
Abstract
It is now almost 70 years since Charles Huggins described the relationship between testosterone and the prostate gland. Arguably defining one of the first targeted therapies, the reduction of testosterone to castrate levels remains unaltered as the standard of care for men with metastatic prostate cancer. The failure of castration to permanently control the growth of prostate cancer leads to a state called castration-resistant prostate cancer (CRPC). Whilst numerous mechanisms have been suggested for the emergence of castration resistance [Scher and Sawyers (J Clin Oncol 23(32):8253–8261, 2005); Chen et al. (Curr Opin Pharmacol 8(4):440–448, 2008), Pienta and Bradley (Clin Cancer Res 12(6):1665–1671, 2006); Feldman and Feldman (Nat Rev Cancer 1(1):34–45, 2001); Mostaghel and Nelson (Best Pract Res Clin Endocrinol Metab 22(2):243–258, 2008)], a greater understanding of prostate cancer biology suggests that many such cancers retain a dependency on androgens and endeavour to increase bioavailable androgens through mechanisms such as AR amplification and intracrine androgen synthesis [Mohler et al. (Clin Cancer Res 10(2):440–448, 2004); Attard et al. (Clin Cancer Res 17(7):1649–1657, 2011); Hu et al. (Expert Rev Endocrinol Metab 5(5):753–764, 2010)]. With the recent approval of abiraterone acetate (Zytiga) and the pending approval of MDV3100, this article previews the future directions in clinical development and issues that will arise with the next generation of androgen-targeted agents.
Similar content being viewed by others


Introduction
Drug development in prostate cancer has been dominated over the last 3–4 years by the emergence of a series of agents that take advantage of the discovery that whilst prostate cancer may become castration independent, it remains largely androgen dependent for ongoing growth. The preclinical basis for this is manifold: (1) a frequent change following the development of metastatic castration resistant disease is the mutation, gene amplification and/or overexpression androgen receptor (AR) [9]; (2) several studies utilising diverse methodologies suggest that it is the intracrine and paracrine effects of in situ androgen synthesis or circulating adrenal derived steroid precursors that significantly contribute to prostate cancer growth [10] and (3) there appear to be multiple abnormalities within the AR pathway, involving coactivators and corepressors (~100% in CRPC metastatic samples) that predispose to AR pathway activation [11]. To date, there are broadly two new classes of hormonally active drugs in development: more effective AR antagonists (such as MDV3100, ARN-509, TOK-001) and inhibitors of the androgen biosynthetic pathway [such as abiraterone, TAK-700 (orteronel)]. The current clinical status of these agents will be reviewed (Table 1) and future developmental challenges outlined.
Androgen Receptor Signalling Agents in Clinical Development
CYP17 Inhibitors
The agent most advanced in clinical development is abiraterone acetate (AA), which was recently approved in the USA, Canada and the European Union. It is an orally administered medication that inhibits the cytrochrome p450 enzyme, CYP17A1. This enzyme has a dual function as a 17a-hydroxylase and C17,20 lyase: both of these enzymes are required to synthesise androgens from cholesterol. AA was first investigated in two phase 1 studies in standard dose escalation schema [12, 13]. These studies clearly demonstrated that the compound was well tolerated and effective with 66% of patients exhibiting a PSA decrease of >30% and 38% experiencing a partial response by RECIST criteria.
Both phase 1 studies recommended a 1,000-mg daily dose on the basis of a plateau effect in the increase of upstream corticosterone and deoxycorticosterone from 750 up to 2,000 mg. Although the numbers were small in these studies, there was no suggestion of clinically relevant differences in the numbers of responders at the lower dose levels, although neither study was designed to explore relative efficacy. Given that the variability between fed patients is comparable to that observed between fasted patients [13], exploring these lower doses, in particular when combined with a meal, may be a useful strategy to explore (currently underway in Clinicaltrials.gov NCT01424930).
Subsequently two well-conducted phase 2 studies were carried out in the pre- and post-docetaxel settings at the 1,000-mg dose, demonstrating prostate-specific antigen (PSA) response rates (50% decline from baseline) and time to PSA progression of 79% and 16.3 months and 36% and 169 days, respectively [14, 15]. Thereafter, two large phase 3 studies completed accrual; the first was a randomised, double-blind, placebo-controlled trial of abiraterone and prednisone that recruited 1,158 patients with CRPC who progressed after docetaxel known as COU301 (Clinicaltrials.gov NCT00638690). In this trial, 1,195 patients were randomised 2:1 in favour of abiraterone, and the study was unblinded after the first interim analysis, with the results [16] demonstrating an improvement in overall survival by approximately 4 months (14.8 vs. 10.9 months; hazard ratio 0.65; 95% confidence interval 0.54 to 0.77; P < 0.001). All secondary end points, including time to PSA progression (10.2 vs. 6.6 months; P < 0.001), progression-free survival (5.6 vs. 3.6 months; P < 0.001) and PSA response rate (29% vs. 6%, P < 0.001), favoured the treatment group. There is also been detailed quality of life analyses that suggest similar trends in favour of abiraterone; for example, time to skeletal-related event (pathologic fracture, spinal cord compression or palliative radiation/bone surgery) was 301 days (AA) vs. 150 days (placebo; P = 0.0006) and symptomatic improvement in pain intensity [155/349 (44%) vs 44/163 (27%)].
A second phase 3 trial completed accrual in mid-2010 known as COU302 (Clinicaltrials.gov NCT00887198) after recruiting approximately 1,000 patients with CRPC who are chemotherapy naive and asymptomatic or minimally symptomatic, with dual end points of radiographic progression-free survival (rPFS) and overall survival (OS). Certainly, demonstration of the original OS end point in this latter trial may be challenging to achieve given the potential for crossover and the current availability of the agent although the currently available expanded access protocol (Clinicaltrials.gov NCT01217697) or compassionate-release financial assistance packages in the USA and Canada although both exclude patients who have been enrolled in COU302 from participating in the trial.
The COU302 trial was open for just over 12 months (April 2009–May 2010), and the FDA approval of abiraterone occurred on April 28, 2011. The critical issue for the FDA in considering the approval of abiraterone in the pre-chemotherapy setting will occur if this OS benefit is not met given the crossover. Undoubtedly, there will be great patient pressure for oncologists to prescribe abiraterone in the pre-chemotherapy setting should the rPFS data from COU302 be robust. Indeed, in the absence of robust evidence suggesting that rPFS does not influence OS, we suspect this prescribing pattern will become standard regardless of the OS results. This is particularly the case, given that a sizeable proportion of patients with CRPC may not be candidates for docetaxel [17]. How a result lacking OS benefit will affect funding decisions with health insurers and in centralised health care countries remains to be seen and may have to await clinical trials examining a sequencing question.
The future development plans for abiraterone remain unclear—there is a phase 2 study in the M0 castration resistant setting (Clinicaltrials.gov NCT01314118), but whether this will lead to a phase 3 study is yet to be determined. Other preliminary studies include a phase 2 in patients with a suboptimal response to androgen deprivation (Clinicaltrials.gov NCT01309672). One outstanding issue with abiraterone is the need for and the consequences of long-term prednisone administration. The 10-mg dose was empirically used based on a number of factors including it being the standard dose on docetaxel; however, in earlier disease, a 5-mg dose may be equally efficacious without longer term consequences.
A direct competitor to abiraterone is TAK700 (orteronel), which is also a CYP 17 lyase inhibitor. There do appear to be some albeit small preclinical differences between this agent and abiraterone that may affect its utility in the clinic. For example, TAK-700 may be more potent 17,20-lyase than 17alpha lyase inhibitor [18] and thus may not lead to as profound mineralocorticoid effects as abiraterone. Data from the recently completed phase 1 study of TAK-700 suggest that schedules both with and without prednisone are feasible [19], which may affect future development plans for this drug given the concerns with long-term steroid administration with abiraterone. The company has embarked on an aggressive set of clinical trials in combination with prednisone. Two phase 3 studies in the post- and pre-docetaxel setting have OS as their primary end point (Clinicaltrials.gov NCT 01193244, Clinicaltrials.gov NCT 01193257), and it will be interesting to see how this can be managed given the regulatory approval of abiraterone. Interestingly, the company has also chosen to pursue two phase 2 studies of interest. The first is a phase I/II in combination with docetaxel (Clinicaltrials.gov NCT01084655) until recently notably absent from the abiraterone development plan (Clinicaltrials.gov NCT0140055), whilst another is a small 42-patient phase 2 study in M0 disease (Clinicaltrials.gov NCT01046916) with the percentage of patients who achieve a PSA <0.2 following 3 months of treatment as the primary end point.
AR-Targeting Agents
The most advanced agent of the AR antagonist class is MDV3100. This novel molecule was developed from the non-steroidal hydantoin RU59063 following optimisation of structure activity relationships [20]. MDV3100 binds ARs with eight times the affinity of bicalutamide and exhibits potent pure androgen antagonist properties that prevent nuclear translocation, co-activator peptide recruitment and DNA binding of the AR, likely by inducing a novel conformational change in the AR distinct from bicalutamide.
The published phase 1–2 results noted antitumor effects at all doses, such as decreases in serum PSA of 50% or more in 78 (56%) patients, responses in soft tissue in 13 (22%) of 59 patients and stabilised bone disease in 61 (56%) of 109 patients [20]. An update to these initial results [21] demonstrated that the median time to PSA progression by Prostate Cancer Clinical Trials Working Group 2 (PCWG2) criteria is 281 days in the chemotherapy naive population (n = 65), whilst in the post-chemotherapy population is 148 days. Interestingly, in an exploratory analysis of 33 ketoconazole naive patients, the time to PSA progression was 677 days by PCWG2 criteria, suggesting that prior exposure to ketoconazole may influence the development of resistance to this agent.
The phase 3 trial of approximately 1,200 patients randomised 2:1 in favour of MDV3100 vs. placebo (AFFIRM, Clinicaltrials.gov NCT 00974311) with a primary end point of overall survival in the post-docetaxel was reported by press release in mid November 2011. MDV3100 produced a 4.8-month advantage in median overall survival compared to placebo with a hazard ratio of 0.631. The estimated median survival for men treated with MDV3100 was 18.4 months compared with 13.6 months for men treated with placebo. The study has now closed, and crossover of remaining patients is allowed.
In the pre-chemotherapy setting similar to COU302, a further phase 3 study of over 1,600 patients (PREVAIL, Clinicaltrials.gov NCT01212991) is currently randomising patients between MDV3100 and placebo in the pre-chemotherapy setting. Of interest, similar to TAK700, there is a strong development program with MDV3100 including trials in metastatic treatment naive (Clinicaltrials.gov NCT01302041), Japanese populations (Clinicaltrials.gov NCT01284920) and against bicalutamide in a randomised phase 2 design (Clinicaltrials.gov NCT01288911).
There are a number of other high potency androgen antagonists in development that parallel those mentioned; ARN-509 is another very potent anti-androgen and has recently entered a phase 1/2 clinical trial (Clinicaltrials.gov NCT01171898) with the objectives to determine the maximum tolerated dose and recommended phase 2 dose in an estimated 132 patients. Dose escalation cohorts that are planned include (1) non-metastatic CRPC docetaxel and abiraterone naive (50 patients), (2) mCRPC docetaxel and abiraterone naive and (3) mCRPC abiraterone pretreated. Similarly, TOK-001 (galeterone) is an interesting molecule with three putative mechanisms of action: at low concentrations, it primarily inhibits CYP17,which is likely its predominant mechanism of action however at moderate concentrations; it is an effective AR antagonist whilst and at higher concentrations, it is capable of inducing AR degradation and ER stress [22]. It is currently in a phase 2 clinical trial (Clinicaltrials.gov NCT00959959) that specifically excludes prior treatment with Provenge, abiraterone or MDV3100. Finally, EZN-4176 is a novel AR mRNA antagonist (Clinicaltrials.gov NCT 01337518).
Future Directions
With the development of these agents, the accepted treatment paradigm has changed in prostate cancer, with a return to hormonal treatment being a robust strategy to extend OS. Understandably, these agents have been developed—at least initially—in the post-chemotherapy setting as this is where regulatory approval based on an OS benefit is easiest to achieve, and unlike their predecessors (e.g. bicalutamide, nilutamide, flutamide), they are required to demonstrate robust end points that correlate with significant clinical end points. Given this shifting paradigm for the approval, we outline below several crucial issues that we feel will arise in the short to medium term with the use of these agents.
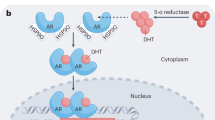
What are the Mechanisms of Resistance and What are the Strategies to Overcome Them (Fig. 1)?
Despite the high rates of clinical activity observed with these novel hormonal agents, prostate cancer progression remains inevitable and clarification of the mechanisms of resistance to these drugs is critical. Broadly, one can hypothesise that similar to tyrosine kinase pathways, resistance to either AA or MDV3100 can arise in either an AR-dependent or independent (bypass) fashion. To date, despite intensive speculation, there are only limited data available to suggest pathways that mediate resistance. Cai et al. characterized abiraterone resistance in a murine model and found that the emergence of resistant VCaP cell lines was associated with an upregulation of the CYP17 enzyme [23]. Overcoming CYP17 upregulation could possibly be achieved with increasing doses of abiraterone or more potent CYP17 inhibitors, however this will incur greater mineralocorticoid side effects, and is likely to be temporary. There is a study underway to explore the efficacy of abiraterone and dutasteride (Clinicaltrials.gov NCT 01393730) that is predominantly biomarker driven, exploring the utility of additional 5-alpha-reducatse inhibition. Alternative agents that block steroid synthesis may also find utility here such as steroid sulfatase inhibitors (no currently open trials in prostate cancer), 3- (trilostane) and 17-beta HSD inhibitors (ASP 9521, Clinicaltrials.gov NCT01352208), these latter adrenolytic agents may also be useful in particular if adrenal-derived steroid precursors are found to be relevant to the aetiology of abiraterone resistance.

Left a representation of canonical androgen synthesis pathways with relevant enzymes (black text) and relevant inhibitors thereof (white text) that are found in the testis, adrenals and prostate cancer cells (indicated on right panel). Other mechanisms of strategies to inhibit the action of the androgen receptor are also illustrated either by pharmacological class, e.g. HSP inhibitors or by name (blue text)
Resistance to MDV3100 has been recapitulated in xenograft-derived LNCAP cell lines by Kuruma et al. [24]. These cell lines are yet to be fully characterized but demonstrate a range of AR and PSA levels of expression that recapitulate clinical findings (e.g. AR/PSA +/+, AR/PSA −/−). It may be that resistance to MDV3100 arises through non-AR mechanisms given the potency of the AR blockade or alternatively through AR splice forms.
The human AR gene is composed of at least eight exons, and it contains several functional domains, including an N-terminal transactivation domain (exon 1), a DNA-binding domain (exons 2 and 3) and a ligand-binding domain (LBD, exons 5 to 8). A series of AR splice variants lacking the LBD are upregulated in hormone-resistant prostate cancer cell lines and tissues [25, 26] and promote castration-resistant growth, however their impact in hormone resistance remains to be defined as they still may be dependent on full-length receptor (and hence sensitive to MDV3100) for function although this remains controversial [27, 28].
Addressing alternative pathways may require further preclinical rationale. Two recent papers [29, 30] have described a preclinical rationale for the dual inhibition of the PI3K and androgen pathways whereby AR transcriptional output is decreased in human and murine tumours with PTEN deletion and PI3K pathway inhibition activates AR signalling by relieving feedback inhibition of HER kinases. Similarly, AR inhibition activates AKT signalling by reducing levels of the AKT phosphatase PHLPP. Therefore, combining any number of current PI3K inhibitors in development with MDV 3100 or abiraterone (and potentially more specific PI3Kbeta inhibitors) may be worthwhile. This will certainly be testable in the clinic with a new generation of PI3K inhibitors (Clinicaltrials.gov NCT01385293, Clinicaltrials.gov NCT01331083) and an ongoing trial of temsirolimus (Clinicaltrials.gov NCT01020305) to reverse androgen insensitivity associated with combined androgen blockade with bicalutamide may suggest the relevance of the AKT/MTOR pathway in a subset of men.
Novel methods of targeting the AR will also be critical. EPI-001 [31] is a peptide that targets the amino terminal domain of the AR. It is one of a number of drug candidates (e.g. sintokamides, decoy peptides [32, 33]) that aim to disrupt AR functioning by disrupting the regulatory component of the protein. Deletion experiments have shown that the N-terminal domain is essential for transcriptional activity of the AR in response to ligand as well as in the absence of ligand [34]. EPI-001 inhibits AR activity induced by a variety of non-canonical activators such as forskolin, IL-6, and by factors secreted by osteoblasts (bone-derived factors [35]). EPI-001 also inhibited constitutively active AR devoid of an LBD [35], which implies that it may have use in combating the splice variants of the AR that may mediate abiraterone resistance. EPI-001 does not reduce levels of AR protein nor does it prevent nuclear translocation of the AR in response to androgen [35]. Other androgen-directed agents that are in development include AR antagonists (ODM-201, Clinicaltrials.gov NCT01317641) and AR production inhibitors (ZD 3514, Clinicaltrials.gov NCT01162395).
One further class of novel agents that may further disrupt androgen signalling are stress chaperone [Clusterin (OGX-011), HSP 27(OGX-427), HSP 90 (e.g. STA-9090)]. The latter two are in phase 2 clinical trials [Clinicaltrials.gov NCT01120470 (OGX-427), Clinicaltrials.gov NCT 01270880 and Clinicaltrials.gov NCT 01368003(STA-9090)]; currently however, the most advanced candidate inhibitor of chaperone proteins is a second-generation oliognucelotide to clusterin (OGX-011). This agent is currently in a phase 3 clinical trial in combination with docetaxel (Clinicaltrials.gov NCT01188187), however recent data suggest that the combination of OGX-011 and MDV3100 shows synergistic effects and more potently suppressed LNCaP cell growth rates in a dose- and time-dependent manner compared to OGX-011 or MDV3100 monotherapy alone. Putative mechanisms for this include accelerated AR degradation, repressed AR transcriptional and the prevention of the induction of autophagy.
How Long Should Abiraterone Acetate be Continued After Progression in CRPC?
Now that the paradigm is established that the androgen axis is essential for prostate cancer growth beyond chemotherapy, it may appear paradoxical to suggest that these agents should be stopped upon progression by standard PSA or clinical progression criteria. However, this assumption is dependent upon the resistance mechanism to abiraterone. For example, if resistance mechanisms involve upregulation of critical enzymatic pathways (i.e. CYP17 in the case of abiraterone), the sudden cessation of these novel anti-androgen agents may conceivably lead to a “disease flare”. Alternatively, if the mechanism of resistance occurs through upregulated formation of non-canonical AR splice forms or activation of the AKT pathway, then ceasing these drugs will make little difference. In the COU301 study, patients were able to continue the drug until the combination of clinical, radiological and PSA progression occurred, a situation unlike clinical reality, where patients may be looking to try the next line of treatment following progression on any one of the former criteria. Indeed, the design of a study to explore whether continuing abiraterone beyond standard progression or ceasing it would be difficult to design, implement and accrue so ultimately, this issue may never be resolved through a formal clinical trial, but rather it may become a de facto standard of care to continue the use of full androgen suppression upon progression in particular in the metastatic setting where the risks of long-term steroid treatment (in the case of CYP17 lyase inhibitors) and cost of continued treatment are somewhat mitigated by the risk of mortality from the disease.
Can We or Should We Combine CYP17 Inhibitors with Chemotherapy?
Aside from the aforementioned TAK700 phase 1/2 study, to date, none of these agents are being explored in combination with chemotherapy. The absence of effort in this setting is telling. Indeed, there is an extensive history of attempting to do the opposite—add androgens to chemotherapy to increase effectiveness—that met with no success [36, 37]. Manni et al. studied exogenous androgens as a means of priming prostate cancer to increase the efficacy of cytotoxic chemotherapy but found that the patients receiving androgens had shortened survival and often exacerbated symptoms [38]. Can these studies therefore allow us to infer anything regarding the converse, i.e. that reducing functional androgen signalling levels further than standard castrate (50 ng/ml) may alter the efficacy of chemotherapy? This strategy certainly has not been beneficial in breast cancer [39, 40], but there is the theoretical potential that exposure to a highly androgen-deficient state, either prior to or simultaneous with chemotherapy, may reduce the efficacy of subsequent taxane-based chemotherapy as taxanes may exert part if not all of their effect [41] by blocking AR nuclear translocation, an effect that may be lost [42] with alternative AR growth pathways activated. To date, this concept has been most closely modelled in the neoadjuvant studies of androgen withdrawal and docetaxel which to date indicate that clinically relevant disease remains despite combination therapies [43, 44], and to date, the histological outcomes are no different from neoadjuvant hormonal therapy alone. Nevertheless, this concept will likely require assessment in prospective well-controlled studies, although it will be difficult to perceive how such a hypothesis will be practically tested given the suggestion that significant numbers of men will start taking abiraterone prior to chemotherapy in the near future and potential crossover will limit an effect on overall survival.
Will These Agents be Used in Earlier Disease States than the Metastatic CRPC Setting?
As discussed, recent studies of prostate cancer biology suggest that with advancing disease, there is an increasing array of mutations affecting androgen signalling. Indeed, a number of retrospective reviews of androgen signalling with oral anti-androgen agents suggest that responses are more likely to occur in relatively localised compared to widely metastatic disease [45]. This principle has also held true in the early studies of both abiraterone and MDV3100 in a comparison of median time to PSA progression for the pre- vs. post-chemo populations. For example, in the recently updated data on the MDV3100 phase 2 trial [21], the median time to PSA progression was 41 weeks for naive and 20 weeks for post-chemo groups and the median time to radiographic progression was 56 weeks for naive and 24 weeks for post-chemo groups. Therefore, moving these agents up into earlier stages of diseases such as CRPC M0 or castration naive M0/M1 would likely prolong the time to PSA and disease progression even further. The crucial issue is an agreed upon regulatory end point of these studies. For example, if the premise of radiographic progression-free survival in prostate cancer is accepted by regulatory authorities on the basis of the results of the COU302 and PREVAIL studies, then this may act as an achievable end point. However, the appearance of two or more metastases in patients that have a rising PSA may take several years and the clinical relevance of this end point is debatable in the absence of data on improvements in overall survival or symptomatic progression. Recent retrospective estimates of this on the basis of control arms of recent clinical trial suggest that approximately 50% of men with CRPC and M0 disease will develop metastases by 2 years [46] whilst in the placebo arm of the denosumab 147 study, the time to bone metastases-free survival was 25.7 months in a population that was a priori defined to be at high risk of bony metastases (high risk for development of bone metastasis defined as PSA value greater than or equal to 8.0 ng/mL, obtained no more than 3 months before randomization OR PSA doubling time less than or equal to 10.0 months). To date, there are two studies that are attempting to examine this issue; Clinicaltrials.gov NCT01288911 is a 370-patient randomised phase 2 of MDV3100 vs. bicalutamide post-LHRH failure trial that is being carried out in European centres. A 370-randomised phase 2 study will only give some indication of the hazard ratios and numbers needed to be achieved to move towards a definitive randomised phase 3 study. Undoubtedly further studies will be planned for these agents; our conservative calculations suggest that a randomised phase 3 trial in this setting could be achieved with approximately 800 patients (i.e. a total of 766 patients in a two-treatment parallel-design study with an 80% power that the study will detect a treatment difference at a two-sided 5.0% significance level, if the true hazard ratio is 0.8). This is based on the assumption that the accrual period will be 2 years, the follow-up period will be 4 years and the median survival is 2.2 years. The total number of events will be 630. Indeed, a similar study is now open with abiraterone: a single-arm, 125-patient phase 2 study (Clinicaltrials.gov NCT01314118) with a PSA end point that presumably may provide some preliminary information to help plan a larger phase trial in the M0 population.
Will These Agents Find a Greater Role with Localised Disease?
Unlike surgery, there is a clear paradigm for the use of androgen deprivation therapies to improve outcomes in the setting of high-risk disease with primary radiation therapy [47]. Whilst this may seem an avenue to expand the approved indication for these agents, the commercial incentive to do so may be limited by the limited remaining patent in abiraterone in the USA (2014–2017). Thus long-term outcomes are unlikely to be available before the drug becomes generic. Nevertheless, at least for abiraterone, there are a number of small collaborative studies in progress examining the paradigm of neoadjuvant treatment that may generate preliminary data to allow a collaborative group trial, for example Clinicaltrials.gov NCT00924469 (A Phase 2 study of Neoadjuvant Abiraterone Acetate Plus Leuprolide Acetate in Men With Localized High Risk Prostate Cancer pre prostatectomy—58 patients), Clinicaltrials.gov NCT01088529 (A Randomized, Open-Label, Neoadjuvant Prostate Cancer Trial of Abiraterone Acetate Plus LHRHa Versus LHRHa Alone pre Prostatectomy—66 patients) and Clinicaltrials.gov NCT01023061 (Abiraterone Acetate, Prednisone, and Leuprolide Acetate or Goserelin Before and During Radiation Therapy in Treating Patients With Localized or Locally Advanced Prostate Cancer—25 patients).
The critical scientific issue is whether the further reduction in functional androgen signalling achievable with CYP 17 inhibition or MDV3100 will further add to the putative mechanisms of synergy with radiation to achieve better outcomes. There is only limited, indirect literature to suggest that this may be an effective strategy. Zagars et al. [48] examined the outcome—local, nodal, distant metastatic and biochemical—for 486 men with clinically localised prostate cancer treated with radiation and in whom testosterone measurements were available. They found a highly significant correlation between testosterone level and metastatic relapse. Patients with testosterone level greater than 500 ng/dL had a markedly higher 6-year metastatic rate (16%) than those with a testosterone level of 500 ng/dL or less (4%, P = 0.001). In multivariate analysis, testosterone level was an independent determinant of metastatic relapse, second only to PSA level. In a more contemporary setting, Roach et al. determined that baseline testosterone affected prostate-specific outcomes in men on the RTOG 9,202 and 9,413 studies that examined the use of androgen deprivation therapies (ADT) and enlarged radiotherapy fields, respectively. There was no discernible effect on outcome [49]. Similarly, Taira et al. [50] examined the effect of baseline serum testosterone on brachytherapy outcomes, again without any discernible effect being noted. Taken together, these trends seem to suggest that further reductions in bioavailable testosterone are unlikely to change OS outcomes when combined with standard hormone withdrawal alone (LHRH agonist plus anti-androgen). Whilst we recognise that standard androgen suppression does not suppress androgen-related gene expression within the prostate completely and this may be more effectively carried out with the new generation of drug [51], the critical issue is the mechanism for the synergy with radiation. Conceivably, this may include a reduction in the size of the gland, a decrease in the hypoxic fraction of the gland [52] or a decrease in the proliferating fraction of prostate cells/an increase in apoptosis. The former two are likely to be minimally affected by more potent androgen suppression, however the latter is the most crucial. If subclinical metastases or relatively radio-resistant disease can be induced to undergo apoptosis by more effective androgen withdrawal alone, then this may indeed be a worthwhile strategy. To date, even the preclinical data on the relative increase in apoptosis between near castrate and absolute castrate levels are limited, however greater insight into this issue may come from the recently completed studies of neoadjuvant abiraterone (Clinicaltrials.gov NCT00924469).
There may be greater value of more profound androgen suppression in the adjuvant setting. The optimal duration of ADT in men undergoing RT has been addressed in at least four trials, all of which suggest benefit from prolonged therapy [47, 53–56]. For example, in the largest trial, RTOG 92-02, 1,554 men with T2c-T4 disease received 4 months of goserelin and flutamide (2 months before and during EBRT) and were then randomly assigned to no further therapy or 24 months of additional goserelin. Long-term ADT significantly reduced 10-year rates of biochemical failure, local recurrence, and distant metastases (52% vs. 68%, 12% vs. 22% and 15% vs. 23%, respectively) but not overall survival (54% vs, 52%). In a post hoc analysis, overall 10-year survival was significantly increased with long-term ADT in the subset of men with a Gleason score of 8 to 10 (45% vs. 32%). In contrast, an overall survival benefit with a longer duration ADT was observed in EORTC trial 22961. In this trial, 970 men undergoing RT for locally advanced or node-positive disease received 6 months of complete androgen blockade and were then randomly assigned to observation or 2.5 years of additional treatment with a gonadotropin-releasing hormone agonist. At a median follow-up of 6.4 years, the prolonged course of ADT was associated with a significant decrease in overall mortality compared to the 6-month course of treatment (5-year mortality rate 15.2% vs. 19.0%, hazard ratio 0.70, 95% CI 0.55–0.92). Therefore, increasing the potency of androgen suppression in this setting may impact OS and PFS when compared to standard therapies.
Given the data in this setting for radiation, the role of more profound androgen deprivation with surgery is more complex. Neoadjuvant hormonal therapies have been extensively attempted in prostate cancer, without success [57, 58], whilst adjuvant hormonal therapy has become a de facto standard of care for men with PSA relapse, or even high risk disease [59] albeit only with a poor evidence base [60, 61]. If more profound androgen deprivation is to find a role, it is provocative to hypothesise that similar to radiation there may be a defined role in high-risk prostate cancer for adjuvant hormonal therapies.
Can We Use Biomarkers to Target Those Most Appropriate to Derive Benefit?
Ultimately, the aim of personalised medicine would be to only give appropriately targeted drugs to appropriate patients. Given the significant overall survival benefit seen from abiraterone, such a biomarker would have to have an excellent negative predictive value to warrant withholding the drug from patients with CRPC. However, to date, biomarker evaluation in this setting is still only in the preliminary stages. Higher pretreatment serum levels of DHEA, DHEA-S and androstenedione as well as estradiol levels are associated (P < 0.05) with a 50% PSA response and time to PSA progression suggesting that the contribution of adrenal androgens to hormone resistance is important [62]. Furthermore, there is an association between the presence of the TMPRSS2-ERG gene rearrangement in circulating tumour cells (CTCs) and both the magnitude of PSA decline and fall in CTC counts following treatment with abiraterone. Evaluation of ERG gene status in 77 patients treated on abiraterone acetate phase I/II clinical trials reported that 12 out of 15 patients who had a 90% PSA decline had an ERG gene rearrangement [63].
Potentially greater insight into prostate cancer biology could be carried out if metastases were easier to biopsy. Unfortunately, given the predominantly bony location, this is often problematic. To date only one study of serial bone marrow biopsies has appeared in the literature. Efstathiou et al. demonstrated no correlation between baseline serum testosterone (median 23.7 ng/dL, range <10–41) and bone marrow aspirate testosterone levels (median 15.4 ng/dL, range <10–81; Spearman’s r 0.28, P = 0.2), however depleted baseline bone marrow testosterone (<10 ng/dL) did correlate with early progression (P = 0.05). In contrast, an “intracrine androgen-signalling signature” (higher BM-T, AR, CYP 17 expression) favoured treatment benefit (P = 0.048). Interestingly, loss of BM CYP17 expression was observed at progression following benefit in 6 out of 11 patients in whom posttreatment biopsies were available [64]. To date, this remains an impractical approach to target therapies to patients and more extensive investigations will require a greater understanding of both de novo and developed mechanisms of androgen resistance and better mechanisms to understand evolving tumour biology such as circulating tumour cells.
Conclusions
The androgen axis remains the most important “targeted therapy” in prostate cancer care, and despite being discovered over 70 years ago, it remains valid to contemporary practice. The recent approval of novel therapeutics targeting this pathway as well as a strong development pipeline suggest that it will remain relevant to prostate cancer therapeutics indefinitely and ultimately may contribute to long-term disease maintenance. Novel targets within the androgen pathway such as AR chaperone proteins, different aspects of the AR receptor and more potent combination therapies are likely to form the development pipeline in the next 5–10 years.
References
Scher HI, Sawyers CL (2005) Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol 23(32):8253–8261
Chen Y, Sawyers CL, Scher HI (2008) Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol 8(4):440–448
Pienta KJ, Bradley D (2006) Mechanisms underlying the development of androgen-independent prostate cancer. Clin Cancer Res 12(6):1665–1671
Feldman BJ, Feldman D (2001) The development of androgen-independent prostate cancer. Nat Rev Cancer 1(1):34–45
Mostaghel EA, Nelson PS (2008) Intracrine androgen metabolism in prostate cancer progression: mechanisms of castration resistance and therapeutic implications. Best Pract Res Clin Endocrinol Metab 22(2):243–258
Mohler JL et al (2004) The androgen axis in recurrent prostate cancer. Clin Cancer Res 10(2):440–448
Attard G, Richards J, de Bono JS (2011) New strategies in metastatic prostate cancer: targeting the androgen receptor signaling pathway. Clin Cancer Res 17(7):1649–1657
Hu R, Denmeade SR, Luo J (2010) Molecular processes leading to aberrant androgen receptor signaling and castration resistance in prostate cancer. Expert Rev Endocrinol Metab 5(5):753–764
Visakorpi T et al (1995) In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet 9(4):401–406
Mellado B et al (2009) Molecular biology of androgen-independent prostate cancer: the role of the androgen receptor pathway. Clin Transl Oncol 11(1):5–10
Taylor BS et al (2010) Integrative genomic profiling of human prostate cancer. Cancer Cell 18(1):11–22
Ryan CJ et al (2010) Phase I clinical trial of the CYP17 inhibitor abiraterone acetate demonstrating clinical activity in patients with castration-resistant prostate cancer who received prior ketoconazole therapy. J Clin Oncol 28(9):1481–1488
Attard G et al (2008) Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol 26(28):4563–4571
Danila DC et al (2010) Phase II multicenter study of abiraterone acetate plus prednisone therapy in patients with docetaxel-treated castration-resistant prostate cancer. J Clin Oncol 28(9):1496–1501
Ryan CJ et al (2011) Phase II study of abiraterone acetate in chemotherapy-naive metastatic castration-resistant prostate cancer displaying bone flare discordant with serologic response. Clin Cancer Res 17(14):4854–4861
de Bono JS et al (2011) Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 364(21):1995–2005
Chen H et al (2003) Can older cancer patients tolerate chemotherapy? A prospective pilot study. Cancer 97(4):1107–1114
Matsunaga N et al (2004) C(17,20)-lyase inhibitors. Part 2: design, synthesis and structure–activity relationships of (2-naphthylmethyl)-1H-imidazoles as novel C(17,20)-lyase inhibitors. Bioorg Med Chem 12(16):4313–4336
Dreicer R, Agus DB, MacVicar GR, Wang J, MacLean D, Stadler WM (2010) Safety, pharmacokinectics, and efficacy of TAK-700 in metastatic castration-resistant prostate cancer: a phase I/II open-label study. In American Society of Clinical Oncology Genitourinary Symposium: San Francisco
Scher HI et al (2010) Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1–2 study. Lancet 375(9724):1437–1446
Higano CS, Beer TM, Taplin M, Efstathiou E, Anand A, Hirmand M, Flesher M, Scher HI (2011) Antitumor activity of MDV3100 in pre- and post-docetaxel advanced prostate cancer: long-term follow-up of a phase I/II study. In 2011 Genitourinary Cancers Symposium, American Society of Clinical Oncology: Orlando, Florida
Swanson HI et al (2010) Targeting drug-metabolizing enzymes for effective chemoprevention and chemotherapy. Drug Metab Dispos 38(4):539–544
Cai C et al (2011) Intratumoral de novo steroid synthesis activates androgen receptor in castration resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res 71:6503
Kuruma H, Matsumoto H, Zoubeidi A, Thomas C, Lamoureux F, Gleave M (2011) Use of MDV3100 to establish androgen-receptor antagonist resistant LNCaP cells for modelling castrate-resistant progression. American Association of Cancer Research, Orlando
Guo Z, Qiu Y (2011) A new trick of an old molecule: androgen receptor splice variants taking the stage?! Int J Biol Sci 7(6):815–822
Haile S, Sadar MD (2011) Androgen receptor and its splice variants in prostate cancer. Cell Mol Life Sci 68(24):3971–3981. doi:10.1007/500018-011-0766-7
Hu R, Isaacs WB, Luo J (2011) A snapshot of the expression signature of androgen receptor splicing variants and their distinctive transcriptional activities. Prostate 71(15):1656–1667
Watson PA et al (2010) Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A 107(39):16759–16765
Carver BS et al (2011) Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 19(5):575–586
Mulholland DJ et al (2011) Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 19(6):792–804
Sadar MD (2011) Small molecule inhibitors targeting the “Achilles’ heel” of androgen receptor activity. Cancer Res 71(4):1208–1213
Andersen RJ et al (2010) Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 17(6):535–546
Quayle SN et al (2007) Androgen receptor decoy molecules block the growth of prostate cancer. Proc Natl Acad Sci U S A 104(4):1331–1336
Jenster G et al (1991) Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol Endocrinol 5(10):1396–1404
Sadar MD et al (2008) Sintokamides A to E, chlorinated peptides from the sponge Dysidea sp. that inhibit transactivation of the N-terminus of the androgen receptor in prostate cancer cells. Org Lett 10(21):4947–4950
Fowler JE Jr, Whitmore WF Jr (1982) Considerations for the use of testosterone with systemic chemotherapy in prostatic cancer. Cancer 49(7):1373–1377
Rathkopf D et al (2008) Phase II trial of docetaxel with rapid androgen cycling for progressive noncastrate prostate cancer. J Clin Oncol 26(18):2959–2965
Manni A et al (1988) Androgen priming and chemotherapy in advanced prostate cancer: evaluation of determinants of clinical outcome. J Clin Oncol 6(9):1456–1466
Boccardo F et al (2011) Chemotherapy versus tamoxifen versus chemotherapy plus tamoxifen in node-positive, oestrogen receptor-positive breast cancer patients. Very late results of the ‘gruppo di ricerca per la chemio-ormonoterapia adiuvante (GROCTA)’ 01-Trial in early breast cancer. Breast Cancer Res Treat 126(3):653–661
Albain KS et al (2009) Adjuvant chemotherapy and timing of tamoxifen in postmenopausal patients with endocrine-responsive, node-positive breast cancer: a phase 3, open-label, randomised controlled trial. Lancet 374(9707):2055–2063
Darshan MS et al (2011) Taxane-induced blockade to nuclear accumulation of the androgen receptor predicts clinical responses in metastatic prostate cancer. Cancer Res 71(18):6019–6029
Gan L et al (2009) Inhibition of the androgen receptor as a novel mechanism of taxol chemotherapy in prostate cancer. Cancer Res 69(21):8386–8394
Tzelepi V et al (2011) Persistent, biologically meaningful prostate cancer after 1 year of androgen ablation and docetaxel treatment. J Clin Oncol 29(18):2574–2581
Mellado B et al (2009) Phase II trial of short-term neoadjuvant docetaxel and complete androgen blockade in high-risk prostate cancer. Br J Cancer 101(8):1248–1252
Ryan CJ, Small EJ (2003) Role of secondary hormonal therapy in the management of recurrent prostate cancer. Urology 62(Suppl 1):87–94
Smith MR et al (2011) Disease and host characteristics as predictors of time to first bone metastasis and death in men with progressive castration-resistant nonmetastatic prostate cancer. Cancer 117(10):2077–2085
Bolla M et al (2009) Duration of androgen suppression in the treatment of prostate cancer. N Engl J Med 360(24):2516–2527
Zagars GK, Pollack A, von Eschenbach AC (1997) Serum testosterone—a significant determinant of metastatic relapse for irradiated localized prostate cancer. Urology 49(3):327–334
Roach M 3rd et al (2010) Baseline serum testosterone in men treated with androgen deprivation therapy and radiotherapy for localized prostate cancer. Int J Radiat Oncol Biol Phys 78(5):1314–1322
Taira AV et al (2011) Serum testosterone kinetics after brachytherapy for clinically localized prostate cancer. Int J Radiat Oncol Biol Phys. doi:10.1016/j.ijrobp.2011.01.027
Mostaghel EA et al (2007) Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer Res 67(10):5033–5041
Milosevic M et al (2007) Androgen withdrawal in patients reduces prostate cancer hypoxia: implications for disease progression and radiation response. Cancer Res 67(13):6022–6025
Laverdiere J et al (1997) Beneficial effect of combination hormonal therapy administered prior and following external beam radiation therapy in localized prostate cancer. Int J Radiat Oncol Biol Phys 37(2):247–252
Hanks GE et al (2003) Phase III trial of long-term adjuvant androgen deprivation after neoadjuvant hormonal cytoreduction and radiotherapy in locally advanced carcinoma of the prostate: the Radiation Therapy Oncology Group Protocol 92-02. J Clin Oncol 21(21):3972–3978
Denham JW et al (2008) Time to biochemical failure and prostate-specific antigen doubling time as surrogates for prostate cancer-specific mortality: evidence from the TROG 96.01 randomised controlled trial. Lancet Oncol 9(11):1058–1068
Horwitz EM et al (2008) Ten-year follow-up of radiation therapy oncology group protocol 92-02: a phase III trial of the duration of elective androgen deprivation in locally advanced prostate cancer. J Clin Oncol 26(15):2497–2504
Aus G et al (2002) Three-month neoadjuvant hormonal therapy before radical prostatectomy: a 7-year follow-up of a randomized controlled trial. BJU Int 90(6):561–566
Klotz LH et al (2003) Long-term follow up of a randomized trial of 0 versus 3 months of neoadjuvant androgen ablation before radical prostatectomy. J Urol 170(3):791–794
Williams SB et al (2011) Utilization and expense of adjuvant cancer therapies following radical prostatectomy. Cancer 117(32):4846–4854
Siddiqui SA et al (2011) Impact of adjuvant androgen deprivation therapy after radical prostatectomy on the survival of patients with pathological T3b prostate cancer. BJU Int 107(3):383–388
Kumar S, Shelley M, Harrison C, Coles B, Wilt TJ, Mason MD (2006) Neoadjuvant and adjuvant hormone therapy for localised and locally advanced prostate cancer. Cochrane Database Syst Rev 4:CD006019
Efstathiou E, Titus M, Tsavachidou D, Tzelepi V, Wen S, Hoang A, Molina A, Chieffo N, Smith LA, Smith MK, Troncoso P, Logothetis CJ (2011) Characterization of castrate resistant prostate cancer treated with abiraterone acetate and prednisone. American Association for Cancer Research, Orlando
Attard G et al (2009) Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol 27(23):3742–3748
Efstathiou E, Tu S, Aparicio A, Hoang A, Wen S, Troncoso P, Smith LA, Chieffo N, Molina A, Logothetis C (2010) Use of “intracrine androgen signaling signature” to predict benefit from abiraterone acetate (AA) in patients with castrate-resistant prostate cancer (CRPC), in 2010 ASCO Annual Meeting. American Society of Clinical Oncology, Chicago
Funding
Funding for this article was supported in part by Prostate Cancer Canada and the Motorcycle Ride for Dad (AMJ).
Declaration of Interest
No author declares any conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
All authors contributed to the instigation, research, writing and review of this paper.
Rights and permissions
About this article
Cite this article
Niraula, S., Chi, K. & Joshua, A.M. Beyond Castration—Defining Future Directions in the Hormonal Treatment of Prostate Cancer. HORM CANC 3, 3–13 (2012). https://doi.org/10.1007/s12672-011-0096-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-011-0096-0