
Abstract
MicroRNAs (miRNAs) are 21–23-nucleotide-long small noncoding RNAs, function as mediators in gene silencing, and play essential roles in gene regulation in development, differentiation, and proliferation. Hundreds of miRNAs have been found, and tissue-specific or organ-specific expression of miRNAs has been detected. Here, I describe procedures for detection of miRNAs in the course of neuronal differentiation of human teratocarcinoma NTera2D1 and mouse embryonic carcinoma P19 cells.
Similar content being viewed by others

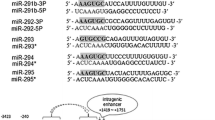

Key words
1 Introduction
MicroRNAs (miRNAs) are small noncoding RNAs, typically 21–23 nt in length, which are processed from primary miRNA transcripts forming stem-loop structure by digestion with Drosha in the nucleus and Dicer in the cytoplasm (1–3). After processing by Dicer, miRNA duplexes undergo strand selection, and single-strand miRNA elements are incorporated into the RNA-induced silencing complex (RISC) and function as mediators in gene silencing (4). miRNAs play important roles in gene regulation by inhibiting translation of messenger RNAs (mRNAs), which are partially complementary to the incorporated miRNAs, and/or by digestion of mRNAs which are nearly complementary to the miRNAs, such as RNA interference (RNAi), in the process of development, differentiation, and proliferation (5–9).
Hundreds of miRNA genes have been identified in plants and animals (1, 5, 10), and most of them appear to be expressed by RNA polymerase II (11). In addition, tissue- or organ-specific and stage-specific expression of miRNAs has been detected. In mammals, a major small RNA class transition from retrotransposon-derived small interfering RNAs (siRNAs) and Piwi-interacting RNAs (piRNAs) to zygotically expressed miRNAs occurs in the course of early development of pre-implantation embryos (12), and tissue- or organ-specific expression patterns of miRNAs are generated thereafter.
Human teratocarcinoma NTera2D1 cells and mouse embryonic carcinoma P19 cells can be induced to differentiate into neurons or neuron-like cells which exhibit differentiated morphologies with long neuritic processes (13). Since miRNAs play essential roles in cell differentiation and proliferation, it is likely that they can participate in the differentiation of NTera2D1 and P19 cells and also maintenance of stemness in an undifferentiated state of the cells. We have investigated the expression profiles of miRNAs and also mRNAs over the course of neuronal differentiation of both NTera2D1 and P19 cells (13, 14). I herein describe procedures allowing for the detection of miRNAs and mRNAs expressed in the course of the differentiation of NTera2D1 and P19 cells.
2 Materials
2.1 Cell Culture
-
1.
Dulbecco’s Modified Eagle’s medium (DMEM) with 4,500 mg/L glucose (Wako Pure Chemical Industries, Osaka, Japan) supplemented with 10% fetal bovine serum (Sigma-Aldrich Corporation, St. Louis, MO, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin (Sigma-Aldrich).
-
2.
Alpha-Minimum Essential medium (α-MEM) (Wako) supplemented with 10% fetal bovine serum (Sigma-Aldrich), 100 U/mL penicillin, and 100 μg/mL streptomycin (Sigma-Aldrich).
-
3.
Neurobasal® medium (Invitrogen Corporation, Carlsbad, CA, USA) supplemented with B-27® supplement (Invitrogen).
-
4.
Dulbecco’s Phosphate Buffered Saline [D-PBS(−)] (Sigma-Aldrich).
-
5.
BD BioCoat™ Poly-D-Lysine plates (BD Biosciences, Franklin Lakes, NJ, USA).
-
6.
6-well plates with Ultra-Low Attachment surface (Corning Incorporated Life Sciences, Lowell, MA, USA).
-
7.
All-trans-retinoic acid (RA) (Sigma-Aldrich) (see Note 1).
-
8.
Dimethyl sulfoxide (DMSO) Hybri-Max™ (Sigma-Aldrich) (see Note 1).
-
9.
Cytosine β-D-arabinofuranoside (Ara-C) (Sigma-Aldrich) (see Note 2).
-
10.
Trypsin-EDTA solution (Sigma-Aldrich).
2.2 Isolation of Total RNAs and Preparation of Small-Sized RNAs
-
1.
TRIzol® reagent (Invitrogen).
-
2.
RNasay® MinElute® Cleanup kit (Qiagen, Venlo, The Netherlands).
-
3.
UltraPure™ DNase/RNase-free distilled water (Invitrogen).
2.3 Detection of MicroRNAs and Messenger RNAs
-
1.
PlatinumBright 647 Infrared nucleic acid labeling kit (Kreatech Diagnostics, Amsterdam, The Netherlands).
-
2.
Genopal ®-MICM07 and -MICH07 DNA chips for mouse and human miRNAs, respectively (Mitsubishi Rayon Co., Lid., Tokyo, Japan) (see Note 3).
-
3.
Genopal ®- DNA chip reader (Yokogawa Electric Corporation, Tokyo, Japan).
-
4.
TaqMan® Universal PCR Master Mix (Applied Biosystems, Faster City, CA, USA).
-
5.
TaqMan® MicroRNA Assays (Applied Biosystems).
-
6.
NanoDrop-3300 fluorospectrometer (Thermo Fisher Scientific, Waltham, MA, USA).
-
7.
AB 7300 Real Time PCR System (Applied Biosystems).
-
8.
AB GeneAmp® PCR System 9700 (Applied Biosystems).
-
9.
TURBO DNA-free™ (Applied Biosystems).
-
10.
Oligo(dT)15 primer (Promega, Madison, WI, USA).
-
11.
SuperScript® III Reverse Transcriptase (Invitrogen).
-
12.
mirVana™ qRT-PCR miRNA detection kit and primer sets (Applied Biosystems (Ambion)).
-
13.
Synthetic DNA oligonucleotides for PCR primers (Sigma-Aldrich).
-
14.
Perfect Real Time Primers (TAKARA BIO INC, Shiga, Japan).
-
15.
TaKaRa LA Taq® DNA polymerase (TAKARA BIO).
-
16.
NuSieve™ 3:1 agarose (TAKARA BIO).
-
17.
Tris-Acetate-EDTA Buffer (50×) (Nacalai Tesque, Inc., Kyoto, Japan).
-
18.
40% Acrylamide/Bis solution, 19:1 (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
-
19.
Tris-Borate-EDTA (TBE) Buffer (10×) (Nacalai Tesque).
-
20.
20× SSC buffer (Bio-Rad).
-
21.
Sodium Dodecyl Sulfate (Wako).
3 Methods
3.1 Culture of Human Teratocarcinoma NTera2D1 Cells in Normal Medium
NTera2D1 cells are grown in DMEM (Wako) supplemented with 10% fetal bovine serum (Sigma-Aldrich), 100 U/mL penicillin, and 100 μg/mL streptomycin (Sigma-Aldrich) at 37°C in 5% CO2-humidified chamber.
3.2 Induction of Neuronal Differentiation of NTera2D1 Cells (Fig. 1)

Time schedule of neuronal differentiation of human NTera2D1 and mouse P19 cells. The start of the neuronal differentiation of the cells by retinoic acid (RA) is indicated by day 0. The schedules of the NTera2D1 and P19 differentiation are shown on the week (w) and day time scales, respectively. Culture medium conditions and culture plates used are indicated.
-
1.
The day before induction, cultured cells are trypsinized, diluted with fresh medium, seeded onto T25 (25 cm2) tissue culture flasks (approximately 2 × 104 cells/cm2), and incubated at 37°C in 5% CO2-humidified chamber.
-
2.
Induction of neuronal differentiation is initiated by changing the medium to fresh medium containing 1 × 10−5 M all-trans-retinoic acid (RA) (Sigma-Aldrich).
-
3.
Culture medium is changed to fresh medium containing RA twice a week.
-
4.
After 3∼4 weeks of RA treatment, RA-treated NTera2D1 cells are trypsinized, diluted with fresh normal medium without RA, seeded onto 12-well BD BioCoat™ Poly-D-Lysine plates (BD Biosciences) (approximately 1 × 105 cells/cm2), and incubated at 37°C in 5% CO2-humidified chamber.
-
5.
After 2-day incubation, medium is changed to fresh medium containing 10 μM cytosine β-D-arabinofuranoside (Ara-C) (Sigma-Aldrich) (see Note 4), and further incubation is carried out.
-
6.
Culture medium is changed to fresh medium containing Ara-C twice a week. Neurons or neuron-like cells with long neuritic processes can be observed during the incubation (Fig. 2).
Fig. 2. 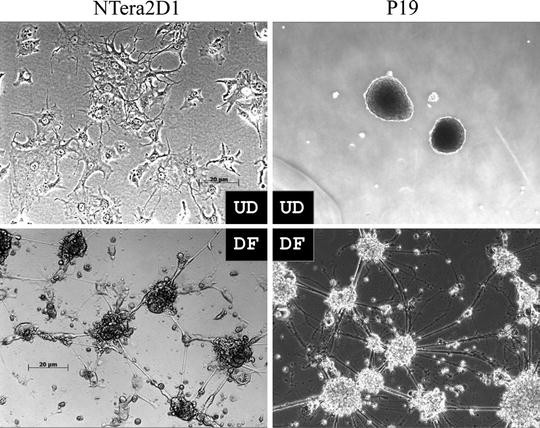
Morphological differentiation of NTera2D1 and P19 cells. Neuronal differentiation of NTera2D1 and P19 cells was carried out. Undifferentiated (UD) and completely differentiated (DF) cells are shown. Undifferentiated P19 cells indicate clumps of the cells which are grown in culture plates with Ultra-Low Attachment surface. The photo of undifferentiated NTera2D1 cells was reprinted from (13).

3.3 Culture of Mouse Embryonic Carcinoma P19 Cells in Normal Medium
P19 cells are grown in α-MEM (Wako) supplemented with 10% fetal bovine serum (Sigma-Aldrich), 100 U/mL penicillin, and 100 μg/mL streptomycin (Sigma-Aldrich) at 37°C in 5% CO2-humidified chamber.
3.4 Induction of Neuronal Differentiation of P19 Cells (Fig. 1)
-
1.
Cultured P19 cells are trypsinized, diluted with fresh medium containing 5 × 10−7 M RA (Sigma-Aldrich) (approximately 1 × 105 cells/mL), seeded into 6-well plates with Ultra-Low Attachment surface (Corning) (2 mL/well) (see Note 5), and then incubated at 37°C in 5% CO2-humidified chamber.
-
2.
After 2-day incubation, culture medium is changed to fresh medium containing RA.
-
(a)
Collect culture medium, where clumps of P19 cells are present (see Note 6), by using a 5 mL measuring pipette and transfer the medium into a 15 mL centrifuge tube.
-
(b)
Stand the tube straight up for 5 ∼ 10 min to precipitate the cells spontaneously.
-
(c)
Remove the supernatant by a pipette (see Note 7).
-
(d)
Add fresh medium containing 5 × 10−7 M RA and pipette twice carefully such that the clumps of P19 cells are not collapsed.
-
(e)
Transfer the medium containing the clumps of the cells into 6-well plates with Ultra-Low Attachment surface (Corning) (2 mL/well).
-
(a)
-
3.
Incubate cells at 37°C in 5% CO2-humidified chamber for two more days.
-
4.
After 4-day induction with RA, clumps of the cells are collected by spontaneous precipitation as described above, and rinsed twice with D-PBS(−) followed by centrifugation.
-
5.
Precipitated cells are subjected to trypsinization as follows:
-
(a)
Add approximately 1/10 packed cell volume of Trypsin-EDTA solution (Sigma-Aldrich) to the precipitated cells, and mix them well by pipetting several times.
-
(b)
Add fresh normal medium (∼ 5 mL) for quenching trypsin activity.
-
(c)
Collect the cells by centrifugation and re-suspend them with fresh normal medium.
-
(a)
-
6.
Trypsinized cells are seeded onto 12-well BD BioCoat Poly-D-Lysine plates (BD Biosciences) (approximately 1 × 105 cells/cm2), and incubated at 37°C in 5% CO2-humidified chamber.
-
7.
After 3-day incubation, medium is changed to Neurobasal medium (Invitrogen) containing B-27 supplement (Invitrogen) and 10 μM Ara-C (Sigma-Aldrich) (see Note 4), and cells are further incubated.
-
8.
Medium is changed to fresh Neurobasal medium containing B-27 supplement and 10 μM Ara-C every 2 or 3 days. Neurons or neuron-like cells with long neuritic processes can be observed during the incubation (Fig. 2).
3.5 Extraction of Total RNAs
-
1.
Total RNAs are extracted from undifferentiated and differentiated cells at various time points in the course of neuronal differentiation by TRIzol reagent (Invitrogen) according to the instructions of the manufacturer.
-
2.
Extracted RNAs are dissolved in DNase/RNase-free distilled water (Invitrogen).
-
3.
Store at −20°C.
3.6 Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
-
1.
Total RNAs are treated with TURBO DNA-freeTM (Applied Biosystems) according to the instructions of the manufacturer.
-
2.
Complementary DNA (cDNA) synthesis is carried out with the treated RNA (1 μg/test) as a template using oligo(dT) primer (Promega) and SuperScript® III reverse transcriptase (Invitrogen), according to the instructions of the manufacturer.
-
3.
The resultant cDNA is subjected to PCR analysis using the AB GeneAmp® PCR System 9700 (Applied Biosystems) with TaKaRa LA Taq® DNA polymerase (TAKARA BIO) and following synthetic primers:
-
Mouse Pou5f1-F; 5′-AAGCTGCTGAAGCAGAAGAGGATC-3′.
-
Mouse Pou5f1-R; 5′-ACCTCACACGGTTCTCAATGCTAG-3′.
-
Mouse Map2-F; 5′-TTAAACAGGCGAAGGATAAAGTCAC-3′.
-
Mouse Map2-R; 5′-TGATTGCAGTTGATCCAGGGGTAG-3′.
Perfect Real Time Primers (TAKARA Bio) against the human POU5F1, MAP2, NEFL, and GAPDH genes, and against the mouse Nefl and Gapdh genes.
-
-
4.
Thermal cycling profile:
-
(a)
Heat denaturation at 95°C at 2 min.
-
(b)
25∼30 cycles of amplification including denaturation at 95°C for 30 s, annealing at 56°C for 30 s, and extension at 72°C for 30 s.
-
(c)
Final extension at 72°C for 5 min.
-
(a)
-
5.
PCR products are examined by agarose gel electrophoresis with 3% NuSieve™ 3:1 agarose gels (TAKARA BIO) (see Note 8) and ethidium bromide staining (Fig. 3).
Fig. 3. 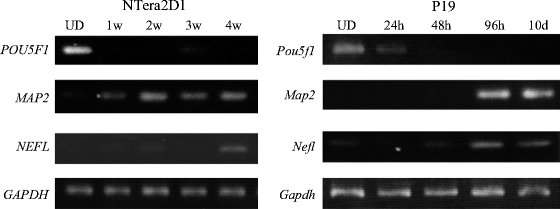
Expression profiles of protein-coding genes related to neuronal differentiation. Total RNAs were prepared from undifferentiated (UD) NTera2D1 and P19 cells and from the cells at various periods (indicated) after induction of differentiation: 1–4 weeks (1w–4w) in NTera2D1 cells and 24 h to 10 days (24 h–10 d) in P19 cells. The RNAs were subjected to RT-PCR analysis to examine the expression of genes indicated. The examined genes were as follows: POU Domain, Class 5, Transcription Factor 1 (POU5F1); Microtubule-Associated Protein 2 (MAP2); Neurofilament Protein, Light Polypeptide (NEFL); and Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) as a control. The results indicate that: (i) the POU5F1 expression was detected in both the undifferentiated NTera2D1 and P19 cells, but rapidly reduced by retinoic acid (RA) treatment, and (ii) the expression of MAP2 and NEFL, which are specific to the neuron, increased after RA treatment and their expression was also detected in the differentiated neuronal cells. The data were reprinted from (13).

3.7 Expression Profile Analysis of miRNAs Using DNA Chips
-
1.
Small-sized RNAs containing miRNAs are prepared from total RNAs by an RNeasy® MinElute® Cleanup kit (Qiagen) according to the instructions of the manufacturer.
-
2.
The small-sized RNAs (∼1 μg) are subjected to direct labeling with a fluorescent dye using a PlatinumBright 647 Infrared nucleic acid labeling kit (KREATECH) according to the instructions of the manufacturer.
-
3.
After fluorescent labeling, the labeled RNAs are purified from free fluorescent substrates by the KREApure columns (KREATECH).
-
4.
The fluorescent intensity of the labeled RNAs is measured by the NanoDrop-3300 fluorospectrometer (Thermo Fisher Scientific).
-
5.
Approximately 105 relative fluorescence units (rfu) of each labeled RNA sample is used in hybridization.
-
6.
Hybridization is carried out with the Genopal ® DNA chips (Mitsubishi Rayon) in 100 μL of hybridization buffer (6× SSC, 0.2% SDS and heat-denatured labeled RNAs (∼105 rfu)) at 42°C overnight using a hybridization chamber specific for the Genopal ®-DNA chip (Mitsubishi Rayon) (see Note 3).
-
7.
After hybridization, the DNA chips are washed twice in 2× SSC containing 0.2% SDS at 50°C for 20 min followed by washing in 2× SSC at 50°C for 10 min.
-
8.
Hybridization signals are examined by a DNA chip reader adopting multi-beam excitation technology according to the instructions of the manufacturer (Yokogawa Electric Corporation, Tokyo, Japan) (see Note 9) (Figs. 4 and 5).
Fig. 4. 
Expression profiles of miRNAs between undifferentiated and differentiated NTera2D1 cells. Small-sized RNAs prepared from undifferentiated and differentiated NTera2D1 cells were examined by Genopal ®-MICH07 DNA chips. MicroRNAs markedly expressed in undifferentiated and differentiated NTera2D1 cells are indicated by arrowheads and arrows, respectively. The data were reprinted from (13).
Fig. 5. 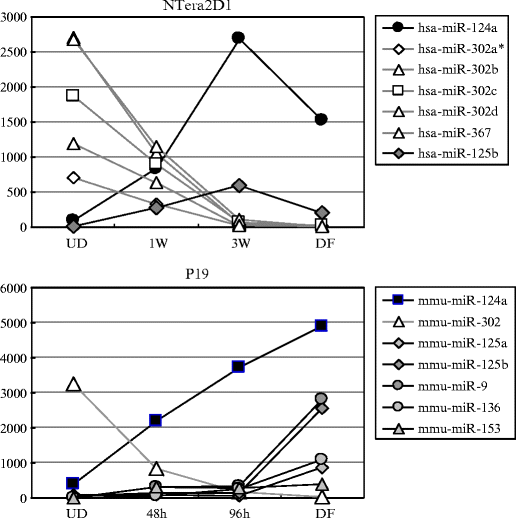
Expression profiles of miRNAs during neuronal differentiation of NTera2D1 and P19 cells. Small-sized RNA samples were extracted from the cells at the indicated time points after retinoic acid (RA) treatment (1 and 3 weeks (W) in NTera2D1; 48 and 96 h (h) in P19) and examined together with the samples prepared from undifferentiated (UD) and differentiated (DF) cells by Genopal ®-DNA chips. The expression levels of miRNAs are represented by their hybridization signal intensities and indicated by arbitrary units. Markedly increased and decreased miRNAs during neuronal differentiation are indicated by solid and open marks, respectively. The data indicated that the expression of the miR-302 members, which are ES-specific miRNAs, markedly decreased after induction of neuronal differentiation by RA, and on the contrary that miR-124a, miR-9, and miR-125, which are brain-specific miRNAs, increased in their levels after RA treatment. The data were reprinted from (13).


3.8 RT-quantitative PCR (RT-qPCR) for miRNAs
-
1.
An aliquot of total RNA isolated is diluted in DNase/RNase-free water, and the concentration of the total RNA is adjusted to 2 μg/mL.
-
2.
10 ng of total RNA per test is used (see Note 10) for synthesizing cDNAs of miRNAs.
-
3.
cDNA synthesis is performed by the AB GeneAmp® PCR System 9700 (Applied Biosystems) with a TaqMan® MicroRNA Reverse Transcription kit together with TaqMan® MicroRNA Assays (Applied Biosystems) according to the instructions of the manufacturer.
-
4.
Real time PCR (qPCR) analysis is subsequently carried out by means of the AB 7300 Real Time PCR System (Applied Biosystems) with a TaqMan® Universal PCR Master Mix and TaqMan® MicroRNA Assays (Applied Biosystems) according to the instructions of the manufacturer (Fig. 6).
Fig. 6. 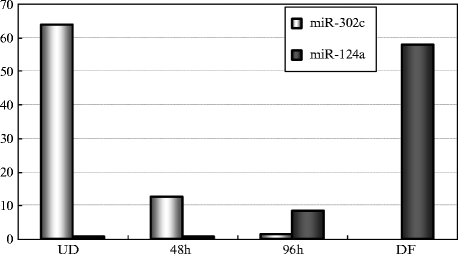
RT-qPCR analysis of miR-124a and miR-302c during neuronal differentiation of P19 cells. Total RNAs were extracted from P19 cells at the indicated time points as in Fig. 5 and examined by RT-qPCR. The expression levels of miR-124a, miR-302c, and U6RNA examined as a control were analyzed by the cycle threshold (Ct) method and plotted when the expression level of U6RNA was 1. The data were consistent with the results of Fig. 5. The data were reprinted from (13).

3.9 End-Point PCR Analysis
End-point PCR analysis for let-7a and 5sRNA as a control is carried out using the AB GeneAmp PCR system 9700 (Applied Biosystems) with a mirVana™ qRT-PCR miRNA detection kit (Ambion) and primer sets (Ambion) according to the instructions of the manufacturer (see Note 11). The resultant PCR products are electrophoretically separated on 12% polyacrylamide gels (see Note 12) and visualized by ethidium bromide staining (Fig. 7).

Expression profile of let-7 miRNA during neuronal differentiation of NTera2D1 and P19 cells. Total RNAs were extracted from NTera2D1 and P19 cells as in Figs. 3 and 5 and subjected to End-point PCR analysis for let-7 and 5sRNA as a control. The PCR products were analyzed by gel electrophoresis with 12% polyacrylamide gels followed by ethidium bromide staining. The data of P19 cells were reprinted from (14).
4 Notes
-
1.
Dissolve RA in DMSO and prepare 1 × 10−2 M RA stock solution. The RA solution is stored in light-blocking tubes at −20°C.
-
2.
Dissolve Ara-C in D-PBS(−) and prepare 1 mM Ara-C stock solution. The Ara-C stock solution is stored at −20°C.
-
3.
Genopal ®- DNA chip is a novel, fibrous DNA chip that allows for real time detection of hybridization signals at every step of washing and thus provides highly reproducible expression profile data, although there is a limit to the number of probes (up to two hundred probes) in the chip (15). The list of the miRNAs, which can be detected by the Genopal ®-MICM07 and -MICH07 DNA chips, can be found at the following address: http://www.mrc.co.jp/genome/pdf/micm07_list.pdf.
-
4.
Ara-C is a pyrimidine analog and interferes with the synthesis of DNA, thereby destroying cells that are dividing rapidly. If Ara-C is absence, glial-like cells could occupy a majority of culture plate.
-
5.
Disposable plastic petri dishes for bacteria can be used in place of culture plates with Ultra-Low Attachment surface.
-
6.
P19 cells aggregate in culture plates with low attachment surface, thereby forming clumps of the cells.
-
7.
Take care not to remove precipitated cells.
-
8.
3% NuSieve™ 3:1 agarose gels are prepared in 1× Tris-Acetate-EDTA (TAE) buffer.
-
9.
When the washing is insufficient (cross hybridization is suspected), re-washing of the chips can be done. This is because hybridization signals can be examined with the chips soaking in washing buffer (15).
-
10.
To obtain reproducible qPCR results, it is important to take and use an exact 5 μL of 2 μg/mL total RNA solution per test. Thus, I recommend using a fine automatic pipette and low retention tips.
-
11.
To perform quantitative analysis, it is important to take and use precise amount of total RNA as a source in the PCR analysis. I recommend using a fine automatic pipette and low retention tips as in see Note 10.
-
12.
12% Polyacrylamide gels (19:1) are prepared in 1× TBE buffer.
References
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ (2004) Processing of primary microRNAs by the Microprocessor complex. Nature 432:231–235
Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S et al (2003) The nuclear RNase III Drosha initiates microRNA processing. Nature 425:415–419
Hutvagner G, Zamore PD (2002) A microRNA in a multiple-turnover RNAi enzyme complex. Science 297:2056–2060
Krichevsky AM, King KS, Donahue CP, Khrapko K, Kosik KS (2003) A microRNA array reveals extensive regulation of microRNAs during brain development. RNA 9:1274–1281
Doench JG, Petersen CP, Sharp PA (2003) siRNAs can function as miRNAs. Genes Dev 17:438–442
Zeng Y, Yi R, Cullen BR (2003) MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proc Natl Acad Sci USA 100:9779–9784
Liu CG, Calin GA, Meloon B, Gamliel N, Sevignani C, Ferracin M, Dumitru CD, Shimizu M, Zupo S, Dono M et al (2004) An oligonucleotide microchip for genome-wide microRNA profiling in human and mouse tissues. Proc Natl Acad Sci USA 101:9740–9744
Cheng AM, Byrom MW, Shelton J, Ford LP (2005) Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res 33:1290–1297
Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T (2002) Identification of tissue-specific microRNAs from mouse. Curr Biol 12:735–739
Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN (2004) MicroRNA genes are transcribed by RNA polymerase II. EMBO J 23:4051–4060
Ohnishi Y, Totoki Y, Toyoda A, Watanabe T, Yamamoto Y, Tokunaga K, Sakaki Y, Sasaki H, Hohjoh H (2010) Small RNA class transition from siRNA/piRNA to miRNA during pre-implantation mouse development. Nucleic Acids Res 38:5141–5151
Hohjoh H, Fukushima T (2007) Marked change in microRNA expression during neuronal differentiation of human teratocarcinoma NTera2D1 and mouse embryonal carcinoma P19 cells. Biochem Biophys Res Commun 362:360–367
Eda A, Tamura Y, Yoshida M, Hohjoh H (2009) Systematic gene regulation involving miRNAs during neuronal differentiation of mouse P19 embryonic carcinoma cell. Biochem Biophys Res Commun 388:648–653
Hohjoh H, Fukushima T (2007) Expression profile analysis of microRNA (miRNA) in mouse central nervous system using a new miRNA detection system that examines hybridization signals at every step of washing. Gene 391:39–44
Acknowledgments
The author would like to thank Akiko Eda and Tatsunobu Fukushima for their helpful assistance. This work was supported by a research grant from the Ministry of Health, Labor, and Welfare of Japan and by a Grant-in-Aid from the Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media, LLC
About this protocol
Cite this protocol
Hohjoh, H. (2013). MicroRNA Expression During Neuronal Differentiation of Human Teratocarcinoma NTera2D1 and Mouse Embryonic Carcinoma P19 Cells. In: Ying, SY. (eds) MicroRNA Protocols. Methods in Molecular Biology, vol 936. Humana Press, Totowa, NJ. https://doi.org/10.1007/978-1-62703-083-0_20
Download citation
DOI: https://doi.org/10.1007/978-1-62703-083-0_20
Published:
Publisher Name: Humana Press, Totowa, NJ
Print ISBN: 978-1-62703-082-3
Online ISBN: 978-1-62703-083-0
eBook Packages: Springer Protocols