mutation rate underlying this repeat expansion locus.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others

References
Nakano KK, Dawson DM, Spence A (1972) Machado disease. A hereditary ataxia in Portuguese emigrants to Massachusetts. Neurology 22:49–55
Woods BT, Schaumburg HH (1972) Nigro-spino-dentatal degeneration with nuclear ophthalmoplegia. A unique and partially treatable clinico-pathological entity. J Neurol Sci 17:149–166
Rosenberg RN, Nyhan WL, Bay C, Shore P (1976) Autosomal dominant striatonigral degeneration. A clinical, pathologic, and biochemical study of a new genetic disorder. Neurology 26:703–714
IJDF: International Joseph Diseases Newsletter (1978)
Coutinho P (1992) Doença de Machado-Joseph- Tentativa de definição. University of Porto Portugal
Coutinho P, Andrade C (1978) Autosomal dominant system degeneration in Portuguese families of the Azores Islands. A new genetic disorder involving cerebellar, pyramidal, extrapyramidal and spinal cord motor functions. Neurology 28:703–709
Romanul FC, Fowler HL, Radvany J, Feldman RG, Feingold M (1977) Azorean disease of the nervous system. N Engl J Med 296:1505–1508
Lima L, Coutinho P (1980) Clinical criteria for diagnosis of Machado-Joseph disease: report of a non-Azorean Portuguese family. Neurology 30:319–322
Healton EB, Brust JC, Kerr DL, Resor S, Penn A (1980) Presumably Azorean disease in a presumably non-Portuguese family. Neurology 30:1084–1089
Ishino H, Sato M, Mii T, Terano A, Hayahara T (1971) [An autopsy case of Marie’s hereditary ataxia]. Seishin shinkeigaku zasshi = Psychiatria et neurologia Japonica 73:747–757
Sakai T, Ohta M, Ishino H (1983) Joseph disease in a non-Portuguese family. Neurology 33:74–80
Yuasa T, Ohama E, Harayama H, Yamada M, Kawase Y, Wakabayashi M, Atsumi T, Miyatake T (1986) Joseph’s disease: clinical and pathological studies in a Japanese family. Ann Neurol 19:152–157
Bharucha NE, Bharucha EP, Bhabha SK (1986) Machado-Joseph-Azorean disease in India. Arch Neurol 43:142–144
Zhao JB, Wang TL, Wang GX (1994) [The pathology of Joseph’s disease in a Chinese family: a report of two autopsy cases]. Zhonghua bing li xue za zhi = Chin J Pathol 23:232–234
Burt T, Blumbergs P, Currie B (1993) A dominant hereditary ataxia resembling Machado-Joseph disease in Arnhem Land, Australia. Neurology 43:1750–1752
Goldberg-Stern H, D’Jaldetti R, Melamed E, Gadoth N (1994) Machado-Joseph (Azorean) disease in a Yemenite Jewish family in Israel. Neurology 44:1298–1301
Sequeiros J, Coutinho P (1993) Epidemiology and clinical aspects of Machado-Joseph disease. Adv Neurol 61:139–153
Fergunson F, Critchley M (1929) A clinical study of an heredo-familial disease resembling disseminated sclerosis. Brain 52:203–225
Harding AE (1982) The clinical features and classification of the late onset autosomal dominant cerebellar ataxias. A study of 11 families, including descendants of the ‘the Drew family of Walworth’. Brain 105:1–28
Giunti P, Sweeney MG, Harding AE (1995) Detection of the Machado-Joseph disease/spinocerebellar ataxia three trinucleotide repeat expansion in families with autosomal dominant motor disorders, including the Drew family of Walworth. Brain 118(Pt 5):1077–1085
Wang J, Shen L, Lei L, Xu Q, Zhou J, Liu Y, Guan W, Pan Q, Xia K, Tang B et al (2011) Spinocerebellar ataxias in mainland China: an updated genetic analysis among a large cohort of familial and sporadic cases. Zhong nan da xue xue bao Yi xue ban = J Cent S Univ Med Sci 36:482–489
de Castilhos RM, Furtado GV, Gheno TC, Schaeffer P, Russo A, Barsottini O, Pedroso JL, Salarini DZ, Vargas FR, de Lima MA et al (2014) Spinocerebellar ataxias in Brazil–frequencies and modulating effects of related genes. Cerebellum 13:17–28
Vale J, Bugalho P, Silveira I, Sequeiros J, Guimaraes J, Coutinho P (2010) Autosomal dominant cerebellar ataxia: frequency analysis and clinical characterization of 45 families from Portugal. Eur J Neurol 17:124–128
Boonkongchuen P, Pongpakdee S, Jindahra P, Papsing C, Peerapatmongkol P, Wetchaphanphesat S, Paiboonpol S, Dejthevaporn C, Tanprawate S, Nudsasarn A et al (2014) Clinical analysis of adult-onset spinocerebellar ataxias in Thailand. BMC Neurol 14:75
Schols L, Amoiridis G, Buttner T, Przuntek H, Epplen JT, Riess O (1997) Autosomal dominant cerebellar ataxia: phenotypic differences in genetically defined subtypes? Ann Neurol 42:924–932
Zhao Y, Tan EK, Law HY, Yoon CS, Wong MC, Ng I (2002) Prevalence and ethnic differences of autosomal-dominant cerebellar ataxia in Singapore. Clin Genet 62:478–481
Tsai HF, Liu CS, Leu TM, Wen FC, Lin SJ, Liu CC, Yang DK, Li C, Hsieh M (2004) Analysis of trinucleotide repeats in different SCA loci in spinocerebellar ataxia patients and in normal population of Taiwan. Acta Neurol Scand 109:355–360
Stevanin G, Durr A, David G, Didierjean O, Cancel G, Rivaud S, Tourbah A, Warter JM, Agid Y, Brice A (1997) Clinical and molecular features of spinocerebellar ataxia type 6. Neurology 49:1243–1246
Maruyama H, Izumi Y, Morino H, Oda M, Toji H, Nakamura S, Kawakami H (2002) Difference in disease-free survival curve and regional distribution according to subtype of spinocerebellar ataxia: a study of 1,286 Japanese patients. Am J Med Genet 114:578–583
van de Warrenburg BP, Sinke RJ, Verschuuren-Bemelmans CC, Scheffer H, Brunt ER, Ippel PF, Maat-Kievit JA, Dooijes D, Notermans NC, Lindhout D et al (2002) Spinocerebellar ataxias in the Netherlands: prevalence and age at onset variance analysis. Neurology 58:702–708
Paradisi I, Ikonomu V, Arias S (2016) Spinocerebellar ataxias in Venezuela: genetic epidemiology and their most likely ethnic descent. J Hum Genet 61:215–222
Moseley ML, Benzow KA, Schut LJ, Bird TD, Gomez CM, Barkhaus PE, Blindauer KA, Labuda M, Pandolfo M, Koob MD et al (1998) Incidence of dominant spinocerebellar and Friedreich triplet repeats among 361 ataxia families. Neurology 51:1666–1671
Pujana MA, Corral J, Gratacos M, Combarros O, Berciano J, Genis D, Banchs I, Estivill X, Volpini V (1999) Spinocerebellar ataxias in Spanish patients: genetic analysis of familial and sporadic cases (The Ataxia Study Group). Hum Genet 104:516–522
Koutsis G, Kladi A, Karadima G, Houlden H, Wood NW, Christodoulou K, Panas M (2014) Friedreich’s ataxia and other hereditary ataxias in Greece: an 18-year perspective. J Neurol Sci 336:87–92
Bauer PO, Zumrova A, Matoska V, Marikova T, Krilova S, Boday A, Singh B, Goetz P (2005) Absence of spinocerebellar ataxia type 3/Machado-Joseph disease within ataxic patients in the Czech population. Eur J Neurol 12:851–857
Votsi C, Zamba-Papanicolaou E, Georghiou A, Kyriakides T, Papacostas S, Kleopa KA, Pantzaris M, Christodoulou K (2012) Investigation of SCA10 in the Cypriot population: further exclusion of SCA dynamic repeat mutations. J Neurol Sci 323:154–157
Sumathipala DS, Abeysekera GS, Jayasekara RW, Tallaksen CM, Dissanayake VH (2013) Autosomal dominant hereditary ataxia in Sri Lanka. BMC Neurol 13:39
Sulek-Piatkowska A, Zdzienicka E, Raczynska-Rakowicz M, Krysa W, Rajkiewicz M, Szirkowiec W, Zaremba J (2010) The occurrence of spinocerebellar ataxias caused by dynamic mutations in Polish patients. Neurol Neurochir Pol 44:238–245
Juvonen V, Hietala M, Kairisto V, Savontaus ML (2005) The occurrence of dominant spinocerebellar ataxias among 251 Finnish ataxia patients and the role of predisposing large normal alleles in a genetically isolated population. Acta Neurol Scand 111:154–162
Dragasevic NT, Culjkovic B, Klein C, Ristic A, Keckarevic M, Topisirovic I, Vukosavic S, Svetel M, Kock N, Stefanova E et al (2006) Frequency analysis and clinical characterization of different types of spinocerebellar ataxia in Serbian patients. Mov Disord: Official J Mov Disord Soc 21:187–191
Pazarci P, Kasap H, Koc AF, Altunbasak S, Erkoc MA (2015) Mutation analysis of 6 spinocerebellar ataxia (SCA) types in patients from southern Turkey. Turk J Med Sci 45:1228–1233
Smith DC, Greenberg LJ, Bryer A (2016) The hereditary ataxias: where are we now? Four decades of local research. S Afr Med J = Suid-Afrikaanse tydskrif vir geneeskunde 106:S38–41
Gaspar C, Lopes-Cendes I, Hayes S, Goto J, Arvidsson K, Dias A, Silveira I, Maciel P, Coutinho P, Lima M et al (2001) Ancestral origins of the Machado-Joseph disease mutation: a worldwide haplotype study. Am J Hum Genet 68:523–528
Nachman MW, Crowell SL (2000) Estimate of the mutation rate per nucleotide in humans. Genetics 156:297–304
Rubinsztein DC, Leggo J, Coetzee GA, Irvine RA, Buckley M, Ferguson-Smith MA (1995) Sequence variation and size ranges of CAG repeats in the Machado-Joseph disease, spinocerebellar ataxia type 1 and androgen receptor genes. Hum Mol Genet 4:1585–1590
Limprasert P, Nouri N, Heyman RA, Nopparatana C, Kamonsilp M, Deininger PL, Keats BJ (1996) Analysis of CAG repeat of the Machado-Joseph gene in human, chimpanzee and monkey populations: a variant nucleotide is associated with the number of CAG repeats. Hum Mol Genet 5:207–213
Djian P, Hancock JM, Chana HS (1996) Codon repeats in genes associated with human diseases: fewer repeats in the genes of nonhuman primates and nucleotide substitutions concentrated at the sites of reiteration. Proc Natl Acad Sci USA 93:417–421
Martins S, Calafell F, Gaspar C, Wong VC, Silveira I, Nicholson GA, Brunt ER, Tranebjaerg L, Stevanin G, Hsieh M et al (2007) Asian origin for the worldwide-spread mutational event in Machado-Joseph disease. Arch Neurol 64:1502–1508
Sequeiros J (1989) Análise genética das causas da vairiação fenotípica na doença de Machado-Joseph. Ph.D. thesis, Universidade do Porto
Martins S, Soong BW, Wong VC, Giunti P, Stevanin G, Ranum LP, Sasaki H, Riess O, Tsuji S, Coutinho P et al (2012) Mutational origin of Machado-Joseph disease in the Australian Aboriginal communities of Groote Eylandt and Yirrkala. Arch Neurol 69:746–751
Chakravarty A, Mukherjee SC (2002) Autosomal dominant cerebellar ataxias in ethnic Bengalees in West Bengal—an Eastern Indian state. Acta Neurol Scand 105:202–208
Jayadev S, Michelson S, Lipe H, Bird T (2006) Cambodian founder effect for spinocerebellar ataxia type 3 (Machado-Joseph disease). J Neurol Sci 250:110–113
Bhargava A, Fuentes FF (2010) Mutational dynamics of microsatellites. Mol Biotechnol 44:250–266
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139:457–462
Putman AI, Carbone I (2014) Challenges in analysis and interpretation of microsatellite data for population genetic studies. Ecol Evol 4:4399–4428
Illarioshkin SN, Slominsky PA, Ovchinnikov IV, Markova ED, Miklina NI, Klyushnikov SA, Shadrina M, Vereshchagin NV, Limborskaya SA, Ivanova-Smolenskaya IA (1996) Spinocerebellar ataxia type 1 in Russia. J Neurol 243:506–510
Saber S, Rostami M, Dehghan MM, Hooshiar KB, Banoie M, Houshmand M (2006) Molecular investigation SCA in 26 patients suspected to SCA in Iran. Eur J Hum Genet 14:275
Kiloh LG, Lethlean AK, Morgan G, Cawte JE, Harris M (1980) An endemic neurological disorder in tribal Australian aborigines. J Neurol Neurosurg Psychiatry 43:661–668
Burt T, Currie B, Kilburn C, Lethlean AK, Dempsey K, Blair I, Cohen A, Nicholson G (1996) Machado-Joseph disease in east Arnhem Land, Australia: chromosome 14q32.1 expanded repeat confirmed in four families. Neurology 46:1118–1122
Buhmann C, Bussopulos A, Oechsner M (2003) Dopaminergic response in Parkinsonian phenotype of Machado-Joseph disease. Mov Disord: Official J Mov Disord Soc 18:219–221
Subramony SH, Hernandez D, Adam A, Smith-Jefferson S, Hussey J, Gwinn-Hardy K, Lynch T, McDaniel O, Hardy J, Farrer M et al (2002) Ethnic differences in the expression of neurodegenerative disease: Machado-Joseph disease in Africans and Caucasians. Mov Disord: Official J Mov Disord Soc 17:1068–1071
Ogun SA, Martins S, Adebayo PB, Dawodu CO, Sequeiros J, Finkel MF (2015) Machado-Joseph disease in a Nigerian family: mutational origin and review of the literature. Eur J Hum Genet: EJHG 23:271–273
Gwinn-Hardy K, Singleton A, O’Suilleabhain P, Boss M, Nicholl D, Adam A, Hussey J, Critchley P, Hardy J, Farrer M (2001) Spinocerebellar ataxia type 3 phenotypically resembling Parkinson disease in a black family. Arch Neurol 58:296–299
Sequeiros J, Suite ND (1986) Spinopontine atrophy disputed as a separate entity: the first description of Machado-Joseph disease. Neurology 36:1408
Taniguchi R, Konigsmark BW (1971) Dominant spino-pontine atrophy. Report of a family through three generations. Brain 94:349–358
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Martins, S., Sequeiros, J. (2018). Origins and Spread of Machado-Joseph Disease Ancestral Mutations Events. In: Nóbrega, C., Pereira de Almeida, L. (eds) Polyglutamine Disorders. Advances in Experimental Medicine and Biology, vol 1049. Springer, Cham. https://doi.org/10.1007/978-3-319-71779-1_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-71779-1_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-71778-4
Online ISBN: 978-3-319-71779-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)