
Abstract
Subcellular fractionation is an important tool used to separate intracellular organelles, structures or proteins. Here, we describe a stepwise protocol to isolate two types of lytic granules, multicore (MCG), and single core (SCG), from primary murine CTLs. We used cell disruption by nitrogen cavitation followed by separation of organelles via discontinuous sucrose density gradient centrifugation. Immunoisolation with a Synaptobrevin 2 antibody attached to magnetic beads was then used to harvest Synaptobrevin 2 positive granules for immunoblotting, mass spectrometry, electron, and light microscopy.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
- Lytic granule
- Subcellular fractionation
- Sucrose density gradient centrifugation
- Immunoisolation
- Magnetic beads
- Nitrogen cavitation
- Mass spectrometry
1 Introduction
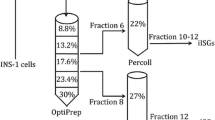
Cytotoxic granules (CGs) are specialized secretory lysosomes of cytotoxic T lymphocytes (CTL) and NK cells [1,2,3,4,5]. Over decades, cell biologists and immunologists have tried to isolate these secretory lysosomes and their luminally active components such as Granzymes and Perforin, to characterize the granules and understand their role in T cell function [6,7,8,9,10,11]. The presence of a dense core was demonstrated using electron microscopy [1, 12]. The protein composition of isolated secretory lysosomes was first reported in human NK cells [10] and later on in human CTLs [8, 9]. Methods for granule isolation differ, with various protocols that result in uneven purity and granule subpopulations, although the typical lysosomal markers such as LAMP-1, CD63, Cathepsin variants, and Granzymes [8,9,10] are present. Electron microscopic analysis revealed the heterogeneity of secretory lysosomes, suggesting subclasses of cytotoxic granules though they all contain cytotoxic molecules and are released at the synaptic cleft adjacent to target cells [8, 13]. Since Synaptobrevin 2 (Syb2) is a specific marker for CGs in mouse CTLs [14], we used Syb2-mRFP knock-in mice (Syb2 KI) for organelle purification and proteomics. CGs are endogenously labeled and can therefore be visualized by red fluorescence during the different isolation steps [15]. To avoid chemical and physical stress on enzymes or subcellular compartments and to preserve the integrity of the organelles, homogenization of the cultivated cells was done by nitrogen cavitation [16]. After several low-speed differential centrifugation steps to separate nuclei and intact cells, cellular fractionation was done by high-speed discontinuous sucrose density gradient centrifugation. We selectively chose the sucrose gradient composition (0.3 M, 0.8 M, 1.0 M, 1.2 M, 1.4 M, and 1.6 M), speed and time of ultracentrifugation were able to enrich Syb2-mRFP positive organelles in two fractions at the interface between 1.0 and 1.2 M (fraction 6) and 1.2 and 1.4 M sucrose (fraction 8) [15]. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed directly on the collected gradient fractions. Western blots were then probed for markers specific for CG (Syb2, Perforin, Granzyme B), plasma membrane (Na+/K+-ATPase), mitochondria (SDHA), and cytoskeleton (β-Actin, α-Tubulin). Magnetic beads coupled to anti-Syb2 antibody were used for immunoisolation of organelles in fractions 6 and 8. After organelle binding, the magnetic beads can be immediately used for high-resolution microscopy, immunoblotting, electron microscopy, and mass spectrometry.
Thus, with this isolation strategy combined with a variety of methods, we were able to characterize two populations of Syb2 organelles, SCG and MCG, the latter containing SMAPs [15, 17].
2 Materials
All solutions were prepared with deionized water and analytical grade reagents. Prepare and store all reagents at room temperature unless indicated otherwise.
2.1 Primary Murine T Cells
-
1.
0.8–1.5 × 108 day 3–4 activated CD8+ T cells isolated from one Syb2 KI mouse (see Note 1).
-
2.
BD FACSAria III analyzer (BD Biosciences) for flow cytometry analysis.
-
3.
Cell surface-specific FITC-, APC-, or PE-conjugated antibodies against CD44, CD62L, and CD25 (BD Pharmingen or eBioscience).
2.2 Cell Homogenization
-
1.
Wash buffer: D-PBS (137.9 mM sodium chloride, 1.47 mM potassium phosphate monobasic, 2.67 mM potassium chloride, 8.09 mM sodium phosphate dibasic) with 0.1% bovine serum albumin (BSA) and 2 mM ethylenediaminetetraacetic acid (EDTA), pH 7.4.
-
2.
Protease inhibitors: 1 mg/mL Pepstatin A (in DMSO), 10 mM E64 (in water), and 400 mM 4-benzenesulfonyl fluoride hydrochloride (Pefabloc SC in water or equivalent). Store separately at −20 °C.
-
3.
Sucrose solubilization buffer: 20 mM HEPES and 5 mM EDTA, pH 7.3 to dissolve sucrose.
-
4.
Homogenization buffer: 300 mM sucrose, 20 mM HEPES, 5 mM EDTA, supplemented with protease inhibitors (2 mM Pefabloc SC, 10 μM E64 and 1 μg/mL Pepstatin A), pH 7.3 (see Note 2). Store at 4 °C.
-
5.
Pre-chilled cell cavitation/disruption bomb (Parr 1019HC T304 SS/Parr Instrument Company) connected to a Nitrogen gas tank.
-
6.
Micromagnetic stir bar.
-
7.
0.4% Trypan blue solution in 0.8X D-PBS or equivalent.
2.3 Subcellular Fractionation
-
1.
Sucrose solutions for gradient: 0.8, 1.0, 1.2, 1.4, and 1.6 M sucrose in 20 mM HEPES with 5 mM EDTA and protease inhibitors (2 mM Pefabloc SC, 10 μM E64, and 1 μg/mL Pepstatin A), pH 7.3 (see Note 3).
-
2.
Refractometer.
-
3.
Beckmann ultracentrifuge with a SW40Ti rotor (or equivalent) and corresponding 14 mL transparent Ultra Clear centrifuge tubes (Beckmann 344,060, or equivalent).
-
4.
10 mL syringes and 20-G needles.
-
5.
1.5 mL low protein binding tubes and pipette tips.
-
6.
Sample buffer for immunoblotting: Lithium dodecyl sulfate (LDS) sample-buffer (4X) and 1 M DTT.
-
7.
Pierce™ 660 nm Protein Assay reagent (see Note 4).
2.4 Immunoisolation of MCG and SCG
-
1.
Dynabeads™ Protein G magnetic beads (Invitrogen).
-
2.
Dynabead wash buffers: D-PBS with 0.01% Tween 20 (pH 7.4).
-
3.
Anti-Syb2 antibody (clone 69.1, Synaptic Systems or equivalent).
-
4.
Dilution buffer: 320 mM KCl, 10 mM HEPES, 5 mM EDTA solution in distilled water (pH 7.3).
-
5.
Wash buffer: 160 mM KCl 10 mM HEPES, pH 7.3.
-
6.
Rotator: Intelli mixer RM-2M (neoLab).
-
7.
1.5 mL low protein binding tubes and pipette tips.
-
8.
Magnet to separate Dynabeads: DynaMag™-2 Magnet (Invitrogen).
-
9.
Sample buffer for immunoblotting: Lithium dodecyl sulfate (LDS) sample-buffer (4X) and 1M DTT.
-
10.
Confocal microscope (LSM 780, Zeiss) or equivalent.
3 Methods
3.1 Cell Homogenization
-
1.
Collect CD8+ cells after expansion to 0.8–1.5 × 108 total CD8+ cells, adequate for granule isolation.
-
2.
Test the quality of the cells and the activation state of the cells by flow cytometry before isolation of cytotoxic granules (see Note 5).
-
3.
Wash the cells in cold wash buffer and resuspend them in 2 mL homogenization buffer.
-
4.
Transfer the cell suspension into the clean and pre-chilled cavitation bomb (4 °C) and add the micro magnetic stir bar.
-
5.
Close the Parr Bomb and place it on top of a magnetic stirrer and connect it to the nitrogen tank.
-
6.
Slowly pressurize to 800 psi with nitrogen gas and stir at 4 °C for 25 min. Open the discharge valve gently and release the pressure to dropwise collect the homogenate in a pre-chilled 15 mL tube (see Note 6; Fig. 1a).
-
7.
Mix 15 μL of the cavitate with trypan blue, add on a microscopy slide, cover with a coverslip, and observe by phase-contrast microscope to judge the effectivity of the cavitation procedure.
-
8.
Centrifuge the cell lysate for 10 min at 1000 × g at 4 °C to pellet unbroken cells, partially disrupted cells, and nuclei. Collect the post nuclear supernatant (PNS, S1) without foam (Fig. 1a).
-
9.
Wash the pellet/foam once in 0.5 mL of homogenization buffer and centrifuge for 10 min at 1000 × g and 4 °C. Add this 0.5 mL supernatant to the PNS from the previous step (~2 mL final volume) and use the pooled supernatant (S1 + S1’) for subcellular fractionation (see Note 7).

Immunoisolation of two classes of cytotoxic granules from Syb2 KI mouse CTLs. (a) Flow chart for CG isolation procedure. (b) Confocal images of immune-isolated organelles of Syb2 KI mouse CTLs. Upper and lower panel represent immune-isolated organelles of fraction 6 (IP6) and fraction 8 (IP8), respectively. Scale bar, 1 μm. (c) Representative TEM images of immune-isolated organelles. Electron micrographs from IP6 (upper image) and IP8 (lower image) are shown. The majority of granules in IP6 contain multi-core granules (MCG), while the majority of granules in IP8 have a single core granule (SCG). Sale bar, 0.2 μm
3.2 Subcellular Fractionation
-
1.
Prepare 10 mL sucrose solutions of different concentrations (0.8, 1.0, 1.2, 1.4, and 1.6 M) from a 2 M sucrose stock solution in sucrose solubilization buffer and add fresh proteinase inhibitors. Keep all solutions on ice and determine the refractive index for each solution using a refractometer (see Note 3).
-
2.
Perform a discontinuous sucrose gradient column from bottom to top in a pre-chilled 14 mL ultra clear centrifuge tube by carefully layering 2 mL of each sucrose solution (see Note 8).
-
3.
Carefully layer the 2 mL of the pooled post-nuclear supernatant (0.3 M sucrose) onto the performed gradient (Fig. 1a).
-
4.
Ultracentrifuge the post-nuclear supernatant at 100,000 × g for 90 min at 4 °C in the SW40Ti rotor with acceleration/deceleration of 8.
-
5.
Slowly and carefully, transfer 12 × 1 mL aliquot from the top of the gradient into protein low binding 1.5 mL tubes. Avoid mixing the layers during this step. Use for each aliquot a new pipette tip (Fig. 1a).
-
6.
Keep the aliquots on ice and add again fresh protease inhibitors to avoid protein degradation (see Note 9).
-
7.
Collect 61 μL of each fraction into a new 1.5 mL tube, add 16 μL LDS-buffer and 3 μL DTT, heat at 96 °C for 7 min, and store at −20 °C for SDS-PAGE and immunoblotting.
-
8.
Additionally, collect 100 μL of each fraction to determine the protein concentration with Pierce™ 660 nm Protein Assay reagent for quantitative western blot (see Note 10).
3.3 Immunoisolation of MCG and SCG
-
1.
Resuspend magnetic Dynabeads and transfer 15 μL per fraction to a 1.5 mL tube, place on magnet, and remove supernatant (see Note 11).
-
2.
Wash beads for 20 min with 300 μL D-PBS with 0.01% Tween 20 in the rotator at 20 ± 2 °C (F1, 5 rpm). Repeat two times.
-
3.
Coat 15 μL beads with 5 μg anti-Syb2 antibody in 200 μL D-PBS with 0.01% Tween 20 (pH 7.4).
-
4.
Rotate 40 min at 20 ± 2 °C (F1, 5 rpm).
-
5.
Wash beads three times using 400 μL D-PBS in the rotator (see Note 12).
-
6.
During the wash time, prepare SCG and MCG from fractions 6 and 8 for immune-isolation. Dilute the fractions 6 and 8 in a 1:1 ratio with dilution buffer (see Note 13, Fig. 1a).
-
7.
Add the diluted fractions to the Syb2 antibody-conjugated Dynabeads and incubate by slow rotation (overnight at 20 ± 2 °C, F1, 5 rpm) (see Note 14).
-
8.
After overnight incubation, place the tube in the magnet and remove the supernatant.
-
9.
Wash the beads by adding 300 μL of 160 mM KCl wash buffer and rotate slowly for 10 min.
-
10.
Add the beads to the magnet and carefully remove the supernatant.
-
11.
Repeat the washing steps five times.
-
12.
Use a few microliter of Dynabeads and check the organelle binding efficiency by mRFP fluorescence (561 nm) with confocal microscopy (Fig. 1b).
-
13.
Use organelle-Dynabead Protein G complexes for TEM and/or SEM analysis (Fig. 1c).
-
14.
For mass spectrometry and western blot mix organelle-Dynabead Protein G complexes add 18 μL LDS-buffer and 2 μL DTT, heat at 96 °C for 10 min, and keep at −80 °C.
4 Notes
-
1.
We find it best to use 0.8–1.5 × 108 activated mouse CD8+ cells. For other cell types, the method has to be adapted.
-
2.
We used an isotonic buffer for granule isolation to avoid osmotic stress and to keep the nuclei intact. 5 mM EDTA is included to chelate the cation to inhibit metalloproteases. Prepare the homogenization buffer with protease inhibitors fresh for each experiment.
-
3.
Prepare a 2 M sucrose stock solution. Weigh 68.46 g sucrose into a glass beaker, fill to 90 mL with buffer (20 mM HEPES, 5 mM EDTA, pH 7.3) and stir with a magnetic stir bar. Once completely dissolved (~1 h), fill up to 100 mL. Store the solution at 4 °C until use. We find it best to prepare the stock solution one day before the subcellular fractionation. Always prepare the sucrose gradient solutions fresh on the day of the experiment and keep on ice.
-
4.
Pierce™ 660 nm Protein Assay for total protein quantitation is compatible with a high concentration of most detergents, reducing agents and other commonly used reagents. The maximum compatible concentration for sucrose in these solutions is 50%, which allows determination of the protein concentration of different sucrose fractions after fractionation.
-
5.
Only intact and healthy cells should be used for organelle isolation (>80%). We find it best to determine the viability of the cells based on their size and granularity. Cell surface-specific antibodies against CD25, CD44, and CD62 were used to determine the activation state of the cells.
-
6.
The finale homogenate should appear milky with foam on top. The yield of the granules is mainly affected by the cavitation process. Try to avoid excessive cavitation and minimize foaming.
-
7.
Repeat the centrifugation step until no pellet is visible. The final post nuclear supernatant should be free of nuclei and whole cells.
-
8.
Start with the highest sucrose concentration (1.6 M) and then gradually add the sucrose solutions of lower concentration using a 10 mL syringe with 20xG needle. Submerge the centrifuge tubes with the sucrose gradient in ice until the homogenate is ready. Do not disturb tubes in any way to avoid mixing of the sucrose layers.
-
9.
We find it best to continue immediately after fractionation with the immunoisolation.
-
10.
Additional aliquots of the fractions can be used for electron microscopy, confocal or structured illumination microscopy to characterize the different fractions and to examine the quality of the isolated organelles. We recommend centrifuging the organelles of the different fractions onto gelatin-coated coverslips for immunostaining and light microscopy.
-
11.
Increase the amount of the magnetic beads depending on the number of fractions that should be analyzed.
-
12.
We suggest to test the binding efficiency of the antibody by western blot.
-
13.
The 1:1 dilution with the 320 mM KCl buffer reduces the sucrose concentration and the viscosity of the solution for better organelle binding. The final 160 mM intracellular KCl concentration stabilizes the organelles.
-
14.
We had less organelle binding efficiency at 4 °C.
References
Peters PJ, Borst J, Oorschot V, Fukuda M, Krahenbuhl O, Tschopp J, Slot JW, Geuze HJ (1991) Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J Exp Med 173(5):1099–1109. https://doi.org/10.1084/jem.173.5.1099
Page LJ, Darmon AJ, Uellner R, Griffiths GM (1998) L is for lytic granules: lysosomes that kill. Biochim Biophys Acta 1401(2):146–156. https://doi.org/10.1016/s0167-4889(97)00138-9
Griffiths GM, Argon Y (1995) Structure and biogenesis of lytic granules. Curr Top Microbiol Immunol 198:39–58. https://doi.org/10.1007/978-3-642-79414-8_3
Clark R, Griffiths GM (2003) Lytic granules, secretory lysosomes and disease. Curr Opin Immunol 15(5):516–521. https://doi.org/10.1016/s0952-7915(03)00113-4
Stinchcombe JC, Griffiths GM (2007) Secretory mechanisms in cell-mediated cytotoxicity. Annu Rev Cell Dev Biol 23:495–517. https://doi.org/10.1146/annurev.cellbio.23.090506.123521
Podack ER, Konigsberg PJ (1984) Cytolytic T cell granules. Isolation, structural, biochemical, and functional characterization. J Exp Med 160(3):695–710. https://doi.org/10.1084/jem.160.3.695
Masson D, Tschopp J (1985) Isolation of a lytic, pore-forming protein (perforin) from cytolytic T-lymphocytes. J Biol Chem 260(16):9069–9072. https://doi.org/10.1016/S0021-9258(17)39328-6
Schmidt H, Gelhaus C, Lucius R, Nebendahl M, Leippe M, Janssen O (2009) Enrichment and analysis of secretory lysosomes from lymphocyte populations. BMC Immunol 10:41. https://doi.org/10.1186/1471-2172-10-41
Schmidt H, Gelhaus C, Nebendahl M, Lettau M, Lucius R, Leippe M, Kabelitz D, Janssen O (2011) Effector granules in human T lymphocytes: the luminal proteome of secretory lysosomes from human T cells. Cell Commun Signal 9(1):4. https://doi.org/10.1186/1478-811X-9-4
Casey TM, Meade JL, Hewitt EW (2007) Organelle proteomics: identification of the exocytic machinery associated with the natural killer cell secretory lysosome. Mol Cell Proteomics 6(5):767–780. https://doi.org/10.1074/mcp.M600365-MCP200
Thiery J, Walch M, Jensen DK, Martinvalet D, Lieberman J (2010) Isolation of cytotoxic T cell and NK granules and purification of their effector proteins. Curr Protoc Cell Biol Chapter 3:Unit3 37. https://doi.org/10.1002/0471143030.cb0337s47
Persechini PM, Liu CC, Detmers PA, Young JD (1989) Heterogeneity of granules of murine cytolytic T lymphocytes. Isolation of a homogeneous population of dense granules. J Immunol Methods 124(1):7–15. https://doi.org/10.1016/0022-1759(89)90179-8
Peters PJ, Geuze HJ, Van der Donk HA, Slot JW, Griffith JM, Stam NJ, Clevers HC, Borst J (1989) Molecules relevant for T cell-target cell interaction are present in cytolytic granules of human T lymphocytes. Eur J Immunol 19(8):1469–1475. https://doi.org/10.1002/eji.1830190819
Matti U, Pattu V, Halimani M, Schirra C, Krause E, Liu Y, Weins L, Chang HF, Guzman R, Olausson J, Freichel M, Schmitz F, Pasche M, Becherer U, Bruns D, Rettig J (2013) Synaptobrevin2 is the v-SNARE required for cytotoxic T-lymphocyte lytic granule fusion. Nat Commun 4:1439. https://doi.org/10.1038/ncomms2467
Chang HF, Schirra C, Ninov M, Hahn U, Ravichandran K, Krause E, Becherer U, Balint S, Harkiolaki M, Urlaub H, Valitutti S, Baldari CT, Dustin ML, Jahn R, Rettig J (2022) Identification of distinct cytotoxic granules as the origin of supramolecular attack particles in T lymphocytes. Nat Commun 13(1):1029. https://doi.org/10.1038/s41467-022-28596-y
Hunter MJ, Commerford SL (1961) Pressure homogenization of mammalian tissues. Biochim Biophys Acta 47:580–586. https://doi.org/10.1016/0006-3002(61)90553-4
Balint S, Muller S, Fischer R, Kessler BM, Harkiolaki M, Valitutti S, Dustin ML (2020) Supramolecular attack particles are autonomous killing entities released from cytotoxic T cells. Science 368(6493):897–901. https://doi.org/10.1126/science.aay9207
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft SFB 894 (E.K., U.B., and J.R). Further, this project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement No 951329 to J.R.). We thank Ulrike Hahn, Keerthana Ravichandran and Momchil Ninov for their participation in the development of these methods. We thank Margarete Klose, Anja Bergsträßer, Nicole Rothgerber, and Anne Weinland for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2023 The Author(s), under exclusive license to Springer Science+Business Media, LLC, part of Springer Nature
About this protocol

Cite this protocol
Schirra, C., Alawar, N., Becherer, U., Chang, HF. (2023). Separation of Single Core and Multicore Lytic Granules by Subcellular Fractionation and Immunoisolation. In: Baldari, C.T., Dustin, M.L. (eds) The Immune Synapse. Methods in Molecular Biology, vol 2654. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-3135-5_11
Download citation
DOI: https://doi.org/10.1007/978-1-0716-3135-5_11
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-3134-8
Online ISBN: 978-1-0716-3135-5
eBook Packages: Springer Protocols