
Abstract
We have developed a DNA assembly platform that utilizes the nonspecific, highly variable sequence signatures of type IIs restriction enzymes to assemble a full-length molecular clone of murine hepatitis coronavirus (MHV) strain A59. The approach also allows changes to be engineered into a DNA fragment by designing primers that incorporate the restriction site and the mutations of interest. By adding the type IIs restriction site in the proper orientation, subsequent digestion removes the restriction site and leaves a sticky end comprising the mutation of interest ready to ligate to a second fragment generated in parallel as its complement. In this chapter, we discuss the details of the method to assemble a full-length infectious clone of MHV and then engineer a specific mutation into the clone to demonstrate the power of this unique site-directed “No See’m” mutagenesis approach.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others



Key Words
- MHV
- coronavirus
- murine
- infectious clone
- No See’m technology
- site-directed mutagenesis
- type IIs restriction enzymes
- reverse genetics
1 Introduction
Mouse hepatitis virus (MHV) strain A59 is an extensively studied group 2 coronavirus (CoV). Its genome is an approximately 31.5-kb single-stranded positive-sense RNA, which contains a 5′ cap and poly(A) tail (1,2). The first two-thirds of the genome encodes the nonstructural proteins required for viral RNA replication and transcription, and the final third comprises six additional open reading frames (ORFs) from which a nested set of 3′ co-terminal subgenomic mRNAs is transcribed (Figs. 1A,B 3C). Upon entry into the cell, the N-encapsidated viral RNA is released from the virion and immediately translated by host machinery, resulting in large polyprotein precursors, which are autocleaved to generate intermediates and eventually 16 mature proteins, most of which are thought to function in viral RNA synthesis (3–6). The subgenomic mRNAs are generated from a similar-sized subgenomic negative strand, which is synthesized only after the viral replication complex is functional. The negative subgenomic RNA serves as a template for transcription of similar-sized subgenomic mRNAs, which contain the leader sequence as well as the ORF that encodes the protein directly downstream of the leader sequence (7–10). All subgenomic mRNAs are co-terminal, with each subsequent mRNA containing the sequence for all downstream ORFs, although only the 5′-most ORF is translated. This allows for rapid detection of viral replication by RT-PCR using primers that anneal to the leader and the N-gene, which results in a ladder of different sized products (Figs. 1B, 3B,C) (7–10).

The MHV genome, subgenomic transcription, and strategy for infectious clone assembly: (A) The MHV genome is ∼31.5 kb, with the first two-thirds comprising the nonstructural replicase genes required for viral RNA synthesis (gray rectangle), and the final third encoding the structural genes important for assembly of the viral particle (large black rectangle). The 5′-end is capped and contains a leader sequence (small black box) and the 3′-end has a poly(A) tail. (B) The structural and accessory genes are encoded as a nested set of co-terminal mRNAs, each of which contains the leader sequence derived from the 5′-end of the genome and a poly(A) tail. (C) For assembly of an infectious clone of MHV, the genome was divided into seven stable contiguous cDNA fragments known as MHV-A, MHV-B, MHV-C, MHV-D, MHV-E, MHV-F, and MHV-G. Unique type IIs restriction sites were used to generate unique junctions, which when ligated together produce a full-length cDNA. This allows manipulation of any region of the genome by targeting the fragment of interest and engineering No See’m sites to incorporate the mutation. In this example we target genomic position 13354-56 (indicated by •), which falls within the MHV-E fragment. Numbers on top of the assembled genome indicate fragment sizes, while bottom numbers represent genomic positions.
In cell culture, murine delayed brain tumor (DBT) cells infected with wild-type MHV experience cytopathology characterized by the formation of syncytia, caused by interaction of spike glycoproteins, which anchor in the cell membrane and interact with other spikes in neighboring cells. Wild-type infections generate uniform circular plaques ∼5 mm in diameter (Fig. 4A).
Prior to the development of an infectious clone of MHV, reverse genetics of the viral coding sequence was restricted to the ORFs downstream of the replicase. Targeted recombination was the primary method for manipulation of the structural and accessory ORFs of MHV, and while extremely powerful, this methodology did not provide an approach to engineer changes into the viral replicase genes (2,1 1). Moreover, RNA recombination can result in aberrant recombination events resulting in second-site changes (1 2), necessitating an approach to manipulate the entire genome as a recombinant molecule.
Full-length cDNA constructs revolutionized reverse genetic applications in the entire MHV genome. Our laboratory developed a full-length molecular clone of MHV-A59 (icMHV) by implementing a No See’m approach (Figs. 1C,2A,B) (1 3). Our strategy was to divide the genome into pieces that could be stably subcloned into E. coli-based plasmids to facilitate targeted mutagenesis studies and would be easy DNA to store and maintain, using the same approach that was successful with the transmissible gastroenteritis virus (TGEV) infectious clone (1 3,1 4) (Figs. 1C,2A,B). However, the MHV molecular clone was difficult to establish as several toxic regions existed in the genome. Consequently, several of the MHV cDNA fragments required subdivision into smaller subclones and cloning into transcription and translation negative vectors (Fig. 2B)

Engineering the clones of MHV: (A) The MHV genome was divided into seven contiguous fragments, named MHV-A through MHV-G, each flanked by unique type IIs restriction endonuclease sites, which when digested leave unique sticky ends that facilitate unidirectional assembly. Top bands represent MHV fragments, while all other bands represent digested vector. (B) The cloning vectors used to stably clone the contiguous fragments of MHV.
To ensure unidirectional ligation of all fragments, native or engineered type IIs restrictions sites were used to form junctions at the ends of each fragment, which allowed the restriction site to be removed by restriction digestion (1 3). A T7 promoter site was added at the 5′-end of the genome to facilitate in vitro transcription of the full-length cDNA fragment after ligation, and a poly(A) tail was included at the 3′-end (1 3) (Fig. 1C). The purpose of this review is to provide a detailed protocol that allows for efficient, systematic assembly and mutagenesis of coronavirus full-length cDNAs using class IIs restriction enzymes and the No See’m based mutagenesis approach. The pros and cons of this approach and an appraisal of what the future holds for this technology are discussed at the end (see Note 8).
2 Materials
2.1 Cell Culture and Lysis
-
1.
Two cell lines are used for MHV full-length RNA transfection experiments: DBT cells and baby hamster kidney cells expressing the MHV receptor (BHK-MHVr) (1 3).
-
2.
Minimum essential medium (Gibco -Invitrogen, Carlsbad, CA) supplemented with 10% tryptose phosphate broth, 10% Fetal Clone II (HyClone, Logan, UT), and 1% gentamicin/kanamycin. In addition, to select for BHK-MHVr cells expressing MHVr, geneticin (0.8 mg/ml) is added to the medium. The resistant clones were selected by 3X cell sorting for CEACAM1a expression and are maintained in geneticin as previously described by our group (1 3).
-
3.
Trypsin (0.25%) for removing cells from bottom of flask and versene (0.5 mM) for washes.
2.2 Transformation and Amplification of Plasmids
-
1.
Seven plasmids containing MHV fragments A-G cloned into pCR-XL-Topo (Invitrogen, Carlsbad, CA) or pSMART (Lucigen, Middleton, WI) vectors, as follows: pCR-XL-Topo-MHV-A, pSMART-MHV-B, pSMART-MHV-C, pSMART-MHV-D, pSMART-MHV-E, pSMART-MHV-F, and pSMART-MHV-G.
-
2.
Chemically competent Top10 cells (Invitrogen) for transformations and maintenance of plasmid DNA.
-
3.
Super Optimal Catabolite-repression (SOC) medium (Invitrogen, Carlsbad, CA) is used to stabilize the transformed chemically competent cells prior to plating them.
-
4.
Luria-Bertani (LB) plates supplemented with ampicillin (75 μg/ml) for pSMART vectors or kanamycin (50 μg/ml) for pCR-XL-Topo for growing colonies of bacteria containing the transformed plasmids.
-
5.
LB broth supplemented with ampicillin (75 μg/ml) for pSMART or kanamycin (50 μg/ml) for pCR-XL-Topo, 5 ml per colony for growing up individual colonies.
2.3 Plasmid Purification, Restriction Digestion Screen, and Digestion
-
1.
Qiaprep Spin Miniprep Kit (Qiagen, Valencia, CA.; cat. no. 27106) is used to purify plasmid DNA according to the manufacturer’s directions.
-
2.
Restriction enzymes and reagents as follows (New England BioLabs, Ipswich, MA): MluI (0.5–1 U/μl final volume), BsmBI (0.5–1 U/μl final volume), BglI (0.5—1 U/μl final volume), AhdI (0.5–1 U/μl final volume), SfiI (0.5–1 U/μl final volume), 10X bovine serum albumin (10 mg/ml), calf intestinal alkaline phosphatase (CIP) (1 U/μl); NEB 10X buffers 1–4 are used to digest the cDNA from the vector.
-
3.
Agarose gel for electrophoresis (0.8–1.0% w/v) of restriction digestions.
-
4.
LB broth supplemented with ampicillin (75 μg/ml) for pSMART or kanamycin (50 μg/ml) for pCR-XL-Topo vectors, 20 ml per culture for growing up larger stocks of colonies that look right by a restriction screen.
2.4 Fragment Purification and Ligation
-
1.
Qiaex II Gel Extraction Kit (Qiagen, Valencia, CA; Cat. no. 20010), 3 M sodium acetate pH 5.2, elution buffer from Qiagen miniprep kit for purifying bands cut out of the agarose gel.
-
2.
Reagents for chloroform extraction/isopropanol precipitation of fragments: chloroform, isopropanol, 70% ethanol, 95% ethanol, and elution buffer.
-
3.
A DNA spectrophotometer for quantifying the concentration of individual purified cDNA fragments.
-
4.
T4 DNA ligase plus 10X ligase buffer (New England Biolabs) for ligating the full-length cDNA.
2.5 In Vitro Transcription and Electroporation
-
1.
MHV N-gene amplified with SP6 promoter on the 5′-end is used as a template for generating N-gene transcripts. Primers for Sp6-N have been published (1 3). Transfecting in parallel with N-gene increases viral replication by roughly 15-fold.
-
2.
mMessage mMachine T7 Transcription Kit (Ambion, Austin, TX; cat. no. 1344) and mMessage mMachine SP6 Transcription Kit (Ambion, Austin, TX; cat. no. 1340) for generating full-length MHV RNA or SP6-N-gene RNA.
-
3.
One 0.4-cm Gene Pulser Cuvette (Bio Rad, Hercules, CA) for each transfection.
-
4.
BHK-MHVr cells are seeded at low density and grown in 150-cm2 flasks to 70% confluence, washed three times in nuclease-free PBS, and resuspended in cold PBS at a concentration of 1.0 ×107 cells/ml, and ∼5 ×106 DBT cells are infected by plating the electroporated BHK-MHVrs on top. We see greatly reduced transfection efficiencies if cells are allowed to approach confluence prior to harvest.
-
5.
Bio Rad (Hercules, CA) Gene Pulser Excel electroporator for doing the transfection.
2.6 Plaque Purification, Harvesting Viral RNA, and RT-PCR
-
1.
2X DMEM medium (Gibco)
-
2.
Fetal Clone II (Hyclone) or fetal bovine serum (Gibco)
-
3.
Low-melting-temperature cell culture grade agarose (Cambrex, Rockland, ME; cat. no. 50000)
-
4.
Phosphate buffered saline (Gibco).
-
5.
Trizol reagents (Invitrogen, Carlsbad, CA) for harvesting total RNA from infected cells.
-
6.
75% ethanol prepared in DepC water (Invitrogen, Carlsbad, CA)
-
7.
PCR/sequencing primers flanking the region of the mutation.
-
8.
SuperScript III Reverse Transcription kit (Invitrogen; cat. no. 18080-044) for reverse transcription of viral RNA to cDNA.
-
9.
pCR-XL-Topo cloning kit (Invitrogen, Carlsbad, CA; cat. no. K4500) for cloning the amplicon for sequencing.
2.7 Designing and Implementing Mutations
-
1.
MHV genomic sequence (Accession no. NC_001846) for designing primers.
-
2.
NebCutter 2.0 restriction digestion tool (New England BioLabs http://tools.neb.com/NEBcutter2/index.php) for analyzing sequences to ensure that the type IIs restriction site selected does not occur naturally in the wild-type sequence and is not introduced by the mutation.
-
3.
Web Primer on-line primer design tool (http://seq.yeastgenome.org/cgi-bin/web-primer) for development of primers for engineering mutations and for sequencing to verify that the correct change was incorporated into the virus.
-
4.
Expand Long PCR Kit (Roche, Basel, Switzerland; Cat. no. 11 681 834 001) for amplification of the mutant fragments.
-
5.
dNTPs (10 mM) prepared in nuclease-free water.
-
6.
Appropriate type IIs restriction enzyme for digestion of the two fragments.
-
7.
T4 DNA ligase for assembling the full-length mutant fragment.
-
8.
pCR-XL-Topo cloning kit for cloning the fragment into the pCR-XL-Topo vector, and for transforming the new plasmid for growth in LB medium.
3 Methods
Assembling the full-length clone of MHV involves a series of steps, each of which is important for amplifying and purifying the reagents necessary for the next step. Briefly, plasmids are transformed into chemically competent E. coli and plated in the presence of appropriate antibiotic selection. Colonies are isolated and grown overnight at room temperature (see Note 6); the DNA is harvested and inserts verified by a screening restriction digestion. It is important to screen the DNA prior to setting up the large-scale preps to ensure plasmid integrity. We typically harvest 50–75 μg of each plasmid DNA, restrict the DNA with the appropriate restriction endonucleases, and purify the viral cDNAs from agarose gels. It is equally important to keep the digested fragments free of contaminants, including residual carbohydrates from the gel extraction procedure or contamination with other fragments, particularly a wild-type version of a mutated fragment (see Note 2). Following gel extraction, chloroform extractions are performed to ensure that all carbohydrate contaminants are removed prior to preparing the ligation reaction (see Note 4).
It is also important to maintain an RNAse-free environment (as much as possible) after ligation of the full-length clone. Always wear gloves, keep the bench top RNAse-free by spraying with RNAse Zap (Ambion, Austin TX; cat. no. 9780), and always use DepC water (see Note 2) and barrier tips (see Note 3). In addition, washing the BHK-MHVr cells with PBS made with DepC water may help prevent degradation of the electroporated full-length viral RNA (see Note 2).
3.1 Transformation and Restriction Screening of the Seven Clones of MHV
-
1.
The seven fragments of MHV are maintained in either pCR-XL-Topo or pSMART vectors, and each vector encodes a different antibiotic resistance cassette. For pCR-XL-Topo cloning vector encodes kanamycin resistance, whereas the pSMART vector encodes ampicillin resistance (pSMART vectors are available with kanamycin or ampicillin resistance; in this case it is ampicillin). LB broth and LB agar plates with each antibiotic are required for growing the newly transformed bacteria. Although other vector-fragment combinations exist, we focus on the following in this report: pCR-XL-Topo-MHV-A, pSMART-MHV-B, pSMART-MHV-C, pSMART-MHV-D, pSMART-MHV-E, pSMART-MHV-F, and pSMART-MHV-G (1 3). Plasmids are stored at approximately 20 ng/μl at –80°C.
-
2.
Thaw each plasmid and the chemically competent Top10 cells (one vial per plasmid) on ice. Add 100 ng of each plasmid to an appropriately labeled vial of cells (50 μl) and incubate on ice for 30 min.
-
3.
Transform the cells by heat shock in a water bath maintained at 42°C for 2 min, followed by 2 min on ice.
-
4.
Add 200 μl SOC medium (without antibiotic) and rock the vials at room temperature for 2 h.
-
5.
Inoculate two LB plates (with appropriate antibiotic) for each plasmid, using 25 μl and 100 μl of culture and incubate the plates at room temperature for 48 h.
-
6.
Pick at least five colonies of each plasmid (from one or both plates) and inoculate each into a 15-ml conical containing 5 ml of LB broth with appropriate antibiotic. Grow the colonies for 16–24 h at 28.5°C in an incubator shaker set at 250 rpm. We have found that plasmid stability is enhanced at the lower temperature.
-
7.
Prepare a library plate by inoculating an LB agar plate (with appropriate selection) with 25 μl of the supernatant of each colony. The library plate should contain all of the colonies for one plasmid, and is grown at room temperature for 16–24 h and then stored at 4°C. Then spin the cultures at 3000 ×g for 10 min at 4°C, aspirate, and discard the supernatant.
-
8.
Plasmids are isolated from E. coli using the Qiagen Miniprep Kit, according to the manufacturer’s instructions. Follow the plasmid DNA purification scheme using the Qiaprep Spin Miniprep Kit and a microcentrifuge protocol, and elute the plasmid DNA in 50 μl of elution buffer preheated to 70°C.
-
9.
Screen each colony by restriction digestion to verify the insert size and restriction map.
-
a.
pCR-XL-Topo-MHV-A: 10 μl plasmid, 1 μl MluI, 1 μl BsmBI, 2 μl NEB Buffer 3, 6 μl water. Incubate at 55°C for 1 h.
-
b.
pSMART-MHV-B: 10 μl plasmid, 1 μl BglI, 2 μl NEB Buffer 3, 6 μl water. Incubate at 37°C for 1 h. Add 1 μl BsmBI and incubate at 55°C for an additional 1 h.
-
c.
pSMART-MHV-C: 10 μl plasmid, 1 μl BglI, 2 μl NEB Buffer 3, 6 μl water. Incubate at 37°C for 1 h. Add 1 μl BsmBI and incubate at 55°C for an additional 1 h.
-
d.
pSMART-MHV-D: 10 μl plasmid, 1 μl BsmBI, 1 μl BSA, 2 μl NEB Buffer 4, 5 μl water. Incubate at 55°C for 1 h. Add 1 μl AhdI and incubate at 37°C for an additional 1 h.
-
e.
pSMART-MHV-E: 10 μl plasmid, 1 μl BsmBI, 2 μl NEB Buffer 3, 7 μl water. Incubate at 55°C for 1 h.
-
f.
pSMART-MHV-F: 10 μl plasmid, 1 μl BsmBI, 2 μl NEB Buffer 3, 7 μl water. Incubate at 55°C for 1 h.
-
g.
pSMART-MHV-G: 10 μl plasmid, 1 μl SfiI, 1 μl BsmBI, 1 μl BSA, 2 μl NEB Buffer 2, 5 μl water. Incubate at 55°C for 1 h.
-
a.
-
10.
Run the restriction digestions out on a 0.8–1.0% agarose gel in TAE buffer (4.84 g/liter Tris base, 2 ml/liter 0.5 M EDTA pH 8.0, 1.14 ml/liter glacial acetic acid; pH to 8.5) at 100 mA, and determine that the plasmid DNAs have the appropriate banding pattern (Fig. 2A). The digests were designed to ensure that the desired MHV insert is the slowest migrating band in the gel (Fig. 2A). MHV-A is 5000 bp, MHV-B is 4672 bp, MHV-C is 1954 bp, MHV-D is 1451 bp, MHV-E is 2792 bp, MHV-F is 6985 bp, and MHV-G is 8711 bp (Figs. 1C,2A).
3.2 Amplification and Digestion of the Seven Fragments of MHV
-
1.
Identify replicates on the library plate for each fragment that looks right by restriction screen.
-
2.
Prepare a 50-ml conical containing 15 ml of LB broth with appropriate antibiotic and inoculate it with a streak from the library plate for each plasmid to be amplified. In general, one 15-ml culture of each plasmid DNA insert is sufficient for assembling at least two full-length MHV clones. Incubate for 12–16 h at 28.5°C in an incubator shaker set at 250 rpm. Then add 5 ml of LB plus antibiotic and allow growth to continue for an additional 4–6 h under the same conditions.
-
3.
Spin the cultures at 3000 ×g for 10 min at 4°C and discard the supernatants. Resuspend the pellet in 750 μl of Qiagen Miniprep P1 Buffer and transfer equally to three 1.5-ml microfuge tubes. Purify plasmids using the Qiaprep Miniprep Kit, and elute to a final volume of 50 μl in elution buffer heated to 70°C. Combine all three 50-μl aliquots of each fragment in a single tube.
-
4.
Digest the plasmids as follows:
-
a.
pCR-XL-Topo-MHV-A: 150 μl plasmid, 10 μl MluI, 20 μl NEB Buffer 3, 20 μl water. Incubate at 55°C for 1.5 h. Add 5 μl of CIP and incubate at 37°C for 1 h. CIP catalyzes the removal of 5′ phosphate groups from DNA, which prevents concatamers of MHV-A from occurring during ligation (1 5). CIP cannot be completely heat inactivated, so the reaction is chloroform extracted and isopropanol precipitated at this point to remove the CIP.
-
i.
For the chloroform extraction all steps are performed at room temperature: Add 25 μl of water, 50 μl 3 M sodium acetate, and 275 μl of chloroform to the reaction. Shake by hand for 2 min, and then spin at full speed in a microcentrifuge for 2 min. Remove the aqueous phase to a fresh tube and add an equal volume of isopropanol. Incubate at room temperature for 10 min, followed by centrifugation at full speed in a microcentrifuge for 10 min. Remove the supernatant and resuspend the pellet in 1 ml of 70% ethanol. Spin for 5 min at full speed, remove the supernatant, and resuspend in 95% ethanol. Spin for 5 min, remove supernatant, and air-dry the pellet for no more than 5 min. Resuspend the pellet in 170 μl elution buffer heated to 70°C.
-
ii.
Add 10 μl BsmBI and 20 μl NEB Buffer 3. Incubate for 2 h at 55°C.
-
i.
-
b.
pSMART-MHV-B: 150 μl plasmid, 10 μl BglI, 20 μl NEB Buffer 3, 10 μl water. Incubate at 37°C for 1.5 h. Add 10 μl BsmBI and incubate at 55°C for an additional 2 h.
-
c.
pSMART-MHV-C: 150 μl plasmid, 10 μl BglI, 20 μl NEB Buffer 3, 10 μl water. Incubate at 37°C for 1.5 h. Add 10 μl BsmBI and incubate at 55°C for an additional 2 h.
-
d.
pSMART-MHV-D: 150 μl plasmid, 10 μl BsmBI, 2 μl BSA, 20 μl NEB Buffer 4, 8 μl water. Incubate at 55°C for 2 h. Add 10 μl AhdI and incubate at 37°C for an additional 1.5 h. AhdI cuts the vector into two fragments, which allows the MHV-D fragment to be the largest band.
-
e.
pSMART-MHV-E: 150 μl plasmid, 10 μl BsmBI, 20 μl NEB Buffer 3, 20 μl water. Incubate at 55°C for 2 h.
-
f.
pSMART-MHV-F: 150 μl plasmid, 10 μl BsmBI, 20 μl NEB Buffer 3, 20 μl water. Incubate at 55°C for 2 h.
-
g.
pSMART-MHV-G: 150 μl plasmid, 10 μl SfiI, 2 μl BSA, 20 μl NEB Buffer 2, 18 μl water. Incubate at 55°C for 1.5 h. Add 5 μl of CIP and incubate at 37°C for 1 h. Do the chloroform extraction as described above (Section 3.2.4.a.i) and resuspend the pellet in 170 μl of elution buffer. Add 10 μl of BsmBI and 20 μl NEB buffer 3 and incubate at 55°C for 2 h.
-
a.
-
5.
Isolate the digested fragments by electrophoresis on a 0.8–1.0% gel in wells capable of holding 75 μl each (three wells per reaction) (Fig. 2A). The restriction products should be run for enough time to allow for high resolution of individual fragments.
-
6.
Excise the top band of the correct size for each fragment from the gel, making sure that it corresponds to the size of the MHV fragment (Fig. 2A) and is not an undigested vector-MHV fragment. Place the excised band in a 1.5-ml microfuge tube.
-
7.
Use the Qiaex II Gel Extraction Kit for extracting the DNA from the agarose as follows:
-
a.
Use the Qiaex II Agarose gel extraction protocol with the following modifications:
-
i.
Resuspend the excised band in 620 μl of QX1 buffer, 12.5 μl 3 M sodium acetate, and 11 μl QIAEX II resin. Incubate in a water bath at 55°C for a total of 12–16 min, agitating every 2 min. For fragments larger than 4 kb (MHV-A, MHV-B, MHV-F, and MHV-G), add 360 μl of water after 6 min of incubation. Follow the protocol for the remaining steps and elute to a final volume of 35 μl. Add the three replicates for each extraction into one tube (∼100 μl).
-
i.
-
a.
-
8.
A final chloroform only extraction is performed to remove any residual impurities that remain after the DNA extraction (see Note 1). Use a 1:1 volume of chloroform, shake for 2 min, and spin at top speed in a microcentrifuge at room temperature for 2 min. Remove the aqueous phase to a fresh tube.
3.3 Assembling the Viral Genome
-
1.
Quantitate each purified MHV fragment with a UV spectrophotometer (optional).
-
2.
Set up a shotgun ligation by adding relatively equivalent amounts of each fragment to a fresh 1.5-ml microfuge tube. For example, add 25 μl of each of the fragments with the lowest concentrations, 20 μl for fragments of intermediate concentrations, and only 15 μl of fragments with the highest concentrations, achieving a goal of relatively equivalent numbers of molecules of each fragment in the ligation mix (1 μg of 1000 bp DNA = 1.52 pmol = 9.1 ×1011 molecules). The reaction size will vary each time but a typical reaction is similar to this:
-
a.
20 μl of MHV-A.
-
b.
25 μl of MHV-B.
-
c.
25 μl of MHV-C.
-
d.
25 μl of MHV-D.
-
e.
25 μl of MHV-E.
-
f.
15 μl of MHV-F.
-
g.
20 μl of MHV-G.
-
h.
15 μl of water.
-
i.
20 μl of 10X ligation buffer.
-
j.
10 μl of ligase.
-
k.
Incubate the ligase reaction overnight at 4°C.
-
a.
-
3.
Chloroform extract and isopropanol precipitate the reaction as described above (Section 3.2.4.a.i), and resuspend the cDNA pellet in 10 μl of DepC water heated to 70°C.
-
4.
In vitro transcription of the full-length cDNA construct is conducted using the mMessage mMachine T7 Transcription Kit using the following recipe per reaction:
-
a.
7.5 μl of GTP (30 mM).
-
b.
25 μl 2X NTP/CAP (15 mM ATP, 15 mM CTP, 15 mM UTP, 3 mM GTP, and 12 mM cap analog).
-
c.
5 μl 10X Buffer.
-
d.
7.5 μl of full-length cDNA template.
-
e.
5 μl enzyme (RNA polymerase).
-
f.
Incubate the reaction at 40.5°C for 25 min, then at 37°C for 50 min, and then at 40.5°C for 25 min additional. Use a PCR-cycler for most consistent results.
-
g.
The remaining 2.5 μl of the ligation reaction and 5 μl of the RNA transcription reaction can be run on a gel to determine if full-length ligation occurred and whether in vitro transcription was successful (Fig. 3A). Use LE Agar (0.5%w/v) for the gel with 10% SDS added to the gel buffer and the running buffer. Treat 5 μl of RNA with DNAse and then add 2 μl of 10% SDS and 4 μl 5 mM EDTA prior to running the gel. For optimal resolution run the gel overnight at 30 mA.
Fig. 3. 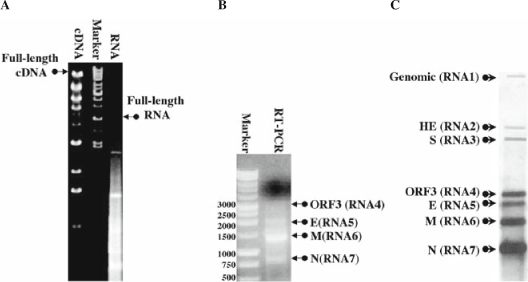
In vitro assembly of full-length cDNAs and transcripts and the recovery of infectious virus: (A) Running the cDNA from an assembly ligation results in a ladder of intermediates, as well as a full-length product ∼31.5 kb. After in vitro transcription, running RNA on a gel results in a smear of RNA including a full-length viral RNA, which runs at 16–17 kb. (B) Detecting viral replication can be accomplished using primers that anneal to the leader sequence and the N-gene. Using the antisense primer to perform reverse transcription and then both primers to amplify the target via RT-PCR generates a series of bands corresponding to several subgenomic mRNAs. (C) A northern blot showing the seven mRNAs of icMHV, using a probe that anneals to the 300 5′ nucleotides of the N-gene allows detection of genomic and all subgenomic mRNAs.
Fig. 4. 
Plaques produced by infection with recombinant MHV and a mutant: (A) DBT cells were infected with recombinant icMHV and overlaid with agar. Plaques formed at 24–36?h, and cells were stained with neutral red. MHV produces clear, uniform plaques of approximately 5?mm in diameter. (B) In contrast, some mutant forms of MHV produce differential plaques sizes. Here we show a mutant with a double amino acid change in Nsp10 (the lysines at positions 24 and 25 in Nsp10 were changed to alanines), which results in smaller overall plaque sizes, as well as a variety of different sizes, some with irregular borders.
-
a.
-
5.
Preparation of SP6-N-gene. Primers for adding the SP6 promoter onto the N-gene have been published previously (1 3). Use these primers (10 mM) along with the 1 μl of purified MHV-G fragment (Section 3.2, step 8) per reaction and reagents from the Expand Long Kit as follows:
-
a.
1 μl cDNA from MHV-G fragment.
-
b.
1 μl sense primer (10 mM).
-
c.
1 μl antisense primer (10 mM).
-
d.
1.75 μl dNTP (10 mM).
-
e.
5 μl Buffer #1.
-
f.
0.75 μl Expand Taq.
-
g.
39.5 μl water.
-
h.
Then run PCR through the following cycles: 1 cycle of 94°C for 5 min, followed by 30 cycles of 94°C for 1 min, 58°C for 1 min, 68°C for 3 min, and then 1 cycle of 68°C for 10 min followed by 4°C until ready for gel electrophoresis.
-
i.
The PCR product is then purified as described above (Section 3.2 steps 5–8).
-
a.
-
6.
In vitro transcription of the N-gene is conducted using the mMessage mMachine SP6 Transcription Kit with the following reaction recipe per reaction:
-
a.
12.5 μl 2X NTP/CAP (10 mM ATP, 10 mM CTP, 10 mM UTP, 2 mM GTP, and 8 mM cap analog).
-
b.
2.5 μl 10X Buffer..
-
c.
3.5 μl SP6-N-gene template
-
d.
2.5 μl enzyme (RNA polymerase).
-
e.
4 μl nuclease-free water.
-
f.
Incubate the reaction at 40.5°C for 25 min, then at 37°C for 50 min, and then at 40.5°C for 25 min more. Use a PCR-cycler for the most consistent results.
-
g.
Do one reaction for each transfection and one additional reaction for the negative control.
-
a.


3.4 Preparation of Cells
-
1.
Cell culture must be planned a day or two in advance, as three T150 flasks of BHK-MHVr cells at ∼70% confluency and one T150 flask of DBTs at 80–90% confluency are required to do one transfection and one negative control (see Note 5).
-
2.
Cells are maintained in the medium described above (Section 2.1, steps 1–3). For passage, the medium is aspirated and the attached cells are washed. Cells are removed from the flask with 0.25% trypsin, and then resuspended and passaged in the medium. Splitting a confluent T150 flask of BHK-MHVr cells (e.g., ∼2–3 ×107 cells) the night before at 1:5 will generally produce a T150 at ∼70% confluency the next day.
-
3.
Preparation of BHK-MHVr cells for transfection (per T150 flask):
-
a.
Remove the medium and wash the cells. Remove the cells from the flask with trypsin. Add 2 ml of trypsin to the flask and incubate for 5 min at 37°C. Remove cells from flask by tapping gently.
-
b.
Resuspend the trypsinized cells in 8 ml of medium and transfer cells from all three flasks to a sterile 50-ml conical.
-
c.
Spin the cells at ∼2000 ×g for 5 min and remove the medium, leaving the pellet in the conical.
-
d.
Resuspend the pellet in 10 ml of ice-cold PBS and spin at 2000 ×g for 5 min.
-
e.
Remove the PBS and resuspend the pellet in an additional 10 ml of ice-cold PBS.
-
f.
Transfer 10 μl of the cell suspension to a hemocytometer for counting, and spin the remaining suspension at ∼2000 ×g for 5–10 min, while counting the cells.
-
g.
Remove the PBS from the cells and resuspend the pellet in the volume of PBS that will give a concentration of 107 cells/ml. Counting 10 μl of cells from a 10-ml preparation translates into 104 cells per 1 mm2 (grid of 16 squares arranged 4 ×4) on the hemocytometer. Count all the cells in three of the 16 square grids and average them. Then, multiply this number by 1 ×104, which gives the concentration in cells/ml. In general, to get to a final concentration of 1 ×107 cells/ml resuspend the final pellet in 1/100 of the final count. For example, if the final average count is 200 cells, this would calculate to 2.0 ×106 cells/ml in a 10-ml volume for a total of 2.0 ×107 cells. Resuspend in 2 ml of PBS to get to a final concentration of 1 ×107 cells/ml. Keep cells on ice until ready for electroporation. A total volume of 1.6 ml of 1 ×107 cells/ml is required for one transfection and one negative control.
-
a.
-
4.
Preparation of DBT cells for infection. Only one confluent T150 flask of DBT cells is required.
-
a.
Remove the medium and wash the cells. Remove the cells from the flask with trypsin.
-
b.
Resuspend the cells in 10 ml of medium and transfer 1 ml of cells to a fresh T75 flask labeled for infection (∼5.0 ×106 cells). Bring up to 9.5 ml total volume with medium. Make one flask per transfection and one for a negative control.
-
a.
3.5 Transfection
-
1.
Add 800 μl of BHK-MHVr cells resuspended in PBS (free of magnesium or calcium) to a concentration of 107 cells to each 0.4-cm Gene Pulser Cuvette labeled for the appropriate transfection or control. Do one for each transfection and one for a negative control.
-
2.
Add 22.5 μl of N-transcript from the SP6-N-gene transcription reaction to each cuvette, including the negative control.
-
3.
Add 45 μl of the full-length RNA from the transcription reaction to the appropriately labeled cuvette.
-
a.
The remaining 5 μl is saved to run on an RNA gel (Fig. 3A).
-
a.
-
4.
Set the Bio Rad Gene Pulser Excel electroporator at 25 μF and 850 V.
-
5.
Place each cuvette into the Shock Pod™ shocking chamber and pulse three times.
-
6.
Allow each cuvette to incubate at room temperature for 10 min.
-
7.
Remove the electroporated cells from the cuvette into the appropriately labeled T75 flask of DBT cells (optional). Alternatively, allow the electroporated MHVr cells to settle onto a T75 flask and incubate at 37°C for 1–2 days to allow for the development of cytopathology.
-
8.
Incubate the flasks at 37°C in an incubator with 5% CO2.
3.6 Detection of Cytopathic Effect
-
1.
Wild-type MHV from the clone will generally produce cytopathic effects (CPE) within 8–12 h, and the syncytium across the monolayer is usually complete by 24 h. We typically recover between 1000 and 10,000 plaques (infectious center assay) following electroporation of transcripts. CPE is very obvious for DBT cells infected with MHV. Excess spike glycoproteins accumulate at and protrude from the cell surface, which facilitates fusion with the neighboring cells. This creates large multinucleated syncytia, which are usually observed by 8–12 h posttransfection. As the cells die, they lift off the flask, and so an efficient transfection of full-length viral RNA will result in a rapid clearing of the monolayer, usually within 16–24 h.
-
2.
Mutants generally come up more slowly and may take up to 72 h for CPE to be obvious. Highly debilitated mutants may not display obvious CPE. Passage usually selects for second-site revertant viruses that replicate efficiently, so extensive passage usually results in recombinant viruses containing multiple mutant alleles scattered across the genome. Reversions at the site of mutation can be reduced by fixing each engineered mutation with two or more nucleotide changes in the codon.
-
3.
Harvest supernatants by spinning the at ∼3000 ×g for 10 min at room temperature and aliquoting to fresh 2-ml screw cap tubes. Store at –80°C. These are labeled as passage zero (P0).
3.7 Plaque Purification
-
1.
Plaque purification allows isolation of single colonies of virus for propagation of stocks and verification of genotype (Fig. 4). This is done by plating a serial dilution of supernatants from P0 onto 60-mm dishes of DBT cells and then adding an agar overlay to prevent diffusion of the virus into the medium.
-
2.
One confluent T150 flask of DBT cells is sufficient to seed twenty 60-mm dishes labeled by dilution and virus. Resuspend washed cells in a total of 61 or 81 ml of medium and aliquot 3 or 4 ml of cells onto each of twenty 60-mm plates. Repeat as necessary for the correct number of plates. The cells are then incubated at 37°C overnight. They are ready to use at ∼95–100% confluency.
-
3.
When the cells are confluent, remove the medium. Dilute the supernatant from P0 using dilutions of one part P0 supernatant to nine parts PBS, ranging from 10–1 to 10–7 dilutions.
-
4.
Add 200 μl of the appropriate dilution onto a 60-mm plate of DBT cells and spread evenly by mixing back and forth every 15 min, while incubating at room temperature for 1 h. This is passage one (P1).
-
5.
Prepare the overlay ahead of time. Make enough to aliquot 5 ml of overlay onto each plate. For 100 ml:
-
a.
Autoclave 49 ml sterile water and 0.8 g of LE agar (low-melt) and incubate in a water bath at 55°C for 15 min.
-
b.
Add 40 ml 2X medium heated to 37°C.
-
c.
Add 10 ml FCII or FBS heated to 37°C.
-
d.
Add 1 ml of Gen/Kan heated to 37°C.
-
a.
-
6.
Carefully add 5 ml of overlay onto each plate. This must be accomplished at close to 40°–42°C as the medium rapidly solidifies below these temperatures and higher temperatures will adversely affect cell viability. Allow the plates to incubate 24–48 h at 37°C.
-
7.
Plaques for ic MHV resemble wt MHV (Fig. 4A), whereas mutant plaques can vary greatly (Fig. 4B). Select five plaques for each virus. For each plaque to be picked: use sterile technique to cut ∼1/3 in. off the end of a P1000 barrier tip, place the tip over the plaque, and push it through the agar, making sure that the surface of the plate and the agar are both manipulated so as to ensure that the entire plaque is extracted without any neighboring plaques. Place the picked medium into 300 μl PBS and incubate at 4°C for 30 min.
-
8.
The 300 μl containing the plaque and PBS is then used to infect a 60-mm plate of DBT cells, brought up as described above. To infect cells, remove the medium and pipette all 300 μl of the plaque and PBS onto a 60-mm plate of DBT cells, spread evenly by mixing back and forth every 10–15 min, and incubate at room temperature for 1 h. Then add 4 ml of medium to each plate. This is passage 2 (P2). Allow the infections to proceed at 37°C until CPE is obvious (usually 24–36 h). We typically only plaque purifiy recombinant viruses 1X as additional passages increase the probability of second-site mutations or reversions (see Note 7).
-
9.
Remove the medium from the 60-mm dishes, centrifuge at ∼3000 ×g for 10 min at room temperature and aliquot the supernatant into 1.5-ml microfuge tubes labeled P2.
-
10.
Resuspend the cells in 1 ml of Trizol reagents and dispense them into a 1.5-ml microfuge tube for isolation of total RNA.
3.8 Harvesting RNA, Reverse Transcription, and Verification of Viral Genotype
-
1.
Total RNA is harvested using Trizol reagents following the standard protocol. Briefly, approximately 4 ×106 cells are harvested in 1 ml of Trizol and the RNA is frozen at –80°C until further use:
-
a.
All steps are performed at room temperature. Start by thawing the frozen cells in Trizol and allowing them to sit at room temperature for 5 min.
-
b.
Add 200 μl chloroform and shake by hand for 15 sec.
-
c.
Spin at 12,000 ×g for 15 min.
-
d.
Remove aqueous phase to a fresh tube.
-
e.
Add 500 μl of isopropanol and incubate for 10 min.
-
f.
Spin at 12,000 ×g for 10 min.
-
g.
Remove supernatants and resuspend the pellet in 1 ml of 75% ethanol diluted in DepC water.
-
h.
Spin at 7500 ×g for 5 min.
-
i.
Remove ethanol and air-dry the pellet for 5–10 min.
-
j.
Dissolve the pellet 100 μl of DepC water heated to 70°C and incubate at 55°C for 10 min.
-
a.
-
2.
Design PCR/sequencing primers flanking the region of the mutation. Use the antisense primer as the gene-specific primer for SuperScript III reverse transcription with modifications to the protocol:
-
a.
Add 2 μl of RNA and 2 μl of antisense primer to a fresh tube and incubate at 70°C for 10 min.
-
b.
Add 2 μl of 0.1 M DTT.
-
c.
Add 2 μl of 10 mM dNTPs.
-
d.
Add 4 μl of 5X first-strand buffer.
-
e.
Add 7 μl of nuclease-free water.
-
f.
Add 1 μl of SuperScript III.
-
g.
Incubate at 55°C for 1 h.
-
h.
Incubate at 70°C for 20 min to inactivate the RT.
-
a.
-
3.
Amplify the region of interest with a 50 μl PCR reaction appropriate for the size and composition of the cDNA target.
-
4.
Isolate the DNA from PCR following the same procedures as described in Section 3.2, steps 5–7.i modifying the recipes for the 50-μl reaction size.
-
5.
Once isolated, clone the target into a vector of choice. We use pCR-XL-Topo, following the manufacturer’s recommended protocol.
-
6.
Transform, grow-up, and screen as described in Section 3.1. Screen by restriction digestion with EcoRI, and sequence clones that are of the appropriate size to verify genotype.

Engineering mutations with the No See’m approach: (A) The position of interest, a glutamate (CAG) to alanine (GTC) mutation at position 13354-56, is targeted and mapped to the MHV-E fragment. The vector sequence, the wild-type fragment, and the mutated fragment are analyzed to determine which type IIs restriction enzymes do not cut any of them, and one of these is selected as the restriction site to add. In this case BbsI is selected. (B) Primers are designed that add the mutation and the restriction site in proper orientation so that upon digestion the restriction site is eliminated and complementing sticky ends are produced. The BbsI cut sites are highlighted in gray. The mutated codon is bold and underlined. (C) PCR is conducted using vector specific primers along with the newly designed primers that incorporate the mutation of interest to produce two amplicons. These are cloned and purified, and then digested with BbsI and BsmBI to produce a mutated MHV-E fragment that can then be incorporated into the full-length clone.
3.9 Designing Mutants Using the No See’m Technology
-
1.
Identify a target of interest. This can be a single/double/triple codon change, a deletion, etc.
-
2.
Determine which type IIs restriction enzymes do not cut the vector, the target fragment, and the fragment with the newly designed mutations incorporated into the sequence. This can be accomplished with any restriction analysis tool; however, NEBcutter2.0 works well enough and is user friendly.
-
3.
Select one of the noncutters to add onto the end of each fragment. In Figs. 1C and 5, we selected a glutamate codon at position 13354-56 (CAG) and changed it to an alanine (GTC). To stabilize the mutation against reversion to the wt genotype, we generally alter the codon at multiple positions. This mutation maps to MHV-E as the fragment target, and enzyme BbsI does not cut the sequence or the pCR-XL-Topo vector.
-
4.
Design primers that incorporate the mutation into the target fragment utilizing the nonspecific site of the restriction cut site to engineer a junction oriented such that upon cleavage the restriction site is removed and complementing sticky ends remain (Fig.5).
-
5.
Amplify both fragments by PCR using the newly designed primers along with vector-specific sequencing primers and reaction conditions appropriate for the size of each target. For MHV-E fragment in pCR-XL-Topo M13F and M13R are used. Run PCR reactions on a gel, extract and purify amplicons of the correct size, clone into pCR-XL-Topo (or another vector of choice), transform, pick colonies and grow larger cultures, screen by restriction digestion, and sequence verify each amplicon (as described above).
-
6.
Digest each amplicon using the appropriate reaction conditions and enzymes. In the case of MHV-E in Fig. 5, we amplified with vector primers M13R and M13F, which ensured that the BsmBI sites were preserved at both ends of the fragment, so each amplicon has a BsmBI site and a newly engineered BbsI site. These amplicons are digested first with BbsI and NEB buffer 2 for 1.5 h at 37°C, followed by digestion with BsmBI at 55°C for 2 h. In addition, we digested wild-type MHV-E with just BsmBI, and purified the vector band (the smaller of the two). All of these digests are purified as described above.
-
7.
With this strategy, both amplicons and the vector are ligated in a single ligation reaction:
-
a.
11 μl of each purified band (amplicon 1, amplicon 2, vector).
-
b.
4 μl ligation buffer.
-
c.
3 μl ligase.
-
d.
Incubate at 4°C overnight.
-
a.
-
8.
Transform 5 μl of the ligation, plate, pick colonies and grow larger cultures, screen by restriction digestion, and sequence verify each amplicon.
-
9.
Build MHV virus using the newly mutated cDNA.
4 Notes
-
1.
Always do a chloroform-only extraction of the viral fragments after digestion as impurities left behind by the extraction kit can interfere with the DNA ligase.
-
2.
Always use PBS prepared in DepC water for washing cells. This reduces the opportunity for RNAses in the medium to degrade the full-length viral transcript prior to electroporation.
-
3.
Always use barrier tips for pipetting, particularly when working with wild-type MHV along side a mutant.
-
4.
Run digestions of wild-type fragment and mutant fragment on different gels with new running buffer each time. Wild-type MHV is so efficient at replication that even very low levels of cross-contamination of fragments will result in wild-type MHV out-competing any mutant.
-
5.
For best results use BHK-MHVr cells at or below 80% confluency.
-
6.
Clone stability is enhanced by growing everything at room temperature.
-
7.
Avoid passage of highly attenuated mutant viruses because reversions to wild-type and second-site reversions occur rapidly upon passage.
-
8.
There are several strengths and a few weaknesses to the No See’m infectious clone approach. Strengths over a single full-length clone include: (a) rapid mutagenesis of independent subclones in parallel; (b) mixing and matching of existing mutants from different subclones; (c) increased stability of smaller cDNAs; (d) safety of molecular clone of highly pathogenic viruses; (e) requirement for high individual expertise, which minimizes the chances of harmful use of the coronavirus molecular clone; (f) high recovery of recombinant viruses; (g) speed of recovery of recombinant virus; and (h) reduced sequencing costs (sequence subclone only). Weaknesses include: (a) technical expertise required for assembly; (b) the difficulty of in vitro transcription of full-length coronavirus RNAs; (c) troubleshooting the lengthy assembly process; and (d) obtaining reliable sequence information for the wild-type genome.
Well over a hundred mutants have been engineered into the clone of MHV using this approach, which far exceeds the number for all other coronavirus reverse genetic systems reported. Moreover, this technology has been used to rapidly develop reagents in response to newly identified emerging coronaviruses. For example, SARS-CoV was identified in the fall of 2002, and a full-length infectious clone was available the summer of 2003 (16). Piecemeal assembly of smaller subclones is within range of commercial DNA synthesis companies, allowing for synthetic reconstruction of coronavirus genomes. However, attempts to reconstruct other coronavirus genomes using this method have proven more difficult. Current problem areas that must be solved for the advancement of this technology include: (a) unreliable sequence information in the public databases; (b) mistake-prone amplification and cloning of AT-rich genomes (such as in human coronavirus NL63); (c) clone stability and sequence toxicity issues with different viruses; (d) size issues of the different fragments for different viruses; and (e) cell tropism issues with newly identified animal viruses (Bat CoV).
Once an infectious clone is established for a virus, rapid production of candidate vaccine strains (1 7– 1 9) and vectors for therapeutic gene delivery in animals and humans (20) is possible. Attenuating mutations can be identified, combinations can be engineered into the infectious clone, and recombinant viruses can be generated and tested. In SARS-CoV, this approach has identified zoonotic candidate vaccine strains (1 7,1 8) and a novel strategy for preventing recombination between wild-type and recombinant viruses by rewiring the viral communication network (1 9). This strategy provides a recombination safe vaccine platform that will likely redefine coronavirus vaccinology.
A tremendous amount of creativity in the coronavirus field has resulted in four independent and unique reverse genetic systems for a single virus family (1 1,1 3,1 4,2 1,2 2), all of which have helped pave the way for the emergence of the golden age of coronavirus genetics. We anticipate the discovery and genetic analysis of a large number of unique genetic functions in the coronavirus genome, and expect that these discoveries will translate to a better understanding of coronavirus replication, transcription, assembly, and mechanisms of pathogenesis of these important human and animal pathogens.
References
Lai, M. M., and D. Cavanagh, (1997) The molecular biology of coronaviruses Adv. Virus. Res. 48, 1–100.
Masters, P. (2006) The molecular biology of coronaviruses Adv. Virus Res. 66, 193–292.
Bost, A. G., et al. (2000) Four proteins processed from the replicase gene polyprotein of mouse hepatitis virus colocalize in the cell periphery and adjacent to sites of virion assembly J. Virol. 74(7), 3379–3387.
Brockway, S. M., et al. (2003) Characterization of the expression, intracellular localization, and replication complex association of the putative mouse hepatitis virus RNA-dependent RNA polymerase J. Virol. 77(19), 10515–10527.
Denison, M. R., et al. (1998) Processing of the MHV-A59 gene 1 polyprotein by the 3C-like proteinase Adv. Exp. Med. Biol. 440, 121–127.
Schiller, J. J., Kanjanahaluethai, A., and Baker, S. C. (1998) Processing of the coronavirus MHV-JHM polymerase polyprotein: identification of precursors and proteolytic products spanning 400 kilodaltons of ORF1a Virology 242(2), 288–302.
Baric, R. S., Curtis, K. M., and Yount, B. (2001) MHV subgenomic negative strand function Adv. Exp. Med. Biol. 494, 459–465.
Sawicki, S. G., and Sawicki, D. L. (1986) Coronavirus minus-strand RNA synthesis and effect of cycloheximide on coronavirus RNA synthesis J. Virol.. 57(1), 328–334.
Sawicki, S. G., and Sawicki, D. L. (1990) Coronavirus transcription: subgenomic mouse hepatitis virus replicative intermediates function in RNA synthesis J. Virol. 64(3), 1050–1056.
Sethna, P. B., Hung, S. L., and Brian, D. A. (1989) Coronavirus subgenomic minus-strand RNAs and the potential for mRNA replicons Proc. Natl. Acad. Sci. USA 86(14), 5626–5630.
Kuo, L., et al. (2000) Retargeting of coronavirus by substitution of the spike glycoprotein ectodomain: crossing the host cell species barrier J. Virol.. 74(3), 1393–1406.
Schickli, J. H., et al. (1997). The murine coronavirus mouse hepatitis virus strain A59 from persistently infected murine cells exhibits an extended host range J. Virol. 71(12), 9499–9507.
Yount, B., et al. (2002) Systematic assembly of a full-length infectious cDNA of mouse hepatitis virus strain A59 J. Virol. 76(21), 11065–11078.
Yount, B., Curtis, K. M., and Baric, R. S. (2000) Strategy for systematic assembly of large RNA and DNA genomes: transmissible gastroenteritis virus model J. Virol. 74(22), 10600–10611.
Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual. 2nd Ed. Cold Spring Harbor Laboratory, New York, Section 5.72.
Yount, B., et al. (2003) Reverse genetics with a full-length infectious cDNA of severe acute respiratory syndrome coronavirus Proc. Natl. Acad. Sci. USA 100(22), 12995–13000.
Baric, R. S., et al. (2006) SARS coronavirus vaccine development. Adv. Exp. Med. Biol. 581, 553–560.
Deming, D., et al. (2006) Vaccine efficacy in senescent mice challenged with recombinant SARS-CoV bearing epidemic and zoonotic spike variants. PLoS Med. 3(12), e525.
Yount, B., et al. (2006) Rewiring the severe acute respiratory syndrome coronavirus (SARS-CoV) transcription circuit: engineering a recombination-resistant genome Proc. Natl. Acad. Sci. USA 103(33), 12546–12551.
Yount, B., et al. (2005) Severe acute respiratory syndrome coronavirus group-specific open reading frames encode nonessential functions for replication in cell cultures and mice. J. Virol., 79(23), 14909–14922.
Almazan, F., et al. (2000) Engineering the largest RNA virus genome as an infectious bacterial artificial chromosome Proc. Natl. Acad. Sci. USA. 97(10), 5516–5521.
Thiel, V., et al. (2001) Infectious RNA transcribed in vitro from a cDNA copy of the human coronavirus genome cloned in vaccinia virus J. Gen. Virol. 82(Pt 6), 1273–1281.
Acknowledgments
We are grateful to Boyd Yount, who developed this protocol and has taught many of us how to use it.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2008 Humana Press, a part of Springer Science+Business Media, LLC
About this protocol
Cite this protocol
Donaldson, E.F., Sims, A.C., Baric, R.S. (2008). Systematic Assembly and Genetic Manipulation of the Mouse Hepatitis Virus A59 Genome. In: Cavanagh, D. (eds) SARS- and Other Coronaviruses. Methods in Molecular Biology, vol 454. Humana Press, Totowa, NJ. https://doi.org/10.1007/978-1-59745-181-9_21
Download citation
DOI: https://doi.org/10.1007/978-1-59745-181-9_21
Published:
Publisher Name: Humana Press, Totowa, NJ
Print ISBN: 978-1-58829-867-6
Online ISBN: 978-1-59745-181-9
eBook Packages: Springer Protocols