
Abstract
Normally, the insulin secretion is increased in response to insulin resistance in order to maintain glucose homeostasis. Pancreatic islet beta-cells respond to glucose, fatty acids, amino acids, autonomic innervation, incretins, and adipokines. Metabolic syndrome is associated with a failure of pancreatic islets to respond appropriately to nutrient, neuronal, and hormonal signals, resulting in glucose intolerance or type 2 diabetes. Pancreatic islet beta-cell dysfunction is characterized by increased glucagon secretion; impaired insulin response to secretagogues, e.g., glucose, arginine, and isoproterenol; blunted first-phase insulin secretion; irregular oscillations of plasma insulin levels; and impaired conversion of proinsulin to insulin. In addition, type 2 diabetes may be associated with reduced beta-cell mass, partly mediated by enhanced islet apoptosis due to glucolipotoxicity. Understanding of normal pancreatic islet physiology and molecular pathways linking islet adaptation to diabetes pathophysiology would facilitate the development of novel treatment modalities.
Similar content being viewed by others


Keywords
Nutrient Sensing in Pancreatic Islets
The prevalence of the metabolic syndrome is dramatically increasing and has emerged as a major threat to public health worldwide. The metabolic syndrome consists of a cluster of metabolic conditions including hypertriglyceridemia, insulin resistance, abnormal glucose tolerance or diabetes , and hypertension (Reaven 1988, 1995). These conditions combined with genetic susceptibility and abdominal obesity are risk factors for type 2 diabetes , vascular inflammation, atherosclerosis, and renal, liver, and heart diseases. Insulin resistance is at the core of metabolic syndrome, because the altered metabolism of nutrients by insulin-sensitive target tissues (muscle, adipose tissue, and liver) can result in high circulating levels of glucose and various lipids , which increase demands on pancreatic islet function to compensate for insulin resistance. Pancreatic islets are highly vascularized structures that monitor the nutrient content of the blood stream and consist of mainly five cell types: alpha cells, beta-cells, delta cells, ghrelin cells, and pancreatic polipeptide (PP) cells which produce the hormones glucagon, insulin, somatostatin, ghrelin, and PP, respectively (Wierup et al. 2014; Newsholme et al. 2014). Insulin -secreting pancreatic beta-cells play a central role in the physiology and in the pathology of obesity and diabetes by regulating glucose homeostasis. A critical role of pancreatic beta-cells is consistent with the observation that diabetes does not develop in obese insulin-resistant persons unless pancreatic beta-cell function or its adaptation is compromised.
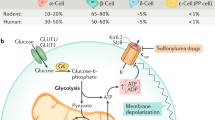
Pancreatic beta-cells account for about 50 % of the islet cell mass in humans and are able to react to elevated dietary nutrients on a moment-to-moment basis and secrete insulin into the blood stream at rates that are appropriate for the maintenance of optimal glucose levels. Carbohydrates are normally the primary source of fuel in food, and glucose is the primary insulin secretagogue. An increase in blood glucose concentration leads to glucose transport into beta-cells by Na+-independent facilitated glucose transporters (GLUTs) with a capacity markedly higher than the beta-cell glycolytic rate. GLUT-2 is expressed in rodent islets (Newgard et al. 2001) and has a Km for glucose and the capacity for glucose transport higher than other members of the family. Human islets express GLUT-2, but at lower levels than rodent islets (Newgard et al. 2001). In addition, human islets express significant levels of the low Km glucose transporters GLUT-1 and GLUT-3. Upon entry of glucose into beta-cells, the glucose is phosphorylated to glucose-6-P by glucokinase which is the rate determinant of glycolysis (Matschinsky 1996). Glucokinase, also known as hexokinase IV, contributes more than 90 % of the glucose-phosphorylating capacity in beta-cells. Enhanced flux through the glycolytic pathway and tricarboxylic acid (TCA) cycle results in elevated mitochondrial ATP generation. ATP can be produced by three different mechanisms in beta-cells (Newgard et al. 2001): (i) a large fraction of NADH produced in the glyceraldehyde phosphate dehydrogenase reaction can be transferred to mitochondria for entry into electron transport chain via alpha-glycerophosphate and aspartate/malate shuttles, (ii) ATP is generated in the phosphoglycerate kinase and pyruvate kinase reaction of glycolysis, and (iii) ATP is produced in mitochondria from oxidation of pyruvate. The increased ATP/ADP ratio induces plasma membrane depolarization by closure of beta-cell KATP-sensitive channels and subsequently leads to the opening of voltage-gated calcium channels. The resultant influx of Ca2+ leads to insulin export through the fusion of a readily releasable pool of insulin-containing vesicles with the plasma membrane (Henquin 2009; Ashcroft et al. 1984; Wollheim and Pralong 1990; Fig. 1). This triggering mechanism of KATP-dependent glucose-stimulated insulin release is responsible for the first phase of insulin secretion (over 5–10 min). The second phase insulin release is longer (30–60 min) and dependent on metabolic stimulus-secretion coupling (Newsholme et al. 2014; Henquin 2000).

Signaling in pancreatic islet beta-cell. Free fatty acid (FFA), glucose, amino acids (AAs), acetylcholine (Ach), and GLP-1 signaling pathways in pancreatic beta-cells are depicted. GPR40 G-protein-coupled receptor for FFA, M 3 R muscarinic receptor type 3, GLP-1R glucagon-like peptide-1 receptor, VDCC voltage-dependent Ca2+ channels, IP3 inositol triphosphate, G-6-P glucose-6-phosphate, Gro3P glycerol-3-phosphate, GL/FFA glycerolipid/FFA cycle, PKC protein kinase C, PKA protein kinase A, Ca2+, ATP adenosine triphosphate, AC adenylate cyclase, phosholipase C, IP 3 inositol triphosphate, MAG monoacylglycerol, DAG diacylglycerol, cAMP-GEF II cAMP-guanine nucleotide exchange factor II, ER endoplasmic reticulum
Pancreatic beta-cells respond to other nutrients such as amino acids, fatty acids (FAs), and ketones. Amino acids administrated alone at physiological concentrations do not affect glucose-stimulated insulin secretion (GSIS), but interact with glucose to increase GSIS (Newsholme et al. 2007a, 2014; Doliba et al. 2007). Amino acids regulate both triggering and amplification pathway of insulin secretion by acting as a substrate for the TCA cycle and/or redox shuttles with subsequent generation of ATP (Newsholme et al. 2007a; Li et al. 2003), direct depolarization of plasma membrane by the transport of positively charged amino acids into the cell via specific amino acid transporter (Newsholme et al. 2007a), and cotransport of Na+ ions resulting in plasma depolarization (Newsholme et al. 2007a) and through amino acid receptors of the plasma membrane, in particular the glutamate receptor expressed in beta-cells (Inagaki et al. 1995; Wollheim and Maechler 2015).
Fatty acids (FAs) alter pancreatic beta-cell function by signaling through the free FA (FFA) receptor: GPR40/FFAR1 (G-protein-coupled receptor 40) that has been shown to be a physiologically relevant receptor for long-chain FAs (Itoh et al. 2003; Soga et al. 2005; Kristinsson et al. 2013). GPR40/FFAR1 belongs to a family of G-protein-coupled receptors that are highly expressed in beta-cells and mediate approximately half of the FFA-induced secretion (Ferdaoussi et al. 2012; Kebede et al. 2008; Latour et al. 2007; Nolan et al. 2006) where it increases insulin secretion by signaling via Gαq and phospholipase C, IP3-mediated Ca2+ release from ER, and stimulation of PKC by increased level of diacylglycerol (DAG) (Kristinsson et al. 2013; Fujiwara et al. 2005; Fig. 1). The second pathway involves FAs entering into the intermediary metabolism of beta-cell (Prentki et al. 2013). At a low glucose concentration, the FAs are converted into long-chain acyl-CoA by the enzyme acyl-CoA synthetase (ACS) and enter the mitochondria through carnitine palmitoyltransferase I (CPT-1), where they are oxidized via the β-oxidation pathway for energy production. When the glucose concentration increases in pancreatic β-cells, FA oxidation is decreased and glucose oxidation fulfills a larger part of the cellular energy needs. The shift in fuel utilization occurs due to a high substrate flux to the TCA cycle that leads to an increase in anaplerosis and efflux of citrate from the mitochondria. Cytosolic citrate is converted into malonyl-CoA by citrate lyase and acetyl-CoA carboxylase (ACC). Malonyl-CoA is a potent allosteric inhibitor of mitochondrial CPT-1 and therefore inhibits the transport of long-chain acyl-CoA into the mitochondria to be oxidized (Prentki et al. 1992; Liang and Matschinsky 1991). The malonyl-CoA/CPT-1/FA-CoA interaction is connected to the glycerolipid/free fatty acid cycle (GL/FFA) (Prentki et al. 2013; Prentki and Madiraju 2008, 2012). At a high glucose concentration, a substantial portion of glucose utilization by pancreatic β-cells (about 30 %) is incorporated into glycerol (GL) via glycerol-3-phosphate (Gro3P) and enters GL/FFA cycling (Peyot et al. 2010; Fig. 1). The GL/FFA cycle consists of lipogenesis (esterification) and lipolysis components that generate many lipid intermediates, some of which serve as signaling molecules, e.g., monoacylglycerol (MAG) , diacylglycerol (DAG) , phosphatidate, lysophosphatidate, FA-CoA, and FFA (Prentki and Madiraju 2008). Specifically, DAG and LC-CoA enhance the exocytotic function of key vesicle priming and docking proteins such as MUNC13, synaptosomal-associated protein 25 (SNAP-25), and synaptotagmin and also modulate signal transduction by PKC activity (Nolan and Prentki 2008; Newsholme et al. 2007b; Rorsman and Braun 2013).
What is the interaction between the GL/FFA cycle and the malonyl-CoA/CPT-1/FA-CoA network? Elevated blood glucose, occurring in the fed condition, enhances GL/FFA cycling by increasing malonyl-CoA that inhibits β-oxidation, providing Gro3P for the esterification arm of the cycle, and activating lipolysis via covalent modification of lipolytic enzymes. Amino acids also enhance anaplerosis and malonyl-CoA production, and exogenous FFAs provide substrate for the GL/FFA cycle. These nutrients act together with glucose to promote GL/FFA cycle activity and production of metabolic coupling factors (Prentki et al. 2013). Thus, the malonyl-CoA/FA-CoA-GL/FFA cycle metabolic signaling network likely plays an integrating role in modulating insulin secretion in response to all classes of fuel stimuli to adjust insulin secretion as a function of the nutritional state (Prentki and Madiraju 2012). Thus malonyl-CoA acts to switch pancreatic islet beta-cell metabolism from FA oxidation to glucose oxidation.
In addition, pancreatic islets express hormone-sensitive lipase (HSL) which may activate endogenous lipolysis and also participate in the regulation of insulin secretion (Prentki et al. 2013). Therefore, triacylglycerol (TAG) stored in pancreatic islet beta-cells plays an important role in stimulus-secretion coupling mechanism of GSIS. It has been shown that both glucose and FA metabolism are needed for normal islet beta-cell function (Prentki et al. 1992). If malonyl-CoA accumulation is blocked by the inhibition of acetyl-CoA carboxylase (ACC) , GSIS is markedly reduced.
The mechanisms described above are related to acute exposure to FFA. In contrast, chronic exposure of pancreatic islet beta-cells to FFA results in inhibition of insulin secretion as shown in vitro in isolated perfused pancreas and islets (Sako and Grill 1990; Zhou and Grill 1995; McGarry 2002) and in vivo studies in humans (Kashyap et al. 2003). This phenomenon has been termed lipotoxicity (Prentki et al. 2002; Poitout and Robertson 2002). Chronic exposure to FFA enhances the basal insulin secretion but decreases the response to glucose. Chronic elevation of FFA also decreases insulin gene expression, proinsulin processing, and induction of islet apoptosis (Newsholme et al. 2007b; El-Assaad et al. 2003; Lupi et al. 2002). Chronic exposure to high glucose and FFA may also lead to ceramide formation and/or NO-mediated apoptosis (Newsholme et al. 2007b; Boslem et al. 2012). This pathway plays a role in the development and pathogenesis of pancreatic islet beta-cell dysfunction in type 2 diabetes (McGarry 2002; McGarry and Dobbins 1999).
FA receptors may also play a role during prolonged exposure to FFA (Kristinsson et al. 2013). Indeed, it was shown that GPR40/FFAR1 knockout (KO) mice generated by Steneberg et al. (2005) did not develop metabolic abnormalities seen in wild-type animals when given a high-fat diet. When islets isolated from the GPR40/FFAR1-KO mice were chronically exposed to elevated levels of FFAs, subsequent GSIS was not impaired. In another study, FFAR1-KO mice generated by another laboratory showed better glucose tolerance after 1 week of high-fat diet compared to wild-type mice (Brownlie et al. 2008). In contrast to these results, FFAR1-KO mice generated by Kebede et al. (2008) and Lan et al. (2008) developed obesity and hyperglycemia. Islets isolated from FFAR1-KO mice were not protected from impairment of GSIS caused by prolonged exposure to elevated levels of FFAs (Latour et al. 2007). Overexpression of FFAR1 in rats was favorable for glycemic control on a high-fat diet (Nagasumi et al. 2009). A number of FFAR1 agonists have been shown to stimulate insulin secretion in a glucose-dependent manner and to lower glucose levels in obese and diabetic rats and mice (Lin et al. 2011; Houze et al. 2012; Luo et al. 2012; Tsujihata et al. 2011; Yashiro et al. 2012). These results demonstrate that pancreatic islet beta-cells are adversely affected by chronic exposure to high glucose and FFA levels which predisposes to impaired insulin secretion and the development of glucose intolerance and type 2 diabetes.
Neuroendocrine Regulation of Pancreatic Islets
In addition to various nutrients, insulin secretion is stimulated by hormones and neurotransmitters. Acetylcholine , the neurotransmitter of the parasympathetic nervous system, plays a key role in the regulation of insulin secretion in pancreatic islet β-cells (Gilon and Henquin 2001; Ahren 2000; Teff and Townsend 1999). Mutant mice lacking the M3 muscarinic acetylcholine receptor subtype in beta-cells display impaired glucose tolerance and reduced insulin release (Gautam et al. 2006; Zawalich et al. 2004). In contrast, transgenic mice overexpressing M3 receptors in β-cells showed an improvement in glucose tolerance and insulin secretion (Gautam et al. 2007). The secretory response of β-cells to fuel stimulation is also markedly enhanced by the gut hormone GLP-1, an incretin released into the portal circulation when a meal is digested (Holz and Habener 1992). These neuroendocrine signals are mediated by specific G-protein-coupled receptors (GPCRs) of the beta-cells (Ahren 2009; Regard et al. 2007; Fig. 1). The binding of various ligands activates specific subgroups of heterotrimeric G proteins, Gs, Gi, and Gq, involved in distinct pathways of signal transmission (Regard et al. 2007; Lagerstrom and Schioth 2008; Kimple et al. 2014).
Signaling by Gs (activated by the GLP1 receptor) (West et al. 2014; Doyle and Egan 2007) and Gq (activated by M3 acetylcholine receptor) (Gilon and Henquin 2001; Ahren 2000; Nakajima et al. 2013; Jain et al. 2013) potentiates fuel-stimulated insulin release during the course of a meal and also stimulates beta-cell proliferation and enhances beta-cell mass (Baggio and Drucker 2006, 2007) as compensation for insulin resistance associated with obesity, the major precipitating factor for type 2 diabetes (Baggio and Drucker 2006). Various lines of evidence suggest that defects of the neuroendocrine regulation of beta-cells play an important role in the molecular pathogenesis of type 2 diabetes (Lee et al. 2012). There is evidence that both release and action of incretin hormones are disrupted in type 2 diabetes (Drucker and Nauck 2006; Drucker 2006; Vilsboll et al. 2001). This defect is partly attributed to reduced expression of GLP1 receptor in pancreatic beta-cells (Rajan et al. 2015). Activation of the vagal afferent pathway is also impaired in a rodent model of type 2 diabetes (Lee et al. 2012; Rocca and Brubaker 1999). Previously we demonstrated in vitro with isolated mouse islets that palmitic acid acutely reduced the glucose-dependent acetylcholine stimulation of insulin release, the total oxygen consumption response to glucose, the Ca2+ response, and cAMP metabolism in isolated mouse islets, while the effects of GLP-1 on these parameters were not altered or potentiated (Doliba et al. 2010). These effects occurred at concentrations of albumin-bound palmitic acid as low as 50 μM, thus implicating the activation of GPR-40 receptors. We have also shown that chronic exposure of pancreatic islets to high glucose and FA concentrations decreased the ability of acetylcholine to potentiate GSIS (Doliba et al. 2015). This effect strongly depends on the glucose concentration in the culture medium with a delayed onset of potentiation at 10 mM glucose and a delayed onset plus reduced maximal effectiveness of the neurotransmitters at 16 and 25 mM concentrations of glucose. Based on the significant contribution of cholinergic regulation to insulin secretion and glucose homeostasis in humans (Gilon and Henquin 2001; Ahren 2000), it was proposed that impairment of this pathway by FA may contribute to the lack of compensatory insulin release in response to insulin resistance.
Pancreatic Islet Adaptation in Pregnancy and Obesity
The endocrine pancreas is a unique organ that can adapt to physiological and pathological conditions by changing its mass and function to ensure glucose homeostasis (Steiner et al. 2010; Lingohr et al. 2002; Karaca et al. 2009). Two types of compensation can occur: a functional one in which beta-cells secrete more insulin and the second one in which there is a change in beta-cell mass (Bonner-Weir 2000). Functional adaptations, including changes in the threshold for glucose-induced insulin secretion (Sorenson et al. 1987) and glucose-induced increase in glucokinase activity (Chen et al. 1994), are involved in the maintenance of glucose homeostasis . After stimulation by high glucose levels, the proinsulin synthesis in beta-cells is increased by more than tenfold, with hormone synthesis approaching 50 % of the total protein production (Schuit et al. 1988). The beta-cell mass is a major determinant of the amount of insulin that can be secreted, and experimental evidence shows that the beta-cell mass can increase or decrease (Bonner-Weir 2000). An adaptive increase in beta-cell mass is well illustrated in pregnancy and obesity (Rhodes 2005; Bernard-Kargar and Ktorza 2001; Sorenson et al. 1997). In mammals, including humans, pregnancy results in profound changes in maternal metabolism and insulin secretion to allow an optimal nutrient supply to the fetus. During the last trimester, there is marked insulin resistance accompanied by a dramatic increase in the insulin response to glucose and doubling of the beta-cells mass (Parsons et al. 1992). Failure to compensate for the high insulin demand during pregnancy leads to gestational diabetes.
Pancreatic beta-cell plasticity also occurs in obesity. Although obesity is associated with insulin resistance, most obese individuals remain normoglycemic because of a compensatory increase in beta-cell function and mass to cope with the increase in metabolic status (Sorenson et al. 1997) (Ackermann and Gannon 2007). This may explain why 70–75 % of obese individuals do not develop diabetes (Mokdad et al. 2001). Pancreatic islet beta-cell adaptation has been documented in several animal models. Placing normal rats on a high-fat diet for 6 weeks results in a modest increase in body weight, mild insulin resistance, and a 30–40 % increase in islet density and beta-cell size (Buettner et al. 2000). Pancreatic islet beta-cell compensation and expansion of beta-cell mass is also well demonstrated in Zucker fatty (ZF) rats, which possess a leptin-receptor defect that results in obesity and insulin resistance (Clark et al. 1983). However, these animals remain normoglycemic via compensatory hyperinsulinemia . Adaptation to insulin resistance occurs via a fourfold increase in the beta-cell mass (Pick et al. 1998) together with enhanced insulin secretion (Milburn et al. 1995; Zhou et al. 1999), thereby allowing the maintenance of glucose homeostasis. However, as the animals get older, more obese, and glucose intolerant, there is a deficit in the beta-cell population that no longer adequately compensates for the insulin resistance. This worsens with age, so the animals eventually become diabetic (Pick et al. 1998; Unger and Orci 2001).
Previously we have studied pancreatic islet adaptation in diet-induced obese (DIO) C57BL/6 J mice (Roat et al. 2014; Imai et al. 2008). This mouse strain mimics human obesity (Surwit et al. 1988) and develops hyperinsulinemia and mildly elevated blood glucose levels indicating a substantial capacity to compensate for insulin resistance (Roat et al. 2014). Hyperinsulinemia in DIO C57BL/6 J mice is associated with an increase in islet mass and size and increased BrdU incorporation in beta-cells, indicating hyperplasia (Roat et al. 2014). Although studies in humans are limited, a morphometric study of autopsy pancreases from diabetic and nondiabetic patients (Kloppel et al. 1985) has showed that the β-cell mass was increased by 40 % in the obese subjects as compared with lean subjects. These results suggest that there is a compensatory growth of beta-cell mass in response to insulin resistance in obesity.
Glucose , FFA, and some hormones and neurotransmitters are the most physiologically relevant stimuli for beta-cell proliferation and mass increase in vitro and in vivo (Baggio and Drucker 2006; Hugl et al. 1998; Shimabukuro et al. 1998; Brubaker and Drucker 2004; Thorens 2014). A prolonged (48-h) glucose infusion in normal rats led to a twofold increase in beta-cell mass as a result of both hypertrophy and hyperplasia (Bernard et al. 1998), and it was associated with a marked increase in islet responsiveness to glucose (Bernard et al. 1998). In the obese and/or insulin-resistant state , chronic hyperglycemia and hyperlipidemia can evoke beta-cell apoptosis leading to decreased beta-cell mass (Pick et al. 1998; Donath et al. 1999). In rodent models, FFAs can cause a modest increase in beta-cell proliferation at basal glucose concentrations (Shimabukuro et al. 1998). However, long-term exposure of beta-cells to FFAs inhibits beta-cell mitogenesis and induces beta-cell apoptosis (Cousin et al. 2001). These adverse effects of FFAs on beta-cell growth could be mediated via intracellular accumulation of long-chain CoA or ceramides (Shimabukuro et al. 1998).
GLP-1 increases beta-cell mass by activating beta-cell proliferation and differentiation and inhibiting beta-cell apoptosis (Baggio and Drucker 2006; Brubaker and Drucker 2004). These actions are associated with GLP-1 receptor agonists and dipeptidyl peptidase-4 (DPP-4) inhibitors (Drucker and Nauck 2006; Drucker 2013). However, it should be noted that the majority of experiments were carried out in younger animals (Campbell and Drucker 2013) whereas older rodent beta-cells exhibit a substantially diminished or absent proliferative response to multiple regenerative stimuli, including GLP-1 receptor agonists (Rankin and Kushner 2009). Human beta-cells are less responsive to the proliferative action of GLP-1 compared to rodent beta-cells (Parnaud et al. 2008). More work is required to understand whether older diabetic human beta cell retain the capacity to proliferate, resist cell death, or retain a functionally differentiated state in response to GLP-1 agonists (Drucker 2013).
It has been also shown that both parasympathetic and sympathetic nervous systems influence the postnatal development and plasticity of the endocrine pancreas (Thorens 2014). Defects in these autonomic pathways impair beta-cell mass expansion during the weaning period and beta-cell mass adaptation in adult life (Thorens 2014). Various growth factors have been implicated in increasing adult pancreatic beta-cell proliferation and beta-cell neogenesis (Lingohr et al. 2002). The best characterized and physiologically relevant growth factors that increase beta-cell proliferation are insulin-like growth factor (IGF-1), growth hormone (GH), and GLP-1. The IGF and GH signal transduction pathway is described in detail in a review article by Lingohr et al. (2002).
Glucolipotoxicity and Diabetes
Hyperglycemia develops when pancreatic islet beta-cells fail to synthesize and secrete sufficient insulin for maintaining the physiological glucose concentration of 5 mM. Glucolipotoxicity, the operationally defined condition resulting from caloric overload, is proposed to worsen or cause beta-cell damage which eventually leads to type 2 diabetes . The term “glucolipotoxicity” implies that repeated or continued exposure to high blood glucose and lipid levels is required for beta-cell damage and functional dysfunction to occur. However, a compelling mechanistic molecular explanation of glucolipotoxicity effecting pancreatic beta-cells is still lacking. In an attempt to model glucolipotoxicity in vitro, pancreatic islets are usually cultured for several days in high glucose and FA concentrations. Studies have described multiple cellular processes involved in the pathogenesis of beta-cell dysfunction, including changes in gene expression (Biden et al. 2004; Gremlich et al. 1997), intermediary metabolism (Iizuka et al. 2002), mitochondrial function (Koshkin et al. 2003), ion channel activity (Branstrom et al. 1997, 1998; Reimann et al. 2003), insulin synthesis, and exocytosis (Somesh et al. 2013). Different molecular mechanisms of FA-induced beta-cell dysfunction have been proposed including accumulation of ceramide (Boslem et al. 2012), apoptosis of beta-cells due to oxidative (Gehrmann et al. 2010; Morgan et al. 2007) and ER stress (Kharroubi et al. 2004; Cnop et al. 2008), as well as others mechanisms (Joseph et al. 2004; Cnop et al. 2005). Many of these mechanisms remain controversial. For example, the exposure of human islets for 24 h in elevated FA and glucose conditions was found in one study to initiate apoptosis (El-Assaad et al. 2003), but other studies have failed to find evidence of any significant apoptosis following long-term exposure to FAs (Kelpe et al. 2003). Olofsson et al. (2007) reported that inhibition of GSIS by long-term exposure to the FAs oleate and palmitate was not related to any signs of increased beta-cell death, reduced insulin synthesis, and impaired glucose metabolism, KATP channel regulation, or Ca2+ signaling. These discrepancies could be due to differences in acute or chronic islet experimentations. In vitro albumin/FA ratios are often not optimal, glucose concentrations are often excessive to be meaningful (i.e., >16 mM), there are significant limitations inherent in animal models, and there is a lack of a clear definition of the glucolipotoxicity phenomenon (Poitout et al. 1801). While various molecular and cellular mechanisms of glucolipotoxicity and their roles in obesity and diabetes have been described (Prentki et al. 2013; Prentki and Madiraju 2012; Poitout et al. 1801; Poitout and Robertson 2008), it is unclear whether these models recapitulate the pathogenesis of human type 2 diabetes.
Pancreatic Islet Bioenergetics and Diabetes
A faulty bioenergetic process is a plausible explanation for defective insulin secretion in type 2 diabetes. Normally the ATP, generated by metabolism of glucose, amino acids, and probably FAs, serves as the obligatory coupling factor in fuel-stimulated insulin release involving beta-cell-specific mechanisms (Ashcroft et al. 1984). The unique role of ATP as a critical messenger in the stimulus-secretion coupling was clearly shown in our previous studies with mouse, rat, and human islets where oxygen consumption, glycolysis, and glucose oxidation were related to insulin secretion (Doliba et al. 2012). Such measurements allowed us to calculate the ATP production rate in beta-cells as a function of the glucose concentration and insulin secretion. Making a reasonable assumption that islet glycogen stores are negligible (Matschinsky et al. 1971) and that coupling of oxidative phosphorylation is intact, we were able to develop an islet “ATP production/insulin secretion” curve (Fig. 2). Despite major differences in insulin profiles (Fig. 2a), the ATP production/insulin secretion curves were similar for mouse, rat, and normal human islets, and the data for all species fit a single sigmoidal curve (Fig. 2b), showing a clear relationship between the pancreatic islet energy (ATP) production rate and insulin secretion (Doliba et al. 2012).

Mouse, rat, and human islet ATP production/insulin secretion curves. Panel A: Secretion profiles of mouse, rat, and human islets as a function of the glucose concentration. Panel B: combined energy production/insulin secretion curve with data on ATP production and insulin release from all three types of islets projected on a single continuous sigmoidal curve. The ATP production was calculated based on oxygen consumption and glucose usage data
The ATP production/insulin secretion curve is steeply sigmoidal and has a threshold for glucose-stimulated insulin secretion at about 15 pmole/islet/min, a Hill coefficient of about 11 – which is an indication of highly cooperative coupling mechanisms – and a half-maximal effective rate (ER50) of about 25 pmole/islet/min. These values imply that ATP turns over about twice at the threshold and about four times at maximal glucose stimulation, but that ATP turnover is changed relatively little (perhaps not more than 30 %) in the physiologically important segment of the curve. We speculate that the “ATP production/insulin secretion” curve has clinical significance comparable to that of the classical “Frank/Starling” curve of the heart. In the heart, it is the venous return and the ensuing muscle fiber stretch which determines cardiac output maintaining the match between the pump rate and body oxygen requirements. In pancreatic islet beta-cells, it is the fuel load and the ensuing ATP turnover that determines the insulin output and maintains glucose homeostasis . We also showed that ATP production/insulin secretion curve is modified by GLP-1 and a glucokinase activator piragliatin (Doliba et al. 2012). We speculate that the “ATP production/insulin secretion” curve is modified in type 2 diabetic islets.
The literature proposes various ways by which mitochondrial energy metabolism can be altered due to fuel overload. It has been shown that FAs may act as uncouplers and inhibitors of mitochondrial respiration (Wojtczak and Schonfeld 1993), operating as protonophors and by inhibiting the electron transport, respectively (Schonfeld and Reiser 2006; Schonfeld and Wojtczak 1767, 2008). In addition, FAs induce uncoupling protein (UCP2) in pancreatic islets (Joseph et al. 2004; Chan et al. 2004; Zhang et al. 2001). FAs may act as complex-I-directed inhibitors (Loskovich et al. 2005) and can also serve as a substrate for transport by the ANT inhibiting ATP and ADP exchanges (Klingenberg 1778). FAs may alter mitochondrial membrane permeability by opening of the permeability transition pore (Scorrano et al. 2001; Bernardi et al. 2002; Penzo et al. 2002; 2004).
Fatty acids (FAs) increase the expression of PGC-1 alpha which may alter bioenergetics in pancreatic beta-cells (Yoon et al. 2003). PGC-1 alpha is elevated in islets from different animal models of diabetes and in human studies (Yoon et al. 2003; Ek et al. 2001; Oberkofler et al. 2004, 2009). PGC-1 alpha promotes mitochondrial biogenesis in brown tissue (Puigserver et al. 1998); however, adenovirus-mediated expression of PGC-1 alpha to levels similar to those present in diabetic rodents produces a marked inhibition of GSIS in isolated islets and in mice (Yoon et al. 2003), by suppressing glucose oxidation or decreasing the cell’s ability to drive ATP production. PGC-1 alpha increases the transcription of UCP2 (Oberkofler et al. 2006) by PGC-1-mediated upregulation of beta-cell sterol element binding protein (SREBP) gene expression. Since UCP2 modulates the efficiency of ATP production (Klingenberg and Huang 1999) by catalyzing the translocation of protons across the mitochondrial membrane, one should expect changes in oxygen consumption and oxidative ATP synthesis. However, such data are limited and related to insulinoma cells (Barlow and Affourtit 2013). Only measurements of total ATP concentration in islets exposed to FA have been reported (Somesh et al. 2013). Since insulin granules contain ATP, which is co-secreted with insulin, it is difficult to dissociate between the effects of FA on ATP syntheses and changes of ATP content in insulin granules. In fact, the insulin content is decreased in islets chronically exposed to FFA (Somesh et al. 2013), which may result in concomitant decreased total ATP concentration which says little about the functional ATP.
In order to access beta-cell bioenergetics and its relationship to insulin secretion , we performed two sets of experiments: (i) the bioenergetics, ionic, and secretion profiles of pancreatic islets isolated from healthy and type 2 diabetic organ donors were examined and (ii) isolated normal human islets were exposed to a glucolipotoxicity condition (high glucose and FFAs) in organ culture, and the bioenergetics and insulin secretion were studied in perifusion experiments. When islets were exposed to a “staircase” glucose stimulus in the perifusion setup, the glucose dependency curves of insulin secretion (Fig. 3a) and respiration (Fig. 3c) of diabetic islets showed decreased maximal rates and a right shift for the oxygen consumption rate (OCR) as compared to the control islets. It is worthy of note that the baselines for both parameters are comparable. In the insulin secretion profiles, the difference is most pronounced at the 6 and 12 mM glucose steps (Fig. 3b) showing decreased rates of rise and a decreased extent and loss of biphasicity. The glucose dependency of the OCR of the diabetic islets was flat and reduced by 50 % at 24 mM glucose (Fig. 3c). Addition of a mitochondrial uncoupler, FCCP (5 μM), blocked insulin release instantly and transiently increased OCR in control and diabetic islets to the same level, indicating that strong coupling exists between islet respiration and oxidative phosphorylation in both types of islets (Fig. 3c).

Impaired insulin release ( A and B ), oxygen consumption ( C and D ), and intracellular calcium ( E ) of isolated islets from control and type 2 diabetic organ donors. Panel A: shows the insulin release patterns with glucose stimulation using stepwise increases of glucose from zero to 3, 6, 12, and 24 mM. Panel B: magnified view of selected section (85–190 min) of the experiment presented in A to show the loss of the first phase of insulin release in type 2 diabetic islets. Panel C: islet respiration during stepwise of glucose concentration followed by treatment with 5 μM of the uncoupler of Ox/Phos FCCP and 1 mM Na-azide. O2 consumption was determined with a method based on phosphorescence quenching of metalloporphyrins by oxygen (Doliba et al. 2006). Panel D: oxygen consumption rate as function of glucose concentration. Panel E: represents corresponding changes in intracellular Ca2+ of human islets due to stepwise increases of glucose from zero to 1, 3, and 9 mM. The Fura-2 method was employed. Typical experiments are presented (n of the series = 3). Hb A1c levels for the pancreas donors with T2DM were 9.3, 11.0, and 7.4 %. Results are presented as means ± SE (SE when applicable) of three experiments
Oxygen consumption (VO2) of healthy and type 2 diabetic human islets increased sigmoidally as a function of a stepwise rise of glucose concentrations (Fig. 3d). In islets from type 2 diabetics, the maximal stimulation of respiration (Vmax) by glucose was reduced from 0.4 ± 0.02 in control to 0.32 ± 0.01 nmol/min/100islets, and the S0.5 rose from 4.39 ± 0.01 in control to 5.43 ± 0.13 mM (Fig. 3d). Panel E of Fig. 3 presents changes in intracellular [Ca2+]i of human islets. 3 mM glucose caused a transient and 9 mM glucose a biphasic sustained increase in [Ca2+]i. of control islets. Diabetic islets responded only to 9 mM and this response was delayed and significantly lower than in controls.
Together, these data indicate that impaired pancreatic islet beta-cell bioenergetics resulting in reduced ATP production is critical in the molecular pathogenesis of type 2 diabetes. Importantly, the glucokinase activator piragliatin was able to correct the defect of respiration and GSIS (Doliba et al. 2012). In our second experiment, the glucolipotoxicity, which is a hallmark of type 2 diabetes , was mimicked in vitro by culturing the islets for 3 or 5 days with 0.5 mM palmitic acid or a mixture of palmitic and oleic acid at 1 % albumin and different concentrations of glucose: 10, 16, and 25 (Doliba et al. 2015). We found that chronic exposure of mouse islets to FA with a glucose leads to bioenergetic failure, as evidenced by decreased OCR and reduced insulin secretion upon stimulation with glucose or amino acids. These changes were associated with reduced islet ATP levels, impaired glucose-induced ATP rise, a trend for reduced mitochondrial DNA, and reduced expression of mitochondrial transcription factor A (Tfam). We also discovered accumulation of carnitine esters of hydroxylated long-chain FA (Doliba et al. 2015) that have been shown to uncouple the heart and brain mitochondria (Tonin et al. 2010, 2013). We propose that mitochondrial accumulation of unsaturated hydroxylated long-chain FA uncouples and ultimately inhibits pancreatic islet beta-cell respiration and that this effect of the toxic FA metabolite causes a slow deterioration of mitochondrial function, progressing to bioenergetic failure as the main cause of impaired insulin secretion and reduced beta-cell mass, both hallmarks of type 2 diabetes.
Effects of Fatty Acids in Humans
FAs play an important role in the regulation of pancreatic beta-cell function in humans (McGarry 2002). In the fasting state, free FAs sustain basal insulin secretion and assume efficient nutrient-stimulated insulin secretion when the fast is terminated. Elevated plasma FFA levels have been reported to play an important role in maintaining chronic hyperinsulinemia in insulin-resistant obese subjects, and removal of this FFA stimulus by overnight reduction of plasma FFAs with nicotinic acid impairs glucose-induced insulin secretion (Dobbins et al. 1998). Despite the evidence from in vivo studies, the effects of prolonged elevation of FFA on insulin secretion in humans remain controversial. Boden and colleagues demonstrated that 48-h elevation of plasma FFA potentiated GSIS in healthy subjects at glucose levels clamped at 8.6 mM (Boden et al. 1995) but insulin secretion was defective in type 2 diabetic patients (Boden and Chen 1999). In contrast, Carpentier et al. (1999) have shown that acute enhancement of insulin secretion by FA in healthy humans is lost with prolonged FA elevation. This loss of insulin secretion was specific to glucose and the response to arginine was normal (Carpentier et al. 2001). Interestingly, obese but not diabetic subjects are more susceptible to the inhibitory effect of lipids on GSIS (Carpentier et al. 2000). Kashyap et al. (2003) have examined insulin secretion and insulin action during a 4-day lipid infusion in nondiabetic subjects with and without a family history of type 2 diabetes. The most striking finding is that a 4-day intralipid infusion enhances insulin secretion in control subjects but inhibits GSIS in individuals with family history of type 2 diabetes (Kashyap et al. 2003). These data suggest that in subjects with a high risk of developing type 2 diabetes, beta-cell lipotoxicity may play an important role in the progression from normal glucose tolerance to overt hyperglycemia. Of note, a reduction in plasma FFA concentration with the antilipolytic agent acipimox enhanced first-phase insulin secretion in nondiabetic patients with a family history of type 2 diabetes (Paolisso et al. 1998).
Recently a new strategy was applied to study the functional impairment of human pancreatic islets (Rosengren et al. 2012). The goal of this approach was to calculate a genetic risk score for islet dysfunction leading to type 2 diabetes that involved impaired insulin exocytosis, decreased docking of insulin-containing secretory granules, and reduced insulin secretion (Rosengren et al. 2012). Such calculations were based on correlation analysis of function and genotype of human islets obtained from diabetic and nondiabetic donors. Rosengren et al. (2012) analyzed a panel of 14 gene variants robustly associated with type 2 diabetes susceptibility. This work resulted in the identification of four genetic variants that confer reduced beta-cell exocytosis and six variants that interfere with insulin granule distribution. It is of interest that this study showed that the negative impact of type 2 diabetes loci on beta-cell function was evident in islets from nonobese individuals. This suggests that the functional effects of the type 2 diabetes-associated SNPs may be more pronounced in lean than in obese individual. It may seem counterintuitive that obese individuals with type 2 diabetes exhibit greater insulin secretion than their lean counterparts. This may reflect an adaptation, albeit insufficient to prevent diabetes, in the obese donors. In addition, the lean donors who developed diabetes were likely to be those with the lowest insulin secretory capacity. These data suggest that there may be considerable heterogeneity in the cellular pathways that lead to reduced insulin secretion, which may explain why the reduction of exocytosis is evident only in genetic subgroups and not in the entire type 2 diabetes cohort (Rosengren et al. 2012).
Conclusions
To conclude, based on the existing literature, it is clear that excessive glucose and FAs levels have time-dependent deteriorating effects on pancreatic beta-cell pathophysiology in diabetes. These effects are different at the various stages of beta-cell dysfunction during the course of type 2 diabetes. When insulin resistance develops, for instance, as a result of obesity, the beta-cells mount a compensatory response that increases beta-cell mass, insulin biosynthesis, and insulin secretion. The magnitude of the compensatory beta-cell response is genetically determined (Kashyap et al. 2003; Rosengren et al. 2012; Fadista et al. 2014), and this is a major determinant of the long-term ability of an individual to maintain glucose homeostasis in the face of insulin resistance. In genetically predisposed individuals, pancreatic beta-cell compensation eventually becomes insufficient to sustain a secretory response that matches the high demand imposed by insulin resistance.
The failure of beta-cells to compensate for insulin resistance is a major component of impaired glucose homeostasis and overt diabetes. This defect is the consequence of a decline of insulin response to glucose due to functional beta-cell deficiency. It is also the consequence of an inability of the endocrine pancreas to adapt the beta-cell mass which eventually leads to a decrease in functional beta-cells. This idea has resulted in considerable attention being paid to the development of new therapeutic strategies aimed toward preserving or regenerating functional beta-cell mass (Karaca et al. 2009). GLP-1 enhances beta-cell survival by activating beta-cell proliferation and differentiation and inhibiting beta-cell apoptosis and thus contributing to the long-term regulation of insulin secretion by maintaining a functional beta-cell mass. It should be pointed out that any intervention to improve insulin secretion should start early in the disease when the endogenous insulin secretion and presumably the number of functional beta-cells have not decreased excessively (Grill and Bjorklund 2002; Karvestedt et al. 2002).
References
Ackermann AM, Gannon M. Molecular regulation of pancreatic beta-cell mass development, maintenance, and expansion. J Mol Endocrinol. 2007;38:193–206.
Ahren B. Autonomic regulation of islet hormone secretion--implications for health and disease. Diabetologia. 2000;43:393–410.
Ahren B. Islet G, protein-coupled receptors as potential targets for treatment of type 2 diabetes. Nat Rev Drug Discov. 2009;8:369–385.
Ashcroft FM, Harrison DE, Ashcroft SJ. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature. 1984;312:446–448.
Baggio LL, Drucker DJ. Therapeutic approaches to preserve islet mass in type 2 diabetes. Annu Rev Med. 2006;57:265–281.
Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–2157.
Barlow J, Affourtit C. Novel insights into pancreatic beta-cell glucolipotoxicity from real-time functional analysis of mitochondrial energy metabolism in INS-1E insulinoma cells. Biochem J. 2013;456:417–426.
Bernard C, Thibault C, Berthault MF, et al. Pancreatic beta-cell regeneration after 48-h glucose infusion in mildly diabetic rats is not correlated with functional improvement. Diabetes. 1998;47:1058-1065.
Bernardi P, Penzo D, Wojtczak L. Mitochondrial energy dissipation by fatty acids. Mechanisms and implications for cell death. Vitam Horm. 2002;65:97–126.
Bernard-Kargar C, Ktorza A. Endocrine pancreas plasticity under physiological and pathological conditions. Diabetes. 2001;50(Suppl 1):S30–S35.
Biden TJ, Robinson D, Cordery D, Hughes WE, Busch AK. Chronic effects of fatty acids on pancreatic beta-cell function: new insights from functional genomics. Diabetes. 2004;53(Suppl 1):S159–S165.
Boden G, Chen X. Effects of fatty acids and ketone bodies on basal insulin secretion in type 2 diabetes. Diabetes. 1999;48:577–583.
Boden G, Chen X, Rosner J, Barton M. Effects of a 48-h fat infusion on insulin secretion and glucose utilization. Diabetes. 1995;44:1239–1242.
Bonner-Weir S. Islet growth and development in the adult. J Mol Endocrinol. 2000;24:297–302.
Boslem E, Meikle PJ, Biden TJ. Roles of ceramide and sphingolipids in pancreatic beta-cell function and dysfunction. Islets. 2012;4:177–187.
Branstrom R, Corkey BE, Berggren PO, Larsson O. Evidence for a unique long chain acyl-CoA ester binding site on the ATP-regulated potassium channel in mouse pancreatic beta cells. J Biol Chem. 1997;272:17390–17394.
Branstrom R, Leibiger IB, Leibiger B, Corkey BE, Berggren PO, Larsson O. Long chain coenzyme A esters activate the pore-forming subunit (Kir6. 2) of the ATP-regulated potassium channel. J Biol Chem. 1998;273:31395–31400.
Brownlie R, Mayers RM, Pierce JA, Marley AE, Smith DM. The long-chain fatty acid receptor, GPR40, and glucolipotoxicity: investigations using GPR40-knockout mice. Biochem Soc Trans. 2008;36:950–954.
Brubaker PL, Drucker DJ. Minireview: glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology. 2004;145:2653–2659.
Buettner R, Newgard CB, Rhodes CJ, O'Doherty RM. Correction of diet-induced hyperglycemia, hyperinsulinemia, and skeletal muscle insulin resistance by moderate hyperleptinemia. Am J Physiol Endocrinol Metab. 2000;278:E563–E569.
Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013;17:819–837.
Carpentier A, Mittelman SD, Lamarche B, Bergman RN, Giacca A, Lewis GF. Acute enhancement of insulin secretion by FFA in humans is lost with prolonged FFA elevation. Am J Physiol. 1999;276:E1055–E1066.
Carpentier A, Mittelman SD, Bergman RN, Giacca A, Lewis GF. Prolonged elevation of plasma free fatty acids impairs pancreatic beta-cell function in obese nondiabetic humans but not in individuals with type 2 diabetes. Diabetes. 2000;49:399–408.
Carpentier A, Giacca A, Lewis GF. Effect of increased plasma non-esterified fatty acids (NEFAs) on arginine-stimulated insulin secretion in obese humans. Diabetologia. 2001;44:1989–1997.
Chan CB, Saleh MC, Koshkin V, Wheeler MB. Uncoupling protein 2 and islet function. Diabetes. 2004;53(Suppl 1):S136–S142.
Chen C, Hosokawa H, Bumbalo LM, Leahy JL. Regulatory effects of glucose on the catalytic activity and cellular content of glucokinase in the pancreatic beta cell. Study using cultured rat islets. J Clin Invest. 1994;94:1616–1620.
Clark JB, Palmer CJ, Shaw WN. The diabetic Zucker fatty rat. Proc Soc Exp Biol Med Soc Exp Biol Med. 1983;173:68–75.
Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54(Suppl 2):S97–S107.
Cnop M, Igoillo-Esteve M, Cunha DA, Ladriere L, Eizirik DL. An update on lipotoxic endoplasmic reticulum stress in pancreatic beta-cells. Biochem Soc Trans. 2008;36:909–915.
Cousin SP, Hugl SR, Wrede CE, Kajio H, Myers MG Jr, Rhodes CJ. Free fatty acid-induced inhibition of glucose and insulin-like growth factor I-induced deoxyribonucleic acid synthesis in the pancreatic beta-cell line INS-1. Endocrinology. 2001;142:229–240.
Dobbins RL, Chester MW, Daniels MB, McGarry JD, Stein DT. Circulating fatty acids are essential for efficient glucose-stimulated insulin secretion after prolonged fasting in humans. Diabetes. 1998;47:1613–1618.
Doliba NM, Qin W, Vatamaniuk MZ, et al. Cholinergic regulation of fuel-induced hormone secretion and respiration of SUR1−/− mouse islets. Am J Physiol Endocrinol Metab. 2006;291:E525–E535.
Doliba NM, Wehrli SL, Vatamaniuk MZ, et al. Metabolic and ionic coupling factors in amino acid-stimulated insulin release in pancreatic beta-HC9 cells. Am J Physiol Endocrinol Metab. 2007;292:E1507–E1519.
Doliba NM, Qin W, Vinogradov SA, Wilson DF, Matschinsky FM. Palmitic acid acutely inhibits acetylcholine- but not GLP-1-stimulated insulin secretion in mouse pancreatic islets. Am J Physiol Endocrinol Metab. 2010;299:E475–E485.
Doliba NM, Qin W, Najafi H, et al. Glucokinase activation repairs defective bioenergetics of islets of Langerhans isolated from type 2 diabetics. Am J Physiol Endocrinol Metab. 2012;302:E87–E102.
Doliba NMLC, Chen P, Chen J, Naji A, Bennet M, Matchinsky FM. Long-Chain Beta-Hydroxylated Fatty Acids as Mediators of Pancreatic Beta-Cell Bioenergetic Failure. Boston, MA: American Diabetes Association; 2015. Diabetes. p. Abstract 2354.
Donath MY, Gross DJ, Cerasi E, Kaiser N. Hyperglycemia-induced beta-cell apoptosis in pancreatic islets of Psammomys obesus during development of diabetes. Diabetes. 1999;48:738–744.
Doyle ME, Egan JM. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol Ther. 2007;113:546–593.
Drucker DJ. Incretin-based therapies: a clinical need filled by unique metabolic effects. Diabetes Educ. 2006;32(Suppl 2):65S–71S.
Drucker DJ. Incretin action in the pancreas: potential promise, possible perils, and pathological pitfalls. Diabetes. 2013;62:3316–3323.
Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696–1705.
Ek J, Andersen G, Urhammer SA, et al. Mutation analysis of peroxisome proliferator-activated receptor-gamma coactivator-1 (PGC-1) and relationships of identified amino acid polymorphisms to Type II diabetes mellitus. Diabetologia. 2001;44:2220–2226.
El-Assaad W, Buteau J, Peyot ML, et al. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology. 2003;144:4154–4163.
Fadista J, Vikman P, Laakso EO, et al. Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc Natl Acad Sci U S A. 2014;111:13924–13929.
Ferdaoussi M, Bergeron V, Zarrouki B, et al. G protein-coupled receptor (GPR)40-dependent potentiation of insulin secretion in mouse islets is mediated by protein kinase D1. Diabetologia. 2012;55:2682–2692.
Fujiwara K, Maekawa F, Yada T. Oleic acid interacts with GPR40 to induce Ca2+ signaling in rat islet beta-cells: mediation by PLC and L-type Ca2+ channel and link to insulin release. Am J Physiol Endocrinol Metab. 2005;289:E670–E677.
Gautam D, Han SJ, Hamdan FF, et al. A critical role for beta cell M3 muscarinic acetylcholine receptors in regulating insulin release and blood glucose homeostasis in vivo. Cell Metab. 2006;3:449–461.
Gautam D, Han SJ, Duttaroy A, et al. Role of the M3 muscarinic acetylcholine receptor in beta-cell function and glucose homeostasis. Diabetes Obes Metab. 2007;9(Suppl 2):158–169.
Gehrmann W, Elsner M, Lenzen S. Role of metabolically generated reactive oxygen species for lipotoxicity in pancreatic beta-cells. Diabetes Obes Metab. 2010;12(Suppl 2):149–158.
Gilon P, Henquin JC. Mechanisms and physiological significance of the cholinergic control of pancreatic beta-cell function. Endocr Rev. 2001;22:565–604.
Gremlich S, Bonny C, Waeber G, Thorens B. Fatty acids decrease IDX-1 expression in rat pancreatic islets and reduce GLUT2, glucokinase, insulin, and somatostatin levels. J Biol Chem. 1997;272:30261–30269.
Grill V, Bjorklund A. Type 2 diabetes–effect of compensatory oversecretion as a reason for beta-cell collapse. Int J Exp Diabetes Res. 2002;3:153–158.
Henquin JC. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000;49:1751–1760.
Henquin JC. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia. 2009;52:739–751.
Holz GG, Habener JF. Signal transduction crosstalk in the endocrine system: pancreatic beta-cells and the glucose competence concept. Trends Biochem Sci. 1992;17:388–393.
Houze JB, Zhu L, Sun Y, et al. AMG 837: a potent, orally bioavailable GPR40 agonist. Bioorg Med Chem Lett. 2012;22:1267–1270.
Hugl SR, White MF, Rhodes CJ. Insulin-like growth factor I (IGF-I)-stimulated pancreatic beta-cell growth is glucose-dependent. Synergistic activation of insulin receptor substrate-mediated signal transduction pathways by glucose and IGF-I in INS-1 cells. J Biol Chem. 1998;273:17771–17779.
Iizuka K, Nakajima H, Namba M, et al. Metabolic consequence of long-term exposure of pancreatic beta cells to free fatty acid with special reference to glucose insensitivity. Biochim Biophys Acta. 2002;1586:23–31.
Imai Y, Patel HR, Doliba NM, Matschinsky FM, Tobias JW, Ahima RS. Analysis of gene expression in pancreatic islets from diet-induced obese mice. Physiol Genomics. 2008;36:43–51.
Inagaki N, Kuromi H, Gonoi T, et al. Expression and role of ionotropic glutamate receptors in pancreatic islet cells. FASEB J. 1995;9:686–691.
Itoh Y, Kawamata Y, Harada M, et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature. 2003;422:173–176.
Jain S, Ruiz de Azua I, Lu H, White MF, Guettier JM, Wess J. Chronic activation of a designer G(q)-coupled receptor improves beta cell function. J Clin Invest. 2013;123:1750–1762.
Joseph JW, Koshkin V, Saleh MC, et al. Free fatty acid-induced beta-cell defects are dependent on uncoupling protein 2 expression. J Biol Chem. 2004;279:51049–51056.
Karaca M, Magnan C, Kargar C. Functional pancreatic beta-cell mass: involvement in type 2 diabetes and therapeutic intervention. Diabetes Metab. 2009;35:77–84.
Karvestedt L, Andersson G, Efendic S, Grill V. A rapid increase in beta-cell function by multiple insulin injections in type 2 diabetic patients is not further enhanced by prolonging treatment. J Intern Med. 2002;251:307–316.
Kashyap S, Belfort R, Gastaldelli A, et al. A sustained increase in plasma free fatty acids impairs insulin secretion in nondiabetic subjects genetically predisposed to develop type 2 diabetes. Diabetes. 2003;52:2461–2474.
Kebede M, Alquier T, Latour MG, Semache M, Tremblay C, Poitout V. The fatty acid receptor GPR40 plays a role in insulin secretion in vivo after high-fat feeding. Diabetes. 2008;57:2432–2437.
Kelpe CL, Moore PC, Parazzoli SD, Wicksteed B, Rhodes CJ, Poitout V. Palmitate inhibition of insulin gene expression is mediated at the transcriptional level via ceramide synthesis. J Biol Chem. 2003;278:30015–30021.
Kharroubi I, Ladriere L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL. Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology. 2004;145:5087–5096.
Kimple ME, Neuman JC, Linnemann AK, Casey PJ. Inhibitory G proteins and their receptors: emerging therapeutic targets for obesity and diabetes. Exp Mol Med. 2014;46, e102.
Klingenberg M. The ADP, and ATP transport in mitochondria and its carrier. Biochim Biophys Acta. 1778;2008:1978–2021.
Klingenberg M, Huang SG. Structure and function of the uncoupling protein from brown adipose tissue. Biochim Biophys Acta. 1999;1415:271–296.
Kloppel G, Lohr M, Habich K, Oberholzer M, Heitz PU. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Pathol Res. 1985;4:110–125.
Koshkin V, Wang X, Scherer PE, Chan CB, Wheeler MB. Mitochondrial functional state in clonal pancreatic beta-cells exposed to free fatty acids. J Biol Chem. 2003;278:19709–19715.
Kristinsson H, Smith DM, Bergsten P, Sargsyan E. FFAR1 is involved in both the acute and chronic effects of palmitate on insulin secretion. Endocrinology. 2013;154:4078–4088.
Lagerstrom MC, Schioth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7:339–357.
Lan H, Hoos LM, Liu L, et al. Lack of FFAR1/GPR40 does not protect mice from high-fat diet-induced metabolic disease. Diabetes. 2008;57:2999–3006.
Latour MG, Alquier T, Oseid E, et al. GPR40 is necessary but not sufficient for fatty acid stimulation of insulin secretion in vivo. Diabetes. 2007;56:1087–1094.
Lee J, Cummings BP, Martin E, et al. Glucose sensing by gut endocrine cells and activation of the vagal afferent pathway is impaired in a rodent model of type 2 diabetes mellitus. Am J Physiol Regul Integr Comp Physiol. 2012;302:R657–R666.
Li C, Najafi H, Daikhin Y, et al. Regulation of leucine-stimulated insulin secretion and glutamine metabolism in isolated rat islets. J Biol Chem. 2003;278:2853–2858.
Liang Y, Matschinsky FM. Content of CoA-esters in perifused rat islets stimulated by glucose and other fuels. Diabetes. 1991;40:327–333.
Lin DC, Zhang J, Zhuang R, et al. AMG 837: a novel GPR40/FFA1 agonist that enhances insulin secretion and lowers glucose levels in rodents. PLoS One. 2011;6, e27270.
Lingohr MK, Buettner R, Rhodes CJ. Pancreatic beta-cell growth and survival--a role in obesity-linked type 2 diabetes? Trends Mol Med. 2002;8:375–384.
Loskovich MV, Grivennikova VG, Cecchini G, Vinogradov AD. Inhibitory effect of palmitate on the mitochondrial NADH:ubiquinone oxidoreductase (complex I) as related to the active-de-active enzyme transition. Biochem J. 2005;387:677–683.
Luo J, Swaminath G, Brown SP, et al. A potent class of GPR40 full agonists engages the enteroinsular axis to promote glucose control in rodents. PLoS One. 2012;7, e46300.
Lupi R, Dotta F, Marselli L, et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes. 2002;51:1437–1442.
Matschinsky FM. Banting Lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes. 1996;45:223–241.
Matschinsky FM, Ellerman JE, Landgraf, Krzanowski J, Kotler-Brajtburg J, Fertel R. Quantitative histochemistry of glucose metabolism in the islets of Langerhans. Curr Prob Clin Biochem. 1971;3:143–182.
McGarry JD. Banting lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes. 2002;51:7–18.
McGarry JD, Dobbins RL. Fatty acids, lipotoxicity and insulin secretion. Diabetologia. 1999;42:128–138.
Milburn JL Jr, Hirose H, Lee YH, et al. Pancreatic beta-cells in obesity. Evidence for induction of functional, morphologic, and metabolic abnormalities by increased long chain fatty acids. J Biol Chem. 1995;270:1295–1299.
Mokdad AH, Bowman BA, Ford ES, Vinicor F, Marks JS, Koplan JP. The continuing epidemics of obesity and diabetes in the United States. JAMA. 2001;286:1195–1200.
Morgan D, Oliveira-Emilio HR, Keane D, et al. Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia. 2007;50:359–369.
Nagasumi K, Esaki R, Iwachidow K, et al. Overexpression of GPR40 in pancreatic beta-cells augments glucose-stimulated insulin secretion and improves glucose tolerance in normal and diabetic mice. Diabetes. 2009;58:1067–1076.
Nakajima K, Jain S, Ruiz de Azua I, McMillin SM, Rossi M, Wess J. Minireview: novel aspects of M3 muscarinic receptor signaling in pancreatic beta-cells. Mol Endocrinol. 2013;27:1208–1216.
Newgard CB, Matschinsky F. Substrate control of insulin release. In: L J, eds. Handbook of Physiology. Oxford, CA: Oxford University Press; 2001.
Newsholme P, Bender K, Kiely A, Brennan L. Amino acid metabolism, insulin secretion and diabetes. Biochem Soc Trans. 2007a;35:1180–1186.
Newsholme P, Keane D, Welters HJ, Morgan NG. Life and death decisions of the pancreatic beta-cell: the role of fatty acids. Clin Sci. 2007b;112:27–42.
Newsholme P, Cruzat V, Arfuso F, Keane K. Nutrient regulation of insulin secretion and action. J Endocrinol. 2014;221:R105–R120.
Nolan CJ, Prentki M. The islet beta-cell: fuel responsive and vulnerable. Trends Endocrinol Metab. 2008;19:285–291.
Nolan CJ, Madiraju MS, Delghingaro-Augusto V, Peyot ML, Prentki M. Fatty acid signaling in the beta-cell and insulin secretion. Diabetes. 2006;55(Suppl 2):S16–S23.
Oberkofler H, Linnemayr V, Weitgasser R, et al. Complex haplotypes of the PGC-1alpha gene are associated with carbohydrate metabolism and type 2 diabetes. Diabetes. 2004;53:1385–1393.
Oberkofler H, Klein K, Felder TK, Krempler F, Patsch W. Role of peroxisome proliferator-activated receptor-gamma coactivator-1alpha in the transcriptional regulation of the human uncoupling protein 2 gene in INS-1E cells. Endocrinology. 2006;147:966–976.
Oberkofler H, Hafner M, Felder T, Krempler F, Patsch W. Transcriptional co-activator peroxisome proliferator-activated receptor (PPAR)gamma co-activator-1beta is involved in the regulation of glucose-stimulated insulin secretion in INS-1E cells. J Mol Med (Berl). 2009;87:299–306.
Olofsson CS, Collins S, Bengtsson M, et al. Long-term exposure to glucose and lipids inhibits glucose-induced insulin secretion downstream of granule fusion with plasma membrane. Diabetes. 2007;56:1888–1897.
Paolisso G, Tagliamonte MR, Rizzo MR, et al. Lowering fatty acids potentiates acute insulin response in first degree relatives of people with type II diabetes. Diabetologia. 1998;41:1127–1132.
Parnaud G, Bosco D, Berney T, et al. Proliferation of sorted human and rat beta cells. Diabetologia. 2008;51:91–100.
Parsons JA, Brelje TC, Sorenson RL. Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology. 1992;130:1459–1466.
Penzo D, Tagliapietra C, Colonna R, Petronilli V, Bernardi P. Effects of fatty acids on mitochondria: implications for cell death. Biochim Biophys Acta. 2002;1555:160–165.
Penzo D, Petronilli V, Angelin A, et al. Arachidonic acid released by phospholipase A(2) activation triggers Ca(2+)-dependent apoptosis through the mitochondrial pathway. J Biol Chem. 2004;279:25219–25225.
Peyot ML, Pepin E, Lamontagne J, et al. Beta-cell failure in diet-induced obese mice stratified according to body weight gain: secretory dysfunction and altered islet lipid metabolism without steatosis or reduced beta-cell mass. Diabetes. 2010;59:2178–2187.
Pick A, Clark J, Kubstrup C, et al. Role of apoptosis in failure of beta-cell mass compensation for insulin resistance and beta-cell defects in the male Zucker diabetic fatty rat. Diabetes. 1998;47:358–364.
Poitout V, Robertson RP. Minireview: secondary beta-cell failure in type 2 diabetes–a convergence of glucotoxicity and lipotoxicity. Endocrinology. 2002;143:339–342.
Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr Rev. 2008;29:351–366.
Poitout V, Amyot J, Semache M, Zarrouki B, Hagman D, Fontes G. Glucolipotoxicity of the pancreatic beta cell. Biochim Biophys Acta. 1801;2010:289–298.
Prentki M, Madiraju SR. Glycerolipid metabolism and signaling in health and disease. Endocr Rev. 2008;29:647–676.
Prentki M, Madiraju SR. Glycerolipid/free fatty acid cycle and islet beta-cell function in health, obesity and diabetes. Mol Cell Endocrinol. 2012;353:88–100.
Prentki M, Vischer S, Glennon MC, Regazzi R, Deeney JT, Corkey BE. Malonyl-CoA and long chain acyl-CoA esters as metabolic coupling factors in nutrient-induced insulin secretion. J Biol Chem. 1992;267:5802–5810.
Prentki M, Joly E, El-Assaad W, Roduit R. Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: role in beta-cell adaptation and failure in the etiology of diabetes. Diabetes. 2002;51(Suppl 3):S405–S413.
Prentki M, Matschinsky FM, Madiraju SR. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013;18:162–185.
Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839.
Rajan S, Dickson LM, Mathew E, et al. Chronic hyperglycemia downregulates GLP-1 receptor signaling in pancreatic beta-cells via protein kinase A. Mol Metab. 2015;4:265–276.
Rankin MM, Kushner JA. Adaptive beta-cell proliferation is severely restricted with advanced age. Diabetes. 2009;58:1365–1372.
Reaven GM. Pathophysiology of insulin resistance in human disease. Physiol Rev. 1995;75:473–486.
Reaven GM. Banting Lecture 1988. Role of insulin resistance in human disease. 1988. Nutrition. 1997;13:65; discussion 4, 6.
Regard JB, Kataoka H, Cano DA, et al. Probing cell type-specific functions of Gi in vivo identifies GPCR regulators of insulin secretion. J Clin Invest. 2007;117:4034–4043.
Reimann F, Huopio H, Dabrowski M, et al. Characterisation of new KATP-channel mutations associated with congenital hyperinsulinism in the Finnish population. Diabetologia. 2003;46:241–249.
Rhodes CJ. Type 2 diabetes-a matter of beta-cell life and death? Science. 2005;307:380–384.
Roat R, Rao V, Doliba NM, et al. Alterations of pancreatic islet structure, metabolism and gene expression in diet-induced obese C57BL/6J mice. PLoS One. 2014;9, e86815.
Rocca AS, Brubaker PL. Role of the vagus nerve in mediating proximal nutrient-induced glucagon-like peptide-1 secretion. Endocrinology. 1999;140:1687–1694.
Rorsman P, Braun M. Regulation of insulin secretion in human pancreatic islets. Annu Rev Physiol. 2013;75:155–179.
Rosengren AH, Braun M, Mahdi T, et al. Reduced insulin exocytosis in human pancreatic beta-cells with gene variants linked to type 2 diabetes. Diabetes. 2012;61:1726–1733.
Sako Y, Grill VE. A 48-hour lipid infusion in the rat time-dependently inhibits glucose-induced insulin secretion and B cell oxidation through a process likely coupled to fatty acid oxidation. Endocrinology. 1990;127:1580–1589.
Schonfeld P, Reiser G. Rotenone-like action of the branched-chain phytanic acid induces oxidative stress in mitochondria. J Biol Chem. 2006;281:7136–7142.
Schonfeld P, Wojtczak L. Fatty acids decrease mitochondrial generation of reactive oxygen species at the reverse electron transport but increase it at the forward transport. Biochim Biophys Acta. 1767;2007:1032–1040.
Schonfeld P, Wojtczak L. Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radic Biol Med. 2008;45:231–241.
Schuit FC, In't Veld PA, Pipeleers DG. Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment of pancreatic beta cells. Proc Natl Acad Sci U S A. 1988;85:3865–3869.
Scorrano L, Penzo D, Petronilli V, Pagano F, Bernardi P. Arachidonic acid causes cell death through the mitochondrial permeability transition. Implications for tumor necrosis factor-alpha aopototic signaling. J Biol Chem. 2001;276:12035–12040.
Shimabukuro M, Zhou YT, Levi M, Unger RH. Fatty acid-induced beta cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci U S A. 1998;95:2498–2502.
Soga T, Ohishi T, Matsui T, et al. Lysophosphatidylcholine enhances glucose-dependent insulin secretion via an orphan G-protein-coupled receptor. Biochem Biophys Res Commun. 2005;326:744–751.
Somesh BP, Verma MK, Sadasivuni MK, et al. Chronic glucolipotoxic conditions in pancreatic islets impair insulin secretion due to dysregulated calcium dynamics, glucose responsiveness and mitochondrial activity. BMC Cell Biol. 2013;14:31.
Sorenson RL, Brelje TC. Adaptation of islets of Langerhans to pregnancy: beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res. 1997;29:301–307.
Sorenson RL, Brelje TC, Hegre OD, Marshall S, Anaya P, Sheridan JD. Prolactin (in vitro) decreases the glucose stimulation threshold, enhances insulin secretion, and increases dye coupling among islet B cells. Endocrinology. 1987;121:1447–1453.
Steiner DJ, Kim A, Miller K, Hara M. Pancreatic islet plasticity: interspecies comparison of islet architecture and composition. Islets. 2010;2:135–145.
Steneberg P, Rubins N, Bartoov-Shifman R, Walker MD, Edlund H. The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metab. 2005;1:245–258.
Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet-induced type II diabetes in C57BL/6J mice. Diabetes. 1988;37:1163–1167.
Teff KL, Townsend RR. Early phase insulin infusion and muscarinic blockade in obese and lean subjects. Am J Physiol. 1999;277:R198–R208.
Thorens B. Neural regulation of pancreatic islet cell mass and function. Diabetes Obes Metab. 2014;16(Suppl 1):87–95.
Tonin AM, Ferreira GC, Grings M, et al. Disturbance of mitochondrial energy homeostasis caused by the metabolites accumulating in LCHAD and MTP deficiencies in rat brain. Life Sci. 2010;86:825–831.
Tonin AM, Amaral AU, Busanello EN, Grings M, Castilho RF, Wajner M. Long-chain 3-hydroxy fatty acids accumulating in long-chain 3-hydroxyacyl-CoA dehydrogenase and mitochondrial trifunctional protein deficiencies uncouple oxidative phosphorylation in heart mitochondria. J Bioenerg Biomembr. 2013;45:47–57.
Tsujihata Y, Ito R, Suzuki M, et al. TAK-875, an orally available G protein-coupled receptor 40/free fatty acid receptor 1 agonist, enhances glucose-dependent insulin secretion and improves both postprandial and fasting hyperglycemia in type 2 diabetic rats. J Pharmacol Exp Ther. 2011;339:228–237.
Unger RH, Orci L. Diseases of liporegulation: new perspective on obesity and related disorders. FASEB J. 2001;15:312–321.
Vilsboll T, Krarup T, Deacon CF, Madsbad S, Holst JJ. Reduced postprandial concentrations of intact biologically active glucagon-like peptide 1 in type 2 diabetic patients. Diabetes. 2001;50:609–613.
West GM, Willard FS, Sloop KW, Showalter AD, Pascal BD, Griffin PR. Glucagon-like peptide-1 receptor ligand interactions: structural cross talk between ligands and the extracellular domain. PLoS One. 2014;9, e105683.
Wierup N, Sundler F, Heller RS. The islet ghrelin cell. J Mol Endocrinol. 2014;52:R35–R49.
Wojtczak L, Schonfeld P. Effect of fatty acids on energy coupling processes in mitochondria. Biochim Biophys Acta. 1993;1183:41–57.
Wollheim CB, Maechler P. Beta cell glutamate receptor antagonists: novel oral antidiabetic drugs? Nat Med. 2015;21:310–311.
Wollheim CB, Pralong WF. Cytoplasmic calcium ions and other signalling events in insulin secretion. Biochem Soc Trans. 1990;18:111–114.
Yashiro H, Tsujihata Y, Takeuchi K, Hazama M, Johnson PR, Rorsman P. The effects of TAK-875, a selective G protein-coupled receptor 40/free fatty acid 1 agonist, on insulin and glucagon secretion in isolated rat and human islets. J Pharmacol Exp Ther. 2012;340:483–489.
Yoon JC, Xu G, Deeney JT, et al. Suppression of beta cell energy metabolism and insulin release by PGC-1alpha. Dev Cell. 2003;5:73–83.
Zawalich WS, Zawalich KC, Tesz GJ, et al. Effects of muscarinic receptor type 3 knockout on mouse islet secretory responses. Biochem Biophys Res Commun. 2004;315:872–876.
Zhang CY, Baffy G, Perret P, et al. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell. 2001;105:745–755.
Zhou YP, Grill V. Long term exposure to fatty acids and ketones inhibits B-cell functions in human pancreatic islets of Langerhans. J Clin Endocrinol Metab. 1995;80:1584–1590.
Zhou YP, Cockburn BN, Pugh W, Polonsky KS. Basal insulin hypersecretion in insulin-resistant Zucker diabetic and Zucker fatty rats: role of enhanced fuel metabolism. Metab Clin Exp. 1999;48:857–864.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this entry
Cite this entry
Doliba, N.M. (2017). Pancreatic Islet Adaptation and Failure in Obesity and Diabetes. In: Ahima, R. (eds) Metabolic Syndrome. Springer, Cham. https://doi.org/10.1007/978-3-319-12125-3_27-2
Download citation
DOI: https://doi.org/10.1007/978-3-319-12125-3_27-2
Received:
Accepted:
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-12125-3
Online ISBN: 978-3-319-12125-3
eBook Packages: Springer Reference MedicineReference Module Medicine
Publish with us
Chapter history
-
Latest
Pancreatic Islet Adaptation and Failure in Obesity- Published:
- 02 August 2023
DOI: https://doi.org/10.1007/978-3-319-12125-3_27-3
-
Pancreatic Islet Adaptation and Failure in Obesity and Diabetes
- Published:
- 18 May 2017
DOI: https://doi.org/10.1007/978-3-319-12125-3_27-2
-
Original
Pancreatic Islet Adaptation and Failure in Obesity and Diabetes- Published:
- 01 September 2015
DOI: https://doi.org/10.1007/978-3-319-12125-3_27-1