
Abstract
Rapid advances in bioengineering and biotechnology over the past three decades have greatly facilitated the production of recombinant proteins in Escherichia coli. Affinity-based methods that employ protein or peptide based tags for protein purification have been instrumental in this progress. Yet insolubility of recombinant proteins in E. coli remains a persistent problem. One way around this problem is to fuse an aggregation-prone protein to a highly soluble partner. E. coli maltose-binding protein (MBP) is widely acknowledged as a highly effective solubilizing agent. In this chapter, we describe how to construct either a His6- or a dual His6-MBP tagged fusion protein by Gateway® recombinational cloning and how to evaluate their yield and solubility. We also describe a simple and rapid procedure to test the solubility of proteins after removing their N-terminal fusion tags by tobacco etch virus (TEV) protease digestion. The choice of whether to use a His6 tag or a His6-MBP tag can be made on the basis of this solubility test.
You have full access to this open access chapter, Download protocol PDF
1 Introduction
A major time-consuming process in nearly all structural and functional studies of proteins is their overproduction and purification. Recombinant protein production in Escherichia coli has become the most popular platform for researchers who require large amounts of protein. Immobilized metal affinity chromatography (IMAC) with a polyhistidine tag (usually six consecutive histidine residues) has emerged as the most common and convenient method for purifying recombinant proteins. However, many His-tagged proteins form insoluble aggregates, especially in E. coli [1]. Before abandoning bacterial expression in favor of more complicated and costly eukaryotic systems, we suggest employing a simple strategy that combines the solubility-enhancing benefit conferred by E. coli maltose-binding protein (MBP) [2, 3] with the powerful advantage of IMAC [4], made possible by the use of a polyhistidine tag in a tandem configuration with MBP (His6-MBP) [5]. In this chapter, we describe how to construct either a His6-tagged or His6-MBP tagged fusion protein and conduct a few simple pilot experiments that are reliable predictors of protein production success, prior to extensive resource investment. The outcome of these pilot experiments dictates which N-terminal tag (His6 or His6-MBP) should be used for large-scale protein production.
2 Materials
2.1 Construction of Expression Vectors by Recombinational Cloning
-
1.
The Gateway ® destination vector pDEST566 (see Addgene plasmid #11517).
-
2.
The Gateway® destination vector pDEST-HisMBP (see Addgene plasmid #11085).
-
3.
The Gateway® destination vector pDEST527 (see Addgene plasmid #11518).
-
4.
PCR reagents, including thermostable DNA polymerase (see Note 1 ).
-
5.
Synthetic oligodeoxyribonucleotide primers for PCR amplification (see Fig. 1).
Fig. 1 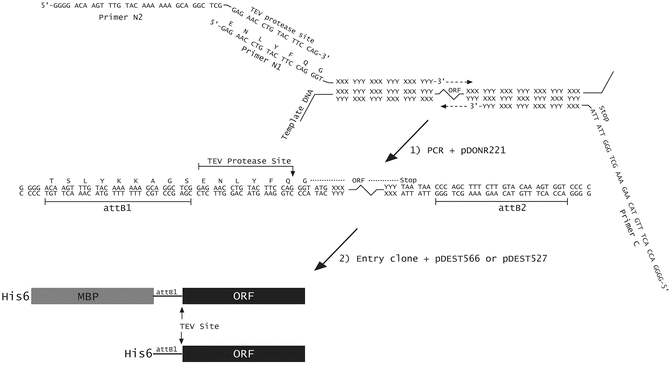
Construction of a His6- or His6-MBP fusion vector using PCR and Gateway ® cloning technology. The ORF of interest is amplified from the template DNA by PCR, using primers N1, N2, and C. Primers N1 and C are designed to base-pair to the 5′ and 3′ ends of the coding region, respectively, and contain unpaired 5′ extensions as shown. Primer N2 base pairs with the sequence that is complementary to the unpaired extension of primer N1. The final PCR product is recombined with the pDONR221 vector to generate an entry clone, via the BP reaction. This entry clone is subsequently recombined with pDEST527, pDEST566 or pDEST-HisMBP using LR Clonase to yield the final His6- or His6-MBP fusion vectors, respectively
-
6.
TE buffer (10 mM Tris–HCl pH 8.0, 1 mM EDTA).
-
7.
Agarose, buffer and an apparatus for submarine gel electrophoresis of DNA (see Note 2 ).
-
8.
MinElute Gel Extraction Kit (Qiagen, Valencia, CA) for the extraction of DNA from agarose gels.
-
9.
Chemically competent ccdB Survival™ 2 T1R cells (Life Technologies, Grand Island, NY) for propagating pDEST566, pDEST527, pDONR221, or any vector with a Gateway® cloning cassette.
-
10.
Competent gyrA + cells (e.g., DH5α, MC1061, HB101) (see Note 3 ).
-
11.
Gateway ® PCR Cloning System (Life Technologies).
-
12.
LB medium and LB agar plates containing ampicillin (100 μg/ml). LB medium: Add 10 g Bacto tryptone, 5 g Bacto yeast extract, and 5 g NaCl to 1 L of H2O and sterilize by autoclaving. For LB agar, also add 12 g of Bacto agar before autoclaving. To prepare plates, allow medium to cool until flask or bottle can be held in hands without burning, then add 1 ml ampicillin stock solution (100 mg/ml in H2O, filter-sterilized), mix by gentle swirling, and pour or pipet ca. 30 ml into each sterile petri dish (100 mm dia.).
-
13.
Reagents for small-scale plasmid DNA isolation (see Note 4 ).
-
14.
An incubator set at 37 °C.

2.2 Pilot Expression, Protease Cleavage, and Solubility Testing
-
1.
Competent Rosetta ™ 2(DE3) (EMD Millipore, Billerica, MA) (see Notes 5 and 6 ).
-
2.
A derivative of pDEST566 and pDEST527 that produces a His6-MBP fusion and His6-fusion protein , respectively, with a TEV protease recognition site in the linker between the N-terminal tag(s) and the passenger protein (see Subheading 3.1).
-
3.
LB agar plates and broth containing both ampicillin (100 μg/ml) and chloramphenicol (30 μg/ml). Prepare a stock solution of 30 mg/ml chloramphenicol in ethanol. Store at −20 °C for up to 6 months. (See Subheading 2.1, item 11 for LB broth, LB agar, and ampicillin stock solution recipes). Dilute antibiotics 1000-fold into LB medium or molten LB agar.
-
4.
Isopropyl-thio-β-d-galactopyranoside (IPTG), dioxane-free, ultrapure (American Bioanalytical, Natick, MA, USA). Prepare a stock solution of 200 mM concentration in H2O and filter-sterilize. Store at −20 °C.
-
5.
Shaker/incubator.
-
6.
Sterile baffled-bottom flasks (Bellco Glass, Inc., Vineland, NJ).
-
7.
Ni-NTA Agarose (Qiagen).
-
8.
AcTEV protease (Life Technologies), or TEV protease produced and purified as described [6].
-
9.
Two IMAC-compatible buffers that contain imidazole at 25 mM (for Buffer A) or 500 mM (for Buffer B) concentration. (For example, Buffer A: 25 mM Tris–HCl, 200 mM NaCl, 25 mM imidazole, pH 7.2; Buffer B: 25 mM Tris, 200 mM NaCl, 500 mM imidazole, pH 7.2.)
-
10.
4× SDS-PAGE sample buffer (Life Technologies) and 2-mercaptoethanol (Sigma Chemical Co., St. Louis, MO, USA).
-
11.
SDS-PAGE gel, electrophoresis apparatus, and running buffer (see Note 7 ).
-
12.
Gel stain (e.g., Gelcode® Blue from Pierce Protein Biology Products, Thermo Fisher Scientific, or PhastGel™ Blue R from GE Healthcare Life Sciences, Piscataway, NJ).
-
13.
Spectrophotometer.
-
14.
1.5 ml microcentrifuge tubes.
3 Methods
3.1 Recombinational Cloning to Generate His6- or His6-MBP Fusion Vector
The Gateway ® recombinational cloning system is based on the site-specific recombination reactions that mediate the integration and excision of bacteriophage λ, respectively, into and from the E. coli chromosome. For detailed information about this system, the reader is encouraged to consult the technical literature supplied by Thermo Fisher Scientific (Waltham, MA) (www.thermofisher.com/gateway).
3.1.1 pDEST566 and pDEST- HisMBP
To utilize the Gateway® system for the production of His6-MBP fusion proteins , one must first construct or obtain a suitable “destination vector.” Two destination vectors that can be used to produce His6-MBP fusion proteins (pDEST566 and pDEST-HisMBP) are available from the authors or the Addgene plasmid repository (www.addgene.org, plasmids #11517 and #11085, respectively).
The Gateway ® cloning cassette in pDEST566 and pDEST-HisMBP carries a gene encoding the DNA gyrase poison CcdB, which provides a negative selection against the destination vector, the donor vector, and various recombination intermediates so that only the desired recombinant is obtained when the end products of the recombinational cloning reaction are transformed into E. coli and grown in the presence of ampicillin. pDEST566, pDEST-HisMBP and other vectors that carry the ccdB gene must be propagated in a host strain with a gyrA mutation (e.g., E. coli DB3.1) that renders the cells immune to the action of CcdB or, alternatively, in a strain that produces the CcdB antidote CcdA (e.g., ccdB Survival™ 2 T1R cells).
3.1.2 pDEST527
A destination vector that can be used to produce His6-fusion proteins (pDEST527) is available from the authors or the Addgene plasmid repository (www.addgene.org, plasmid #11518). This vector is used for expression and affinity purification of proteins that are inherently soluble without the aid of solubility enhancers like MBP . It is a common practice in our laboratory to check the solubility of passenger proteins both with and without MBP and use the pDEST527-derived expression vector for large-scale expression and purification if the His6-tagged passenger proteins do not form insoluble aggregates in E. coli.
3.1.3 Gateway® Cloning Protocol
To construct a His6- or a His6-MBP fusion expression vector, we amplify the target open reading frame (ORF) by PCR, incorporating into the primers elements that are necessary for Gateway® cloning and downstream protein production. Next we perform successive BP and LR reactions. The 3′ ends of the primers include a sufficient number of nucleotides that are complementary to the template sequence to result in a 69 °C melting temperature (by modified Breslauer’s method, see http://www.thermoscientificbio.com/webtools/tmc/). This enables two-step PCR cycling using 72 °C as both the annealing and extension temperature. Proximal to the ORF-specific part of the forward primer, we add a sequence that encodes a TEV protease cleavage site preceded by an attB1 site to enable recombination. Because shorter primers are less expensive and because the TEV- and attB1-containing sequences are common to many of our experimental designs, we often use two overlapping forward primers, only one of which is ORF-specific (Fig. 1). An attB2 recombination site is added to the 5′ end of the ORF-specific portion of the reverse PCR primer. During early rounds of cycling, the inner, ORF-specific forward primer (N1) acts with the reverse primer (C) to create a template amplified by N2 and the same reverse primer in later rounds. To favor full-length product accumulation, the concentration of N1 is 20-fold lower than that of N2 and C (see Note 8 ).
-
1.
The PCR reaction mix is prepared as follows (see Note 9 ): 10–25 ng template DNA, 10 μl 2× Phusion Flash PCR Master Mix (contains all necessary reaction components except primers and template), 0.025 μM primer N1, 0.5 μM primer N2, 0.5 μM primer C, H2O (to 20 μl total volume).
-
2.
The reaction mixture is placed in a thin-walled tube in a thermal cycler with an appropriate program, such as the following: initial denaturation for 3 min at 98 °C; 30 cycles of 98 °C for 10 s and 72 °C for 15 s, and final extension at 72 °C for 60 s (see Note 10 ); hold at 4 °C.
-
3.
Purification of the PCR amplicon by agarose gel electrophoresis (see Note 2 ) is recommended.
-
4.
To create the His6-MBP fusion vector, the PCR product is recombined first into a donor vector, such as pDONR221, to yield an entry clone intermediate (BP reaction), and then into pDEST566 (LR reaction; see Note 11 ). Similarly, to create the His6-fusion vector, the PCR product is recombined first into a donor vector, such as pDONR221, to yield an entry clone intermediate (BP reaction), and then into pDEST527 (LR reaction) as detailed below.
-
(a)
Add to a microcentrifuge tube: 100 ng of the PCR product in 1–5 μl TE or H2O, 1.3 μl of 150 ng/μl pDONR vector DNA, and enough TE to bring the total volume to 12 μl. Mix well.
-
(b)
Thaw BP Clonase II enzyme mix on ice (2 min) and then vortex briefly (2 s) twice (see Note 12 ).
-
(c)
Add 3 μl of BP Clonase II enzyme mix to the components in (a) and vortex briefly; incubate the reaction at room temperature for at least 4 h (see Note 13 ).
-
(d)
Add to 10 μl of BP reaction: 2 μl of 150 ng/μl destination vector (pDEST566 or pDEST527) and 3 μl of LR Clonase II enzyme mix (see Note 12 ). Mix by vortexing briefly.
-
(e)
Incubate the reaction at room temperature for 2 h.
-
(f)
Add 2 μl of the proteinase K stop solution and incubate for 10 min at 37 °C.
-
(g)
Transform 1 μl of the reaction into 50 μl of appropriate competent E. coli, such as electrocompetent DH5α cells (see Note 3 ).
-
(h)
Spread the cells on an LB agar plate containing ampicillin (100 μg/ml), the selective marker for pDEST566, pDEST-HisMBP, and pDEST527. Incubate the plate at 37 °C overnight (see Note 14 ).
-
(a)
-
5.
Plasmid DNA is isolated from saturated cultures started from individual ampicillin-resistant colonies and screened by sequencing putative clones to ensure that there are no PCR-introduced mutations.
3.2 Pilot Fusion Protein Expression, TEV Protease Cleavage, and Solubility Testing
Before investing time and resources in the large-scale expression and purification of a protein, we perform a series of pilot experiments to assess protein production, TEV protease cleavage, and target protein solubility. First, we transform the sequence-verified expression plasmid into an appropriate expression strain and induce production of the fusion protein. Following ultrasonic disruption of the cells, we confirm that the fusion protein is present in the soluble fraction of the crude cell lysate. After passing this checkpoint, we check for successful TEV cleavage and sustained solubility of the protein of interest following its liberation from His6-MBP or His6 tag in the crude lysate. A problem at any of these steps can be addressed before scaling -up.
3.2.1 Protein Expression
-
1.
Transform competent Rosetta ™ 2(DE3) cells (see Notes 5 and 6 ) with the His6-MBP or His6 fusion protein expression vector and spread them on an LB agar plate containing ampicillin (100 μg/ml) and chloramphenicol (30 μg/ml). Incubate the plate overnight at 37 °C.
-
2.
Inoculate 5 ml of LB medium containing ampicillin (100 μg/ml) and chloramphenicol (30 μg/ml) in a culture tube with a single colony from the plate. Grow to saturation overnight at 37 °C with shaking.
-
3.
The next morning, inoculate 50 ml of the same medium in a 250 ml baffled-bottom flask with 0.5 ml of the saturated overnight culture.
-
4.
Grow the cells at 37 °C with shaking to mid-log phase (OD600nm ~ 0.5).
-
5.
Adjust the temperature to 30 °C (see Note 15 ) and add IPTG (1 mM final concentration).
-
6.
After 4 h, measure the OD600nm of the cultures (dilute cells 1:10 in LB to obtain an accurate reading). An OD600nm of about 3–3.5 is normal, although lower or higher densities are possible.
-
7.
Transfer 10 ml to a 15 ml conical centrifuge tube and pellet the cells by centrifugation (4000 × g) at 4 °C.
-
8.
Resuspend the cell pellets in 2–4 ml of lysis buffer and then transfer the suspensions to a 1.5-ml microcentrifuge tube. Normalize the cell suspensions using absorbance values (OD600) for comparisons.
Store the cell suspensions at −80 °C. Alternatively, the cells can be disrupted immediately and the procedure continued without interruption, as described below.
3.2.2 Sonication and Sample Preparation
-
1.
Thaw the normalized cell suspensions (expressing either a His6-tagged or His6-MBP tagged protein) at room temperature , then place them on ice.
-
2.
Lyse the cells by sonication (see Note 16 ).
-
3.
Prepare samples of the total (T) intracellular protein from the IPTG-induced cultures for SDS-PAGE by mixing 30 μl of each sonicated cell suspension with 10 μl of 4× SDS-PAGE sample buffer containing 10% (v/v) 2-mercaptoethanol.
-
4.
Pellet the insoluble cell debris (and proteins) by centrifuging the sonicated cell suspension from each culture at maximum speed in a microcentrifuge for 10 min at 4 °C.
-
5.
Prepare samples of the soluble (S) intracellular protein from the IPTG-induced cultures for SDS-PAGE by mixing 30 μl of each supernatant from step 4 with 10 μl of 4× SDS-PAGE sample buffer containing 10% (v/v) 2-mercaptoethanol.
3.2.3 TEV Protease Cleavage
To the soluble crude lysate prepared from induced cells (see Subheading 3.2, step 2) add approximately 0.05–0.10 mg/ml final concentration of pure TEV protease [6]. Mix and remove an aliquot for overnight incubation at room temperature ; incubate remaining reaction at 4 °C overnight. Spin these tubes at maximum speed in a microcentrifuge for 5 min and analyze the supernatant (TEV+).
3.2.4 SDS-PAGE
We typically use precast NuPAGE gradient gels for SDS-PAGE to assess the yield and solubility of fusion proteins (see Note 7 ). The reader may choose any appropriate SDS-PAGE formulation appropriate for the protein size and laboratory preference.
-
1.
Prepare new samples by mixing 30 μl of each solution with 10 μl of 4× SDS-PAGE sample buffer containing 10% (v/v) 2-mercaptoethanol.
-
2.
Heat the T, S, and TEV+ samples at 90 °C for about 5 min and then spin them at maximum speed in a microcentrifuge for 5 min.
-
3.
Assemble the gel in the electrophoresis apparatus, fill it with SDS-PAGE running buffer, load the samples (5–20 μl) and carry out the electrophoretic separation according to standard lab protocols. T, S, and TEV+ samples are loaded in adjacent lanes to allow easy assessment of solubility. Molecular weight standards may also be loaded on the gel, if desired.
-
4.
Stain the proteins in the gel with GelCode® Blue reagent, PhastGel™ Blue R, or a suitable alternative.
3.2.5 Interpreting the Results
The overexpressed fusion proteins should be apparent as the predominant protein present on the gel. Examining the heaviest band relative to a molecular weight standard should confirm that the fusion is about the size of the protein of interest for His6 tagged or plus 42 kDa (the approximate size of MBP ) for His6-MBP fusions. Placing the total and soluble fractions next to each other on the gel allows easy comparison and determination of solubility. A side-by-side analysis of His6 tagged versus His6-MBP tagged proteins will help to choose which tag to use for large-scale expression and purification.
Figure 2 illustrates the benefit of using MBP as a solubility enhancer . Lane 1 indicates that, upon induction, the Rosetta 2(DE3) expression strain was able to produce Chikungunya virus (ChikV) protease from a plasmid encoding the His6-tagged protein. However, lane 2 reveals that most of the His6-tagged protein is not found in the soluble fraction. In contrast, lanes 3 (total protein) and 4 (soluble protein) clearly demonstrate that a significant portion of the His6-MBP-ChikV protease fusion protein is soluble when produced in the same strain. Lanes 6 (total protein) and 7 (soluble protein) illustrate the solubility of catalytically inactive Middle East Respiratory Syndrome coronavirus 3C-like protease (MERS-CoV 3CLpro C148A) as a His6-MBP tagged protein. Lanes 8 (total protein) and 9 (soluble protein) reveal that a very similar solubility is obtained even with His6-fusions of MERS-CoV 3CLpro. Hence, MERS-CoV 3CLpro is an example of a protein that does not require a solubility enhancer for overproduction in E. coli.

Comparison of the solubility of His6-tagged and His6-MBP-tagged fusion proteins . Lanes 1–4 of the SDS-PAGE gel represent protein extracted from Rosetta 2(DE3) cells expressing either His6-tagged ChikV protease or His6-MBP-ChikV protease from the appropriate plasmids. Lane M: SeeBlue Plus2 pre-stained marker standards. Lane 1: His6-ChikV total protein. Lane 2: His6-ChikV soluble protein. Lane 3: His6-MBP-ChikV total protein. Lane 4: His6-MBP-ChikV soluble protein. Lanes 6–9 represent protein extracted from Rosetta 2(DE3) cells expressing either His6-tagged or His6-MBP tagged MERS-CoV 3CLpro from the appropriate plasmids. Lane 6: His6-MBP-MERS-CoV 3CLpro total protein. Lane 7: His6-MBP-MERS-CoV 3CLpro soluble protein. Lane 8: His6-MERS-CoV 3CLpro total protein. Lane 9: His6-MERS-CoV 3CLpro soluble protein
The soluble crude lysate fractions can be used to test the ability of TEV protease to cleave the tags in vitro. In lanes 1 and 2 of Fig. 3, which correspond to the soluble lysate and soluble products of the cleavage reaction, respectively, the band representing the fusion protein has largely disappeared, and three significant new bands have appeared: a 42-kDa band for His6-MBP, 28-kDa band for His-TEV protease, and 37-kDa band migrating at the expected size of ChikV protease. The prominent band corresponding to ChikV protease indicates that it remains soluble after cleavage from MBP , suggesting that it is probably properly folded. Similarly, the cleavage of His6-tagged MERS-CoV 3CLpro was conducted and lanes 3 and 4 indicate the soluble protein and soluble products of TEV digestion, respectively. The cleaved His6-tag and the MERS-CoV 3CLpro (33-kDa) are clearly visible on the gel. Had the TEV protease failed to cleave the fusions or had the target protein become insoluble after cleavage, troubleshooting would have been necessary. Otherwise, having successfully passed these diagnostic tests, the production and purification of the protein may now be scaled up as described [4].

Small-scale pilot expression and digestion of fusion protein with TEV protease. ChikV protease and MERS-CoV 3CLpro were expressed from derivatives of pDEST-HisMBP and pDEST527, respectively, in Rosetta 2(DE3) cells as described (see Subheading 3.2) and analyzed by SDS-PAGE. Lane M: SeeBlue Plus2 pre-stained marker standards. Lane 1: His6-MBP-ChikV protease soluble protein. Lane 2: TEV protease digest of lane 1 sample, soluble protein. Lane 3: His6-MERS-CoV 3CLpro soluble protein. Lane 4: TEV protease digest of lane 3 sample, soluble protein (see Subheading 3.2, steps 4 and 5). Slanted arrows indicate the positions of the liberated passenger proteins
A typical large-scale purification profile of a His6-tagged protein (MERS-CoV 3CLpro) and a His6-MBP tagged protein (ChikV protease) are shown in Fig. 4. The bands representing fusion proteins and their tagless forms during the purification process are indicated in the figure.

Purification of fusion proteins by immobilized-metal affinity chromatography (IMAC). Purification monitored by SDS-PAGE at different stages. In panels A (ChikV protease) and B (MERS-CoV 3CLpro), the following gel-loading pattern applies. Lane M: SeeBlue Plus2 pre-stained marker standards. Lane 1, soluble lysate (crude); lane 2, flow-through from first IMAC column (unbound); lane 3, eluate from first IMAC column; lane 4, products of TEV protease digest; lane 5, flow-through from second IMAC column; lane 6, final sample after size exclusion chromatography
3.2.6 Troubleshooting
Not every fusion protein (His6-tagged or His6-MBP tagged) will be highly soluble. However, solubility usually can be increased by reducing the temperature of the culture from 30 to 18 °C during the time that the fusion protein is accumulating in the cells (i.e., after the addition of IPTG). In some cases, the improvement can be quite dramatic. It may also be helpful to reduce the IPTG concentration to a level that will result in partial induction of the fusion protein. Under these conditions, longer induction times (18–24 h) are required to achieve a reasonable yield.
Occasionally, a passenger protein may accumulate in a soluble but biologically inactive form after intracellular processing of an MBP fusion protein. Exactly how and why this occurs is unclear, but we suspect that fusion to MBP somehow enables certain proteins to evolve into kinetically trapped folding intermediates that are no longer susceptible to aggregation. Therefore, although solubility after intracellular processing is generally a useful indicator of a passenger protein’s folding state, it is not absolutely trustworthy. For this reason, we strongly recommend employing a biological assay (if available) at an early stage to confirm that the passenger protein is in its native conformation. For those proteins that are soluble as His6-tagged fusions, there is no need to use a solubility enhancer for large-scale expression and purification.
When fusion proteins are resistant to digestion by TEV protease, longer incubation times, higher protease concentrations, and/or higher temperature (up to 30 °C) may be helpful. In especially problematic cases, the efficiency of protease digestion can often be improved by inserting additional amino acid residues between the TEV protease recognition site and the N terminus of the passenger protein. We have used both polyglycine (Gly3) and a FLAG-tag epitope in this position with good results [7].
Occasionally, the His6-MBP moiety may exhibit a tendency to “stick” to the cleaved passenger protein and co-purify with it during the second IMAC step, as occurred with the ChikV protease (Fig. 4a). This problem most likely could be alleviated by increasing the salt concentration in the IMAC buffer. However, in this case the final size exclusion chromatography step separated the ChikV final product from the His6-MBP tag.
4 Notes
-
1.
We recommend a processive, high-fidelity polymerase such as Phusion (Thermo Fisher or New England Biolabs, Ipswich, MA, USA) to reduce cycling times and minimize the occurrence of mutations during PCR.
-
2.
We typically purify fragments by horizontal electrophoresis in 1–2% Certified Molecular Biology Agarose (Bio-Rad, Hercules, CA) gels run in sodium borate solution [8] using standard submarine equipment. DNA fragments are extracted from slices of ethidium bromide-stained gel using a MinElute Gel Extraction Kit (Qiagen) in accordance with the instructions of the manufacturer.
-
3.
Any gyrA + strain of E. coli can be used. We prefer ElectroMAX™ DH5α-E™ Competent Cells (Life Technologies) because they are easy to use and very efficient.
-
4.
We prefer the QIAprep™ Spin miniprep kit (Qiagen), but similar kits can be obtained from a wide variety of vendors.
-
5.
Chemically competent cells are transformed according to the manufacturer’s instructions. Electrocompetent cells can be purchased or prepared. Briefly, the cells are grown in 1 L of LB medium (with antibiotics, if appropriate) to mid-log phase (OD600 ~ 0.5) and then chilled on ice. The cells are pelleted at 4 °C, resuspended in 1 L of ice-cold H2O and pelleted again. After several such washes with H2O, the cells are resuspended in 3–4 ml of 10% glycerol, divided into 50 μl aliquots, and immediately frozen in a dry ice–ethanol bath. Competent cells are stored at −80 °C. Electrotransformation procedures can be obtained from the electroporator manufacturers (e.g., Bio-Rad, BTX, Eppendorf). Immediately prior to electrotransformation, the cells are thawed on ice and mixed with 10–100 ng of DNA (e.g., a plasmid vector or a Gateway® reaction). The mixture is placed in an ice-cold electroporation cuvette and electroporated according to the manufacturer’s recommendations (e.g., 1.5 kV pulse in a cuvette with a 1 mm gap). One milliliter of SOC medium [9] is immediately added to the cells and they are allowed to grow at 37 °C with shaking (ca. 250 rpm) for 1 h. 5–200 μl of the cells are then spread on an LB agar plate containing the appropriate antibiotic(s).
-
6.
If the open reading frame encoding the passenger protein contains codons that are rarely used in E. coli (http://www.doe-mbi.ucla.edu/cgi/cam/racc.html), this can adversely affect the yield of a protein. In such cases, it is advisable that the expression strain carries an additional plasmid that codes for rare-codon-tRNA genes. The pRIL plasmid (Stratagene, La Jolla, CA) is a derivative of the p15A replicon that carries the E. coli argU, ileY, and leuW genes, which encode the cognate tRNAs for AGG/AGA, AUA, and CUA codons, respectively. pRIL is selected for by resistance to chloramphenicol. In addition to the tRNA genes for AGG/AGA, AUA, and CUA codons, the pRARE accessory plasmid in the Rosetta ™ host strain (Novagen, Madison, WI) also includes tRNAs for the rarely used CCC and GGA codons. Like pRIL, the pRARE plasmid is a chloramphenicol-resistant derivative of the p15A replicon. Both of these tRNA accessory plasmids are compatible with derivatives of pDEST566, pDEST-HisMBP or pDEST527. Another option is to prepare the insert (cDNA of interest) synthetically, using E. coli-preferred codons.
-
7.
We find it convenient to use precast SDS-PAGE gels, running buffer, molecular weight standards, and electrophoresis supplies from Life Technologies.
-
8.
Alternatively, the PCR reaction can be performed in two separate steps, using primers N1 and C in the first step and primers N2 and C in the second step. The PCR amplicon from the first step is used as the template for the second PCR. All primers are used at the typical concentrations for PCR in the two-step protocol.
-
9.
The PCR reaction can be modified in numerous ways to optimize results, depending on the nature of the template and primers. See Ref. 9 (Vol. 2, Chapter 8) for more information.
-
10.
PCR cycle conditions can also be varied based on reagents and consumables chosen, template complexity and gene length. For example, when using Phusion Flash High-Fidelity PCR Master Mix, extend the cycle for 15 s per kb of DNA. Consult the directions provided by the manufacturer of your thermostable polymerase.
-
11.
This “one-tube” Gateway ® protocol bypasses the isolation of an “entry clone” intermediate. However, the entry clone may be useful if you intend to experiment with additional Gateway® destination vectors, in which case the BP and LR reactions can be performed sequentially in separate steps; detailed instructions are included with the Gateway® PCR kit. Alternatively, entry clones can easily be regenerated from expression clones via the BP reaction, as described in the manual.
-
12.
Clonase enzyme mixes should be thawed according to the manufacturer’s directions.
-
13.
At this point, we remove a 5 μl aliquot from the reaction and add it to 0.5 μl of proteinase K stop solution. After 10 min at 37 °C, we transform 2 μl into 50 μl of competent DH5α cells (see Note 3 ) and spread 100–200 μl on an LB agar plate containing kanamycin (25 μg/ml), the selective marker for pDONR221. From the number of colonies obtained, it is possible to gauge the success of the BP reaction. Additionally, entry clones can be recovered from these colonies in the event that no transformants are obtained after the subsequent LR reaction.
-
14.
If very few or no transformants are obtained after the BP or LR reactions, the efficiency of the process can be improved by incubating the reactions overnight.
-
15.
We have found that decreasing the induction temperature to 30 °C increases the quality and solubility of the fusion protein without significantly decreasing the yield, especially in the presence of glucose. We also test 18 °C inductions if necessary, in which case the inductions are usually longer (18–24 h).
-
16.
We routinely disrupt cells in 1.5-ml microcentrifuge tubes on ice with two or three 30 s pulses using a VCX600 sonicator (Sonics and Materials, Inc.) with a microtip at 38% power. The cells are cooled on ice between pulses.
References
Waugh DS (2005) Making the most of affinity tags. Trends Biotechnol 23:316–320
Kapust RB, Waugh DS (1999) Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci 8:1668–1674
Fox JD, Routzahn KM, Bucher MH et al (2003) Maltodextrin-binding proteins from diverse bacteria and archaea are potent solubility enhancers. FEBS Lett 537:53–57
Tropea JE, Cherry S, Nallamsetty S et al (2007) A generic method for the production of recombinant proteins in Escherichia coli using a dual hexahistidine-maltose-binding protein affinity tag. Methods Mol Biol 363:1–19
Routzahn KM, Waugh DS (2002) Differential effects of supplementary affinity tags on the solubility of MBP fusion proteins. J Struct Funct Genom 2:83–92
Kapust RB, Tozser J, Fox JD et al (2001) Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic efficiency. Protein Eng 14:993–1000
Fox JD, Waugh DS (2003) Maltose-binding protein as a solubility enhancer. Methods Mol Biol 205:99–117
Brody JR, Kern SE (2004) Sodium boric acid: a Tris-free, cooler conductive medium for DNA electrophoresis. BioTechniques 36:214–216
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
Acknowledgments
We thank Karina Keefe and Danielle Needle for constructing the ChikV protease and MERS-CoV 3CLproC148A expression vectors, respectively. This research was funded by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media LLC
About this protocol
Cite this protocol
Raran-Kurussi, S., Waugh, D.S. (2017). Expression and Purification of Recombinant Proteins in Escherichia coli with a His6 or Dual His6-MBP Tag. In: Wlodawer, A., Dauter, Z., Jaskolski, M. (eds) Protein Crystallography. Methods in Molecular Biology, vol 1607. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-7000-1_1
Download citation
DOI: https://doi.org/10.1007/978-1-4939-7000-1_1
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-6998-2
Online ISBN: 978-1-4939-7000-1
eBook Packages: Springer Protocols