
Abstract
Light-sheet microscopy is a powerful method for imaging small translucent samples in vivo, owing to its unique combination of fast imaging speeds, large field of view, and low phototoxicity. This chapter briefly reviews state-of-the-art technology for variations of light-sheet microscopy. We review recent examples of optogenetics in combination with light-sheet microscopy and discuss some current bottlenecks and horizons of light sheet in all-optical physiology. We describe how 3-dimensional optogenetics can be added to an home-built light-sheet microscope, including technical notes about choices in microscope configuration to consider depending on the time and length scales of interest.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key words
1 Imaging Translucent Organisms
Larval fish, flies, and worms are popular model organisms in developmental biology [1] and, increasingly, in systems neuroscience [2,3,4]. Optical translucency make these organisms well-suited to visualize physiological functions using high-resolution fluorescence imaging with sub-cellular spatial resolution. Small size and their ability to thrive when immersed in water make it possible even to image embryonic development and behaviors in toto over hours or days [5].
Light-sheet microscopy, also known as Selective Plane Illumination Microscopy (SPIM), has emerged as the method of choice for imaging smaller organisms, offering a number of advantages over point-scanning microscopy in speed, accessible volume, and phototoxicity. The light-sheet revival over the last two decades is tightly associated with important milestones in live tissue imaging, including the iconic example of whole-brain imaging in larval zebrafish (Danio rerio). Launched by early examples of calcium imaging of fictive activity [6], several research groups worldwide now routinely record calcium activity from the nearly 105 neurons of the young awake, behaving zebrafish. The resulting avalanche of data is beginning to lead to new insights about the communication between different brain areas (see [3, 7] for recent reviews).
A natural extension to such imaging studies is the integration of optical methods for perturbation, such as optogenetics [8, 9], optopharmacology [10], and cell ablation [11, 12]. The expanding toolkit of molecular probes offers many optogenetic actuators to remotely activate or inactivate cellular processes. In the context of controlling neural activity, this progress in engineering molecular probes, together with the development of suitable optical methods [13], makes possible to photostimulate action potentials in cells expressing photosensitive channels and read out the affected neural activity using fluorescent reporters for calcium [14] or voltage [15]. Many of these tools have been developed into transgenic animal strains [16, 17]. Early demonstrations of optogenetic manipulations in combination with light-sheet microscopy include optogenetic control over a variety of physiological phenomena, especially in larval zebrafish, from the beating of the heart [18] to cellular control of reflexive behaviors [19].
In this chapter, we describe how optogenetics can be added to a home-built microscope inspired by the open-source project OpenSpim [20]. As an example, we describe in detail a microscope configuration suitable for cellular or sub-cellular optogenetics in larval zebrafish. The method involves adding two-photon photostimulation shaped by computer-generated holography (2P-CGH). The stimulation module exploits the high numerical aperture (NA) detection objective of the light-sheet microscope to simultaneously excite multifocal points targeted either to sub-cellular regions or to multiple somata. The light-sheet module provides flexibility to readout neural activity in tiny organisms from small volumes to whole brain. Volumetric imaging is achieved with an electrically tunable lens, allowing independent control of imaging depth without moving the detection objective and consequently the axial location of the stimulation foci.
We provide technical notes on optical alignment, alternative configurations for different applications, and limitations and challenges of combining optogenetics with light-sheet microscopy. Finally, we offer some perspectives on extending all-optical physiology to higher spatial resolution in vivo.
1.1 Light-Sheet Technologies
The functioning principle of light-sheet microscopy is to illuminate the sample with a thin sheet of light while collecting the fluorescent signal at an angle (usually orthogonal) relative to the illuminated plane. The illuminated plane is aligned with the focal plane of the detection objective enabling an image to be collected by a camera, as in a widefield microscope. Whereas laser-scanning confocal microscopy achieves optical sectioning through rejection of photons generated outside of the excitation focus, light-sheet microscopy avoids generating out-of-focus fluorescence. This approach provides optical sectioning while minimizing photobleaching and photoxicity.
1.1.1 Light-Sheet Configurations
Light-sheet microscopes can be implemented in a variety of configurations, distinguished by the position and number of microscope objectives, the sheet-forming illumination optics, and the detection optics. Readers are referred to several excellent review articles focused on developmental biology and high-resolution applications [21, 22]. With respect to brain imaging, variations of light-sheet microscopy have been driven by two main challenges:
-
(i)
Balancing optical sectioning with a large field of view (FOV).
-
(ii)
Maximizing imaging speed to resolve fast dynamics from fluorescent sensors.
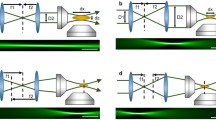
Here, we briefly compare light-sheet configurations used for whole-brain imaging in zebrafish, as shown in Fig. 1.

Selection of light-sheet microscope configurations used for whole-brain imaging in larval zebrafish. (a) Fluorescence is collected along a sheet of light formed by side-ways illumination. (b) In variations of multi-view light-sheet microscopy, additional objectives illuminate or collect fluorescence simultaneously allowing for either greater uniformity of illumination or multiplexing image formation from different angles. (c) In variations of single-objective LS, a tilted sheet is swept laterally across the sample while collecting a tilted epifluorescence image. (d) Selective volume illumination microscopy is a hybrid of light-sheet illumination and extended depth of field detection
Selective Plane Illumination
The basic SPIM design (Fig. 1a) uses two orthogonal microscope objectives for illumination (I) and detection (D) of fluorescence. The illumination light is typically shaped into a two-dimensional sheet with a cylindrical lens [23, 24]. To image a volume, the sample is either translated with respect to the detection objective, or the light sheet is scanned with a galvanometric mirror while also keeping the illuminated plane conjugated to the camera with either a piezo objective or an electrically tunable lens.
Later it was demonstrated that rapid scanning of a pencil beam (a long, thin illumination profile in one dimension) could generate a “virtual” sheet, as seen by the camera, with the major advantage of reducing light exposure to the sample [25]. This approach is alternately called digitally scanned light-sheet microscopy (DSLM). In this case, volume acquisition requires a second scanning mirror.
For (relatively) small, translucent samples, where light can enter the sample from any side, the SPIM design is convenient. Because two (or more) objective lenses are used, this design decouples the axial (△z) and lateral (△x, y) (△x, △y) resolutions that scale as the inverse of the numerical aperture for excitation objective, △z ∼NAI−1, and the detection objective, △x,△y ∼NAD−1.
The constraints on axial resolution limit the usable FOV. The usable length of a Gaussian light sheet is proportional to its thickness. For this reason, longer Gaussian profiles have poor optical sectioning and increased phototoxicity by illuminating a sample slice thicker than the detection depth of field. The most immediate improvement for obtaining longer 1-D uniform excitation profiles comes as a trade-off with temporal resolution: tiling the excitation light sheet [26] allows, in principle, to select only the central uniform region of the excitation profile, stitching together multiple images over an arbitrary FOV. However, in all-optical physiology experiments, the decreased temporal resolution may be unacceptable.
An alternative way to obtain uniform illumination over a larger FOV is to illuminate the sample from two sides using an additional illumination objective opposite of the first (dual-sided light sheet). Depending on how the sample is mounted in the microscope, it may also be possible to illuminate from additional angles [27].
Multiview Illumination
In the case of multiple illumination sheets, each sheet needs to cover only half of the total FOV, so a lower NA sheet can be used to better preserve optical sectioning. For instance, the IsoView [28] light-sheet microscope employs two different DSLM geometries to simultaneously illuminate the sample from two opposite sides, collecting the view with two cameras (Fig. 1b). In this method, combining the overlapping images from multiple angles, it is possible to achieve isotropic spatial resolution [28]. This parallel excitation also represents a robust solution against sample opacity. Moreover, employing two sets of galvanometric mirrors in each illumination arm (one for scanning, one for correcting incidence angle on the sample) allows to employ online optimization algorithms [29] to partially correct for low-order sample-induced aberrations. These improvements come at a cost in terms of both hardware (the number of parts and alignment difficulty) and software complexity. Additionally, because every image is collected twice, the amount of data collected necessitates high-end storage capabilities and lengthy analysis pipelines in order to properly fuse the views into a final volume. An added benefit provided by this geometry is the sample that can be left stationary, while the scanning is performed by the galvanometric mirrors (to move the excitation profile in 3D) coupled with piezo motors that keep the detection objective focused on the illumination plane.
Swept Plane (Single Objective)
A single-objective light-sheet configuration, employing epifluorescence, can be obtained in several ways [30, 31] by generating a tilted elongated focus (Fig. 1c). The tilted sheet is swept laterally across the sample to image a volume. Swept plane approaches are gaining ground in neuroimaging because they facilitate high volume speeds, as reviewed recently by Hillman [32]. The single-objective geometry has the advantage of using the same sample preparation as confocal or two-photon microscopy and, uniquely, can also be extended to samples of arbitrary size [33]. While the swept plane approach partially sacrifices resolution because every image is formed collecting planes from regions far from the optimal focus, re-imaging the tilted plane (and some post-processing) can recover diffraction-limited resolution. Higher NA objectives with short working distances can also be used when applying light-sheet microscopy to small samples (e.g., single cells) [34, 35].
Hybrid Light-Sheet Microscopes
A clever approach to improve imaging speed is to increase out-of-focus contributions in a principled way. Manipulating the detection point-spread function, for instance by adding spherical aberration [36] or a cubic phase profile [37], extends the effective depth of field of the detection objective so that information can be harvested from a thicker illuminated volume. Other approaches for simultaneous volume acquisition borrow from light-field microscopy. For instance, an exciting direction of hybrid imaging called selective volume illumination (SVIM) [38] merges light-sheet excitation with light-field microscopy techniques to allow extremely fast (tens of Hz) volumetric imaging. The trade-off for resolution is acceptable for somatic imaging [39], and SVIM significantly improves contrast over light-field microscopy with widefield illumination.
1.1.2 Engineering Illumination Improves Resolution and Photodamage
Light-sheet microscopy is intrinsically efficient with photons. The local intensity required for light-sheet imaging is smaller than that for confocal techniques [40], including spinning disk confocal microscopy. In fact, with scanning light sheet, the total energy deposited at each point of a 3-dimensional (3D) sample is reduced by a factor equal to the total number of sections obtained during the imaging [41]. This minimizes photodamage to the specimen and also has a positive effect on the imaging speed. Moreover, detectors used in light sheet, as CCDs or sCMOS cameras provide a better dynamical range rather than single-pixel detectors used in point-scanning approaches (e.g., avalanche photodiodes or photomultiplier tubes). A poor dynamic range causes problems of detector saturation that translates to trade-offs in smaller volume or slower acquisition time. Light-sheet illumination is less prone to excitation saturation compared to point-scanning techniques, so it is not necessary to compromise imaging speed with long frame exposure times.
To further improve on minimizing illumination intensity and photodamage, engineering the excitation beam to produce quasi-non-diffracting beams, in particular, Bessel beams [42, 43], has strongly impacted the field. Bessel-like beams preserve a small-beam waist over a longer distance compared to beams with a Gaussian amplitude profile (Table 1), translating to more uniform illumination over the FOV of the detection objective.
Moreover, when propagating through inhomogeneous samples, Bessel-like beams have reduced scattering and beam spreading due to their self-healing property; namely the beam recovers the initial intensity profile after an obstacle. However, Bessel beams have a major downside: a large portion of energy resides in side lobes, which can spread out for a tens of microns beyond the central peak. In fact, they may generate fluorescence signal from out-of-focus planes, preventing the theoretical gain in optical sectioning while increasing phototoxicity. For this reason, researchers have introduced methods to reduce the contribution of the side lobes to the image:
-
–
Prevention: Multiphoton excitation can prevent excitation of fluorescence in the side lobes because of their lower instantaneous intensity [44] (as also discussed in Chapter 10 by Ji).
-
–
Displacement: Interference between multiple illumination rays can diminish illumination intensity in the side lobes. For example, lattice light-sheet microscopy [45] employs a spatial light modulator as active optical element to superimpose an array of Bessel beams that destructively interfere and consequently reduce the impact of undesired side lobes. This solution has been proven effective not only in increasing resolution when imaging cellular samples, but also when studying larger animals (e.g., zebrafish embryos), if combined with adaptive optics techniques to compensate for the aberrations introduced by the morphology of larger organisms [46].
-
–
Rejection: Electronically, fluorescence generated by side lobes can be mitigated by only collecting signal confocal to the scanned beam [47]. This approach only slightly increases system complexity since modern CCD and CMOS cameras already allow to calibrate an internal rolling shutter modality, where only a few lines of pixels are active at a time, following the central lobe of the Bessel excitation.
1.2 Design Choices
1.2.1 Prioritize Scale, Resolution, or Speed
Optimal choice of the light-sheet microscope configuration depends on the research questions of interest, particularly with respect to the spatial and temporal resolutions required. Small organisms tend to have smaller cells than mammalian tissues, so resolution is of particular concern in both imaging and photostimulation. The choice of light-sheet approach is often a matter of choosing the best trade-off between speed and resolution, given the dimensions and transparency of the sample.
In designing the system described below, we considered applications involving the larval zebrafish. For this sample, the SPIM design is convenient. The larval zebrafish brain occupies a volume of approximately 500×800×300 μm, and neuronal somata are typically 5–10 μm in diameter. The whole brain can be measured in a single FOV of a 10x detection objective, with which cellular resolution is easily achieved. However, we also wanted the flexibility to image sub-cellular resolution in smaller brain regions, so we have used higher magnification to achieve sampling of better than 0.2 μm per pixel on the camera. This lateral resolution is achieved with a high-NA water-dipping detection objective (practically limited to NA< 1.1 by commercial objectives). To achieve sub-cellular axial resolution over most of the brain, we chose to apply the superior optical sectioning of a Bessel beam.
For example, considering illumination with a laser source with λ = 488 nm, and an excitation objective with NA= 0.29, a Bessel profile can be generated to cover uniformly 160 μm with a central peak width of ≃ 0.6 μm. To reach the same FOV, a Gaussian beam would have more than double thickness (around 10 μm, as calculated following precisely the Rayleigh length formula): of course, this value is chosen following a trade-off between length and the acceptable divergence that can be tolerated at the edges of the FOV.
1.2.2 Type of Photostimulation
The experimenter has many options to add photostimulation optics to a light-sheet microscope, including the variety of methods discussed in Chapters 1, 3–5, and 11 of this book. Both scanning and parallel approaches to photostimulation can be applied to small, translucent samples. In the first case, resonant scanners or galvanometric mirrors steer a focused beam across multiple regions of interest (ROIs), whereas, in the latter case, all the ROIs are illuminated simultaneously by using computer-generated holograms (CGHs) projected through spatial light modulators (SLMs).
The same excitation strategies applied in living animals require increased optical sectioning and penetration depth, both provided by two-photon (2P) illumination. For example, Dal Maschio et al. [48] have integrated a 2P-CGH module with a two-photon-scanning microscope, generating an instrument capable of identifying behavior-related neural circuits in living zebrafish larvae. The stimulation is targeted to single soma with a diameter of ∼6 μm and an axial resolution of ∼9 μm over a volume of ∼ 160×80×32 μm. Comparable lateral and axial resolutions for circuit optogenetics are achieved by McRaven and colleagues [49], in their 2P-CGH setup coupled to a 2P scanning microscope with remote focusing, to discover cellular-level motifs in awake zebrafish embryos. On the other hand, De Medeiros et al. [12] have combined a scanning unit with a multiview light-sheet microscope. This is a flexible instrument to perform ablation of single cells in zebrafish embryos and also localized optogenetic manipulations with concurrent in toto imaging in Drosophila. Here, the effect of the optogenetic manipulation can be monitored at the embryo scale with cellular resolution. Another example of SPIM integrated with 2P scanning stimulation is presented in [27], where whole-brain imaging and brain-wide manipulations in larval zebrafish reveal causal interaction. All these works demonstrated that in the small brain of the larval fish (∼ 0.1 μm3), it is possible to both record and stimulate with millisecond temporal resolution and single-cell precision over the full volume of the engaged neural circuit.
1.2.3 One Photon or Two?
As mentioned in other chapters, multiphoton excitation is the simultaneous absorption of n lower-energy photons to electronically excite a higher-energy single-photon transition. For visible-absorbing optogenetics chromophores, near infrared (NIR) wavelengths (700–1100 nm) are typical for two-photon absorption. NIR has the added advantage of high penetration depth in biological tissues [50]. Transparent tissue might seem to obviate the need for multiphoton microscopy. On the contrary, there are several arguments for the use of two-photon excitation light-sheet microscopy.
On the imaging side, scanned beam approaches also made two-photon excitations feasible because the spherical focus can generate the highest peak intensity for a given power, extending light-sheet microscopy to imaging in highly scattering samples [51, 52]. Even for optically translucent samples such as larval zebrafish, scattering is noticeably reduced. For brain imaging in larval zebrafish, two-photon microscopy is often preferred because it is more orthogonal to the visual system [2]. Imaging with visible light impacts general brain activity, visual sensitivity, and even innate motor behaviors due to non-visual opsins [53], though by carefully avoiding direct illumination of the eyes, it is possible to deliver visual stimuli and even virtual reality [27].
For photostimulation with cellular, or sub-cellular, precision in 3D tissues, it is crucial to exploit multiphoton absorption for optical sectioning. Since 2P absorption is a non-linear process, its probability depends quadratically on the intensity of the excitation light. The main consequence is an improved optical sectioning because the stimulation is generated only in the vicinity of the geometrical focus where the light intensity is the highest [54]. The resolution of the multiphoton excited fluorescence is described as the full-width half maximum (FWHM) of the three-dimensional point-spread function (3D-PSF) of the fluorescence intensity hi of the excitation. The following equations describe the dependency of the 3D-PSF hi on the 3D-PSF of the illumination intensity of two-photon excitation.
where u and v are, respectively, the axial and lateral coordinates of the optical system.
It should be noted that all-optical experiments require careful control to avoid heating effects induced by NIR stimulation [55]. Compared to experiments in rodents or organotypic tissues, small translucent organisms such as zebrafish larvae are fragile and easily burned. Their small size, typically smaller than the beam waist exiting the microscope objective, means that a significant fraction of the animal’s skin is exposed to defocused light, even while a tight, diffraction-limited focus may be achieved beneath the skin. When exposed to NIR light, even small amounts of dark skin pigment can lead to unintentional burning. Furthermore, ectotherms such as zebrafish and Drosophila lack the ability to maintain a constant internal body temperature and typically regulate temperature behaviorally (e.g., by heat-seeking or heat avoidance behavior [3]). Even a 1–2 K rise in temperature may have a notable effect on the physiology under study. In two-photon imaging, intensity is typically kept around 0.1–0.5 mW/μm2 [56]. In both 2-photon imaging and photostimulation, a potential control experiment to evaluate the 1-photon NIR effect is to test photostimulation with a lower peak power density, e.g., in mode-locked Ti:Sapphire lasers, switching into continuous-wave mode provides a means to obtain the same average power with orders of magnitude lower than energy density.
2 Materials
All-optical physiological experiments require an imaging system that combines a fluorescence microscope with a light path for photostimulation. The microscope serves dual purpose: first to acquire a baseline fluorescence image or movie to identify the spatial location of ROIs and second to acquire a continuous readout of the effect of photostimulation. Here we describe an example of a Bessel-beam light-sheet microscope and a 2P-CGH module.
2.1 Light-Sheet Module
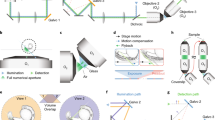
The microscope schematic shown in Fig. 2 is a digitally scanned light sheet implemented by scanning a Bessel beam in a 2D plane. The combination of an axicon with a plano-convex lens generates a beam shaped as a hollow cylinder. Since this beam is collimated, any subsequent conjugate plane can be chosen as the entry pupil of the microscope. The collimated ring is conjugated with two galvanometric mirrors (G1, G2, Thorlabs, ax1210-A) and the back aperture of the excitation objective (EO; Nikon, 10X/NA 0.3 CFI Plan Fluorite).

Example configuration of light-sheet microscope with optogenetics (a) The system consists of a light-sheet imaging module (left, blue lines) and an optogenetics module (right, red lines) that share a common light path through the high-NA detection objective (DO). In the imaging module, a continuous-wave visible wavelength laser is shaped in a Bessel beam by an axicon (A) and scanned onto the sample through galvo mirrors G1 and G2 and excitation objective (EO). Fluorescence collected through the detection objective (DO) is transmitted through a low-pass dichroic mirror (DM) and imaged onto a sCMOS camera by an electrically tunable lens (ETL).The fluorescent signal is recorded with the confocal slit detection approach schematically shown in panel b. In the 2P-DH module, a Ti:Sa pulsed laser beam is magnified through a telescope (L1, L2), impinges on the SLM, placed in a plane where the wavefront is flat. Lenses (L3, L4) relay the wavefront from the SLM to the pupil of the DO. In the focal plane of lens L3, the inverse pinhole (IP) blocks the zero-order diffraction spot. (b) Left: Scheme of confocal slit detection. An active window (yellow line) on the sCMOS camera is rolled synchronously with the scanning of the Bessel beam illumination (cyan line). In this way, we minimize the detection of fluorescence excited by the side lobes of the Bessel beam (light cyan halo). Right: Bessel beam projected into uniform fluorescent solution. The zoom in on the middle region of FOV shows the central lobe of the Bessel beam as it appears with confocal slit detection
Fluorescence is collected through the detection objective (DO; Olympus, 20X/NA 1.0 XLUMPLFLN), transmitted through a low-pass dichroic mirror, and finally, the image is formed by a 300 mm tube lens (L5). The image is relayed to the sCMOS camera (Andor, Zyla 4.2) by a 1:1 telescope (L6 and L7, focal 150 mm). An electrically tunable lens (ETL; Optotune, EL-16-40-TC-VIS-20D), positioned in the common focus of the telescope, can scan the signal at different depths.
To effectively eliminate the out-of-focus emission excited by the side lobes of the Bessel beam, the design implements confocal slit detection of the fluorescent signal, as shown in the inset of Fig. 2b. The slit is virtually created using an active window of the sCMOS camera that is rolled synchronously with scanning of the Bessel-beam illumination. In this way, we minimize the detection of fluorescence excited by the side lobes. The cost is a slower imaging speed.
The ETL in the detection path allows independent control of photostimulation in three dimensions and volumetric imaging, without moving either the objective lens or the sample. The accessible volume is determined by the choice of the tube lens L5. In fact, given the objective the focal of the tube, lens is directly proportional to the image magnification. For such a reason, longer focal lengths give access to a smaller volume, while they achieve better sampling of the fluorescence signal at the camera pixel. The ETL in the configuration shown in Fig. 2a allows to image a volume extending over 500 μm. The photostimulation can be performed on a volume extending over 200 μm in depth, with the SLM being the limiting factor in the theoretical axial FOV.
2.2 2P-CGH Module
We implement computer-generated holography for spatial patterning of the photostimulation light. As shown in Fig. 2a, the ultra-short pulses of NIR light, emitted by the Ti:Sa laser (Coherent, Mira-900F), pass a combination of half-wave plate (HWP) and Glan–Thompson Polarizer (GTP) to control the average power. A mechanical shutter (S; Uniblitz, LS2S2Z1) allows to block or transmit the light. Then the beam, magnified through a telescope (L1 and L2, focal 25.4 mm and 150 mm), impinges on the SLM (Meadowlark optics, P1920-600-1300-HDMI) placed in a plane where the wavefront is flat (where the Gaussian laser beam is collimated). Subsequent lenses relay the wavefront from the SLM to the pupil of the DO: here, the Fourier lens L3 (focal 250 mm) and lens L4 (focal 500 mm) magnify the beam to fill the back aperture of the objective. In the focal plane of lens L3, an inverse pinhole (IP, diameter 1.4 mm) blocks the zero-order diffraction spot.
The light paths of the light-sheet microscope and the 2P-CGH module join at a dichroic mirror (DM; Semrock, FF01-720/SP-25), with a cutoff wavelength at 720 nm. This mirror reflects the NIR stimulation light to the sample and transmits the fluorescent readout to the sCMOS. Two-photon-excited (TPE) epifluorescence can be recorded, and this is useful to characterize the photostimulation beam as described in Subheading 3.1.
The choice of focal lengths of lenses L3 and L4 is important in the design of the experiment. If the SLM image at the objective back aperture is smaller than the aperture itself, the NA of the objective is not fully exploited. This might be an intentional choice. Otherwise, if the SLM image is larger, some stimulation light gets lost. The best option is to choose a telescope magnification that matches the dimension of the SLM image at the back aperture to the aperture itself. In this case, the lateral resolution is only limited by the objective NA.
The pixelated structure of the SLM chip introduces in the reconstructed hologram a zero-order diffraction spot that appears as a bright spot in the center next to the hologram. To separate the hologram from the zero-order illumination, we can either proceed algorithmically, or we can block the zero-order illumination physically. Since the result given by the second method can strongly degrade the quality of the image, it is preferable to implement a combination of the two approaches to preserve the image quality. The hologram can be displaced from the zero order algorithmically by introducing a constant defocus. Once CGH and zero order lay at different depths, the zero-order beam is blocked by means of an inverse pinhole without impairing the quality of the CGH.
In high-resolution applications, the spatial accuracy of photostimulation is paramount. As reported in [57], the precision to address a CGH to a specific target depends on the number of pixels in the SLM and gray scale values available. Several companies produce SLM with over 1000 pixels in the shorter axis, which provide the spatial accuracy required for sub-cellular manipulations and also a high number of degrees of freedom if the SLM is used as an adaptive element to correct for high-order aberrations. The accessible lateral and axial FOV for a CGH is inversely proportional to the SLM pixel size. However, fairly large pixel size (in the order of 10 μm) is preferred because, assuming a constant inter-pixel gap, the fill factor increases with the pixel size. A larger pixel size also reduces the cross-talk between pixels. Cross-talk acts as a low-pass filter on the CGH, and it is due to fringing field effect that causes gradual voltage changes across the border of neighboring pixels and by elastic forces in the liquid crystal material [58]. In the setup in Fig. 2a, we use a 1920×1152 pixel liquid crystal on silicon (LCoS) SLM with a pixel pitch of 9.2 μm. This device guarantees 95.7% fill factor and a CGH switching rate of 31 Hz.
2.3 Sample Preparation
In SPIM, samples are usually mounted in tubes made of fluorinated ethylene propylene (FEP), a plastic with refractive index similar to water. The tube is filled with a solution of water and low-melting-temperature agarose (∼1.5–2%) for short-term imaging (1-3 hours). For larval zebrafish, these conditions ensure stability of the sample while maintaining good physiological conditions given the time frame of the experiment. However, as reported in [59] for longer experiments (over 1 day), it is recommended to use lower agarose percentage (∼0.1%) or methylcellulose solutions (∼3%) to ensure stability but also proper growth of the sample especially between 24–72 h post-fertilization.
3 Methods
3.1 Microscope Alignment
3.1.1 Align the Light-Sheet Module
The microscope is aligned by visualizing the Bessel-beam profile. By using the galvanometric mirror G1, responsible for the planar scan, we can image the full profile of the Bessel beam at specific locations:
-
1.
Prepare a low concentration solution of fluorescent dye such as fluorescein (Invitrogen, F1300) diluted in demineralized water.
-
2.
Fill the sample chamber with the fluorescent solution and turn on the excitation laser at low power (1 mW).
-
3.
Optimize the beam thickness and uniformity in the center of the FOV by setting the galvanometric mirror G1 at 0 V.
-
4.
Optimize the beam focus at the edges of the FOV by setting the galvanometric mirror G1 at its extrema, e.g., ±1 V. For steps 3 and 4, the galvanometric mirror G2 and the ETL voltages are kept to 0 V. These settings ensure that the axial scanning range is centered. As schematically shown in Fig. 3a, a properly aligned Bessel beam appears as a tiny, bright, and straight line profile across the FOV. The inset shows a zoom-in on a good example of Bessel beam where a sharp profile with side lobes can be observed. Contrarily, Fig. 3b shows an example of a misaligned Bessel beam. It is prominently tilted at the edges of the FOV, and its profile is enlarged.
Fig. 3 
Main steps of system alignment. (a) Example of good planar alignment. This image shows an overlay of the Bessel profile at different positions across the planar FOV. Inset shows a shallow Bessel beam with its side lobes. (b) Example of misaligned beam across the planar FOV. (c) Affine transformation between light-sheet and 2P-CGH module: (1) 3D plot of the coordinates fed into the algorithm to generate a point cloud CGH of random points spanning over a 3D volume. Those coordinates are defined in the coordinate system x′y′z′ of the 2P-CGH module. (2) 3D plot of the coordinates measured on the TPE image of the CGH. Those coordinates are defined in the coordinate system xyz of the light-sheet module. (3) Result of the affine transformation between 2P-CGH coordinates and light-sheet coordinates
-
5.
Perform an automated calibration to acquire a planar scan. The planar calibration correlates the vertical displacement of the Bessel beam across the planar FOV and the voltage given to the galvanometric mirror G1. During this calibration, sequentially change the voltages driving G1 and for each voltage and measure the position of the Bessel beam onto the camera. The resulting data are voltages as a function of position. The slope of the linear fit on these data yields the relation between voltage and displacement.
-
6.
Perform the volumetric calibration. This step allows to synchronize the displacement of the Bessel beam in the volumetric FOV with the ETL focal plane. This is achieved by sequentially changing the voltages fed to both the galvanometric mirror G2 and the ETL. For each ETL voltage, scan the full range with G2 and collect an image at each step. For each image, measure the maximum intensity of the central lobe of the Bessel beam. Select the value of G2 corresponding to this maximum to get the excitation profile in focus.

3.1.2 Align the 2P-CGH Module
The 2P-CGH module is aligned by projecting the zero-order diffraction beam into the same fluorescent solution used for the light-sheet alignment:
-
1.
Align the NIR beam to impinge on the SLM with a small angle and to uniformly illuminate the whole SLM chip.
-
2.
Check that the reflection of the zero-order beam from the SLM is well-separated from the incoming beam and that these beams have the same height.
-
3.
Center the zero-order diffracted beam onto the irises placed in front of the downstream optics.
-
4.
Check the position of the beam at the sample using the sCMOS camera. The beam must appear as a bright spot in the middle of the camera FOV. If this spot appears far away from the center of the camera, adjustments in the upstream optics are needed.
-
5.
Install the inverse pinhole in the optical path and visualize a CGH projected into the sample.
-
6.
Calibrate the 2P-CGH module with the ETL voltages. For this, load a series of single-point CGHs with different z positions onto the SLM. For each CGH, adjust the voltage on the ETL and acquire an image of the TPE fluorescence produced by the CGH. Record the second moment of the spot intensity as a function of the voltage on each image series. The voltage corresponding to the minimum second moment is considered the right voltage to have the point in focus. This procedure links the z position of a CGH to the optimal voltage needed for an image in focus.
3.1.3 Registration Between Light-Sheet Module and 2P-CGH Module
To obtain good fidelity between the stimulation target and the holographic illumination, it is critical that the light-sheet module and 2P-CGH module share a common coordinate system. This is accomplished by first calibrating the scan volume of the LS and then calibrating a 3D hologram. Both volumetric scans are achieved by incrementing voltage on the ETL. Once the 2P-CGH and light-sheet volumes are calibrated with the ETL, the two coordinate systems are then aligned through an affine transformation (see Note 1). This calibration is carried out before each experiment, and the resulting affine transformation matrix is applied to the ROIs location selected on the image to get the input coordinates for the holographic stimulation:
-
1.
Compensate for possible rotation and flip between the 2P-CGH and the light-sheet coordinate system. In the setup shown in Fig. 2, the 2P-CGH coordinate system is rotated by −90 ° and flipped vertically with respect to the imaging system because of the orientation of the SLM and the odd number of mirrors between the SLM and the DO back aperture.
-
2.
Calculate a point-cloud CGH of random spots spanning the volume relevant to the experiment (see Note 1) by using x′y′z′ coordinates defined in the 2P-CGH system. In the specific case of the setup described here, the volume is covering 140 × 140 × 200 μm (Fig. 2b).
-
3.
Project the CGH into a concentrated solution of fluorescent dyes.
-
4.
Collect images of the TPE fluorescence induced by the CGH and measure the xyz spots position in the light-sheet system (Fig. 2b).
-
5.
Calculate the affine transformation matrix through least square minimization between the xyz and the x′y′z′ coordinates.
-
6.
Align the xyz coordinates to the x′y′z′ by applying the affine transformation matrix.
3.2 Workflow of Light-Sheet Optogenetics Experiment
The general workflow for all-optical physiology experiment using the system described above would be similar to other methods described in this volume. After alignment of the system, it is typical to first acquire a baseline fluorescence image or a 3D movie to characterize anatomical structure and possibly baseline activity. Then, the ROIs to be stimulated are selected. The criteria to choose the ROIs from the baseline image are set by the experimenter and fed into an algorithm for targeting light, e.g., CGH calculation. Subsequently, we program a pulse sequence encoding for the CGH where the stimulation light is temporally gated by an external shutter. Afterward, the fluorescence signal is recorded over time.
3.2.1 Imaging Larval Zebrafish
After microscope alignment, mount a zebrafish in FEP tubing. Use brightfield illumination to orient and position the sample appropriately. Obtain a baseline fluorescence image. A high-resolution volume is recommended to help with brain registration later in image analysis. Based on anatomical or functional criteria, select regions of interest (ROIs) for photostimulation. Generate the hologram(s) and set a gating sequence or method to trigger the photostimulation sequence of interest.
3.2.2 Analysis of Large-Scale Ca2+ Data Set
Figure 4 shows the analysis of a representative data set measuring spontaneous neural activity in a 5 days past fertilization (dpf) zebrafish larvae expressing nuclear localized GCaMP6s in neurons, as acquired by volumetric calcium imaging on the described Bessel-beam light-sheet microscope. Fluorescence was captured in 20 volume sections of the forebrain, spaced 8 μm apart, with an acquisition rate of 0.67 Hz. Motion correction and the extraction of activity traces from individual neurons were implemented with the open-source calcium image analysis package CaImAn [60]. Motion artifacts were corrected through piecewise-rigid registration. Active neurons were then detected in the motion-corrected data through constrained non-negative matrix factorization (CNMF). To initialize CNMF, the images were first filtered with a Gaussian kernel. Thereafter, the Pearson correlation with neighboring pixels and the peak-to-noise ratio was calculated for each pixel (Fig. 4a). Local maxima in the pointwise product of the peak-to-noise ratio image and correlation image were used as initialization positions. With CNMF, the contours of found neurons (Fig. 4b) and activity traces (Fig. 4c) were then extracted. In total, 2077 active neurons were detected in the imaged brain volume (Fig. 4d,e).

Analysis of light sheet calcium imaging data from the larval zebrafish forebrain with the open-source calcium image analysis package CaImAn. (a) Images were obtained from the forebrain of a larval zebrafish (5 dpf) expressing nuclear localized GCaMP6s. A total of 20 volume sections were imaged with Bessel beam light-sheet microscopy at an acquisition rate of 0.67 Hz. To detect neurons, CNMF was initialized by filtering the motion corrected timeseries data with a Gaussian kernel and calculating the peak-to-noise ratio and Pearson correlation with 4 nearest neighboring pixels. Local maxima in the point-wise product of the peak-to-noise ratio image and correlation image were then used as initialization positions. (b) Contours of neurons extracted with CNMF in a single section. (c) The fluorescence (△F∕F) signals of 10 randomly selected neurons. (d) Maximum intensity projection of the measured volume. (e) Activity map of 2077 fluorescence traces from neurons found in all sections clustered by correlation coefficient
3.3 Choice of CGH Algorithm
The 2P-CGH module shown in Fig. 2a is based on Fourier holography and requires an appropriate algorithm to calculate the desired phase hologram. The aim of CGH algorithms is to retrieve the phase mask, namely the phase of the hologram field Uh, to address the SLM by knowing the field Uo of the target at the image plane, where these fields are one and the Fourier transform of the other. To generate a hologram of ROIs distributed in 3D, we compute a 3D hologram corresponding to a field Uo defined at different depths z as schematically described in Fig. 5a. Optically, the Fourier transform of the field Uh is realized through the objective lens when the SLM is conjugated to the objective back aperture through a telescope (L3 and L4 in Fig. 2a).

CGH approach. (a) Optical transformation needed to project a 3D digital hologram. Uh represents the spatial Fourier transform of the desired pattern Uozi defined at different depths. f is the focal length of the lens (middle element). (b) Scheme of algorithms for extended CGH. Phase masks producing different features at focal planes I, II, III are combined by superposition principle at the SLM. Each of the three phase masks is calculated based on the corresponding image at the focal plane. (c) Scheme of algorithms for point-cloud CGH. Phase masks producing different features at focal planes I, II, III are combined by superposition principle at the SLM. Each of the three phase masks is calculated based on the coordinates xyz of the spots at the focal plane. (d) 3D view of image-based CGH of a grid 160 × 160 × 100 μm. (e) 3D view of point-cloud CGH of the same grid
The choice of the algorithm to calculate CGHs depends on several considerations. Ideally, the algorithm provides high diffraction efficiency, uniformity over the volume of interest, and accuracy between the target and the reconstructed hologram. The algorithm should also be fast given the real-time nature of optogenetics experiments. The shape and dimensions of the target photostimulation pattern also influence the choice of CGH algorithm. When the target object has a complex and extended lateral shape (> 1 μm), image-based algorithms are employed. They enable the generation of extremely complex illumination patterns in very short times; however, they are limited to illumination light focused on a limited number of two-dimensional planes [61]. Conversely, point-cloud algorithms allow one to target diffraction-limited spots arbitrarily distributed in the three-dimensional field of view of the optical system.
Many variations of these algorithms have been developed (Table 2). Here we classify them into two categories (see Note 2): classic algorithms, and more recent algorithms developed specifically for optogenetics applications. Random superposition and Gerchberg–Saxton [62, 63] are the most known algorithms. The first offers high speed but poor quality of the CGH in terms of uniformity and efficiency, whereas the second provides improved quality with an increased computational time. Recently, new algorithms have been developed that are optimized for speed, such as a compressive-sensing version of the Gerchberg–Saxton algorithm [64]. Here the compressive-sensing method allows to reduce the computational time by a factor of 10 without impairing the quality of the CGH achieved with the standard implementation of Gerchberg–Saxton. Another advanced algorithm recently available is computer-generated holography by non-convex optimization [61]. Arbitrary 3D holograms are generated through non-convex optimization of custom cost functions. Another advanced algorithm recently developed, called DeepCGH [65], is based on unsupervised convolutional neural networks. Holograms computed via DeepCGH showed improved computational times and high efficiency suitable for neurostimulation experiments.
As an example, in the high-NA configuration hereby described, we implement weighted Gerchberg–Saxton algorithm to generate stimulation ROIs with a lateral dimension between ∼6 and 10 μm, on the order of the size of small neurons in the larval zebrafish brain. The principle of this approach is schematically described in Fig. 5b, while Fig. 5d shows a 3D view of an extended CGH projected into a thick Rhodamine slide. On the other hand, when targeting sub-cellular regions, the compressive-sensing weighted Gerchberg–Saxton algorithm is faster, but the lateral extension of the CGH depends on the numerical aperture of the detection objective. In the case that NA = 1.0, the lateral extension of the spots is on the order of ∼1 μm. Similarly to the previous case, Fig. 5c illustrates the principle of point-cloud CGH, and panel 5e shows a 3D view of the point-cloud CGH. The compressive-sensing weighted Gerchberg–Saxton algorithm provides comparable results in terms of spot brightness and uniformity as the point clouds generated by the GS and WGS approaches while reducing drastically the computational time [64].
3.4 Effect of Aberrations on CGH
All algorithms previously described compute CGHs based on the assumption that the sample has a uniform index of refraction. This assumption is rarely true in neuroscience since brain tissue is generally optically turbid and non-uniform. Optical inhomogeneities of samples cause a distortion of the stimulation pattern known as optical aberrations. In Fig. 6, we show the effect of optical aberrations on a point-cloud CGH. Specifically to the computed CGH phase, we summed a phase encoding for horizontal coma aberration with progressively increasing coefficient. The aberrated CGH has a decreased intensity, and it is displaced from its original position. Sample induced aberrations can be compensated through adaptive optics techniques using the SLM as an adaptive element.

Effect of coma on CGH: (a) TPE fluorescence induced onto a rhodamine slide by a one-point hologram with progressively increasing horizontal coma coefficient. Subscripts on the images indicate the corresponding peak-to-valley (PV) coefficient in units of the wavelength of horizontal coma. The additional coma phase introduces a dramatic drop in the fluorescence intensity compared to the case without aberrations (0 μm PV coefficient). (b) Bar plot showing the maximum intensity of each aliased fluorescent spot in a. Intensities are normalized to the maximum intensity of the aberration-free spot (0 μm PV coefficient). The maximum intensity is reduced below 50% compared to the intensity of the aberration-free spot. (c) Same fluorescence images showed in (a) where the aberrated spots intensity is enhanced by a constant factor. (d) Maximum intensity projection of the fluorescence images in (b) showing the position of each point across the camera chip. The aberration besides the loss of intensity introduces a loss of the CGH fidelity
4 Summary
Optogenetic manipulation coupled to light-sheet imaging is a powerful tool to perturb and monitor living translucent samples such as zebrafish larvae. Light-sheet imaging can be realized in different flavors with emphasis on balancing optical sectioning with the dimensions of the FOV and maximizing the imaging speed to resolve fast dynamics as reported in Subheading 1.1. Here, the light sheet is realized by scanning a Bessel beam in a 2D plane. This illumination offers a more uniform illumination over the FOV compared to Gaussian beams and a reduced scattering thanks to the self-healing properties of Bessel beams. However, a major drawback is the presence of the side lobes of Bessel profiles. In our microscope design, the side lobes are electronically rejected by implementing a confocal slit detection of the fluorescent signal. Unfortunately, this solution sacrifices the imaging speed. To improve this aspect, an interesting future upgrade of our imaging system is the implementation of a NIR light source. In fact, two-photon excitation can successfully mitigate the fluorescence induced by the side lobes as reported in [44]. Moreover, NIR wavelengths are often preferred for brain imaging of zebrafish larvae due to their orthogonality to the visual system of the animal [2]. The photostimulation module exploits the high NA of the light-sheet detection objective to focus on the sample two-photon computer-generated holograms. Those offer flexibility to illuminate either sub-cellular ROIs with dimensions in the order of the light diffraction limit or whole cell bodies extending over several microns (≈ 6–10 μm). In the first case, optical aberrations due to tissue inhomogeneities are a limiting factor to reach diffraction-limited resolution. Hence, a next step in further developing the 2P-CGH technique is to include adaptive optics methods to compensate for tissue-induced aberrations using the SLM as corrective element. Another improvement could be to include temporal focusing in the 2P holography module [66] to increase the axial confinement of the stimulation light as also reported in Chapters 1, 6, 7.
5 Notes
Note 1
Linear affine transforms will account for all linear transformations between coordinates, including lateral and axial shifts, magnification mismatches (independently for all axes), rigid rotations, and coordinate shearing. Non-linear distortions, such as barrel or pincushion distortion, would require a more complex non-linear transform. Given that the FOV is limited to hundreds of microns, non-linear effects rarely appear in a well-designed system, and a linear transform provides sufficient accuracy even for photostimulation of sub-micron structures. The linear affine transform matrix between homogeneous coordinates is represented by a 4x4 matrix [67]; however, it is determined by only 12 values as the last row in the matrix is always defined as {0, 0, 0, 1}. Hence, the matrix can be determined through least square minimization between vectors Xi of the image coordinates and Xi vectors of 12 SLM coordinates. The accuracy of the matrix estimation is improved using more than 12 coordinates see Subheading 3.1.3.
Note 2
Algorithms for CGH.
Problem Description
The mathematical approach to calculate a CGH differs based on whether we deal with image-based or point-cloud CGHs. To target complex and extended shapes spanning over few focal planes, the electric field of the desired pattern per each focal plane is modeled as
where uo is the square root of the desired intensity at depth z and ϕz is a random phase. The hologram field is defined as the fast Fourier transform (FFT) of the field Uo at each focal plane. Then the phase delay between the fields at different depths is optimized to get an interference pattern at the SLM plane as close as possible to the target illumination pattern. On the other hand for diffraction-limited spots arbitrarily distributed in the 3D FOV, the hologram field is defined as the wavefront generated by the coordinates x, y, z of the single spots in the focal planes. Hence, the field at the SLM is calculated as the superposition of the fields generating each single spot:
where uo,n is the square root of the desired intensity of each spot at the focal plane, f is the focal length of the optical system, λ is the wavelength of the light source, xn, yn, zn are the coordinates of the n desired spots in 3D space, x′ and y′ are the coordinates of each SLM pixel, and ϕn is a constant term. This field can be related to the field at the focal plane via a discrete Fourier transform (DFT) of the single spots. Also in this case, the aim is to optimize the phase delay between the fields generating each single spot.
Classic CGH Algorithms
-
Random Superposition (RS)
The electric field of the desired pattern per each focal plane is modeled as in Eq. 2. The hologram is calculated as the inverse Fourier transform of the field Uo, and it is backpropagated to the SLM plane according to the Fresnel propagator in Fourier Optics [68]. Then the interference field of all the holograms is computed. The phase of this interference field is the hologram retained as the phase mask to load onto the SLM. The average of the hologram fields at different depths is based on the assumption of independence between the depths. Thus, this method does not take into account the interference of the fields at different planes. This results in a degradation of the reconstructed holograms, which is evident when we project holograms with more than 2-3 depths [61]. This method is very fast compared to iterative algorithms.
-
Gerchberg–Saxton (GS)
The electric field Uo of the desired pattern is modeled as in the previous algorithm. Then the hologram is calculated as the inverse Fourier transform of the object field Uo. In the resulting hologram field, the phase is retained, while the amplitude is substituted with a Gaussian amplitude. This new field undergoes another Fourier transform to produce the object field at the focal plane. The pattern produced at the focal plane is similar to the desired one, but it has a decreased efficiency; hence, the phase is kept, while the amplitude is replaced with the one of the desired patterns. At this stage, the inverse Fourier transform of the field is calculated to retrieve the phase mask at the SLM plane. This procedure is repeated iteratively until the algorithm converges to phase mask that produces the best intensity pattern. This algorithm can be implemented for 3D holograms including in the calculation the propagation of the fields to different depth levels according to the diffraction theory [68].
-
Weighted Gerchberg–Saxton algorithm (WGS)
This is a modified version of GS where all the features in the CGH have a better uniformity. For this purpose, an extra degree of freedom is introduced in the CGH calculation that is the intensity weight of every feature in the pattern. At each iteration, the intensity of each feature is optimized comparing the intensity at the last iteration and the intensity of the target. The computational cost remains the same of GS.
Recent Algorithms
-
Compressive-Sensing Gerchberg–Saxton (CS-GS)
This algorithm is a faster version of the GS for sparse diffraction-limited spots in 3D. The field at the SLM is defined as reported in Eq. 3, and it is iteratively calculated for a subset of cM (where c is the compression factor and M is the number of SLM pixels) randomly distributed pixels of the SLM with GS algorithm. The last iteration is a standard GS calculation over all the M pixels in the SLM. The computational cost scales as Ni × Np × cM + Np × M, and this cost for cNi << 1 can be approximated to M × Np, where Ni is the number of iterations, Np is the number of points, and M is the number of SLM pixels. It has been demonstrated [64] that a speed up of over one order of magnitude can be achieved without compromising the quality of the obtained hologram compared to regular multi-spot GS.
-
Compressive-Sensing Weighted Gerchberg–Saxton (CS-WGS)
This algorithm is a faster version of the WGS for sparse diffraction-limited spots in 3D. WGS is incompatible with the compressive-sensing approach leading quickly to divergence, but it is possible to run CS-GS for k − 1 iterations and then perform a final iteration with WGS. This method, known as CS-WGS, has an approximately double computational cost compared to CS-GS when c × Ni << 1. As reported in [69], both CS-WGS and CS-GS can be implemented with GPU calculations on a low-cost graphic card. This upgrade permits to calculate a CGH ten times faster compared to the standard version using the FFT algorithm.
-
Non-convex Optimization for VOlumetric CGH (CGH-NOVO)
This iterative algorithm is to produce phase masks based on the non-convex minimization of a custom-defined cost function [61]. Given the intensity distribution of the target pattern V at different depths z, the corresponding hologram is calculated via the RS algorithm. A cost function is defined as
$$ L\left(I\left(\left({\phi}_s\right),V\right)\right), $$(4)where I(ϕs) is the distribution generated by the random phase mask ϕs at each plane and V is the intensity of the desired pattern at the focal plane. The minimization of the cost function in Eq. 4 gives the optimal phase ϕ∗. When I(ϕ) and the cost function have a well-defined derivative, the problem is solved with a gradient descendant approach.
CGH-NOVO offers the advantage of choosing a cost function tailored for the specific application. For instance, one might be interested in stimulating a subset of neurons, while minimizing the intensity received by off-target neurons below some illumination threshold, cost functions optimized for binary patterns are suitable. Another example of the flexibility of the CGH-NOVO is the possibility to implement a cost function optimized for 2P illumination. This algorithm can be implemented using a GPU to speed up the calculations.
-
DeepCGH
The DeepCGH algorithm [65] uses convolutional neural networks (CNNs) with unsupervised learning to compute 3D holograms with the image-based approach. The distribution of amplitudes A(x, y, z) of the 3D target pattern at the focal planes is the input of the CNN. The CNN provides in output an estimate of the complex field P(x, y, z = 0) at depth z = 0. This field backpropagated to the SLM plane via 2D inverse Fourier transform gives the phase map Φ of the CGH. During unsupervised training of the CNN, the goal is to minimize a loss function \( L\left(A,\tilde{A}\right) \), where A is the intensity distribution of the target and \( \tilde{A} \) is the intensity associated with the predicted SLM phase. As shown in [65], this algorithm offers a 10 times faster computational cost and up to 41% increased accuracy compared to iterative methods such as CGH-NOVO and GS. Moreover, experimentally, DeepCGH holograms can elicit two-photon absorption with 50% higher efficiency compared to GS or CGH-NOVO algorithms, given the same experimental conditions [65]. However, this algorithm requires a high-performance graphic card for GPU calculations.
References
Lemon WC, Pulver SR, Höckendorf B, McDole K, Branson K, Freeman J, Keller PJ (2015) Whole-central nervous system functional imaging in larval Drosophila. Nature Communications 6(Article number 7924)
Vanwalleghem GC, Ahrens MB, Scott EK (2018) Integrative whole-brain neuroscience in larval zebrafish. Curr Opin Neurobiol 50:136–145
Loring MD, Thomson EE, Naumann EA (2020) Whole-brain interactions underlying zebrafish behavior. Curr Opin Neurobiol 65:88–99
Eschbach C, Zlatic M (2020) Useful road maps: studying Drosophila larva’s central nervous system with the help of connectomics. Curr Opin Neurobiol 65:129–137
Keller PJ, Ahrens MB (2015) Visualizing whole-brain activity and development at the single-cell level using light-sheet microscopy. Neuron 85:462–483
Ahrens MB, Orger MB, Robson DN, Li JM, Keller PJ (2013) Whole-brain functional imaging at cellular resolution using light-sheet microscopy. Nature Methods 10(5):413–420
Lovett-Barron M (2021) Learning-dependent neuronal activity across the larval zebrafish brain. Curr Opin Neurobiol 67:42–49
Fenno L, Yizhar O, Deisseroth K (2011) The development and application of optogenetics. Annu Rev Neurosci 34:389–412
Johnson HE, Toettcher JE (2018) Illuminating developmental biology with cellular optogenetics. Curr Opin Biotechnol 52:42–48
Kramer RH, Mourot A, Adesnik H (2013) Optogenetic pharmacology for control of native neuronal signaling proteins. Nature Neuroscience 16(7):816–823
Hill RA, Damisah EC, Chen F, Kwan AC, Grutzendler J (2017) Targeted two-photon chemical apoptotic ablation of defined cell types in vivo. Nature Communications 8:15837
de Medeiros G, Kromm D, Balazs B, Norlin N, Günther S, Izquierdo E, Ronchi P, Komoto S, Krzic U, Schwab Y, Peri F, de Renzis S, Leptin M, Rauzi M, Hufnagel L (2020) Cell and tissue manipulation with ultrashort infrared laser pulses in light-sheet microscopy. Scientific Reports 10:1–12
Emiliani V, Cohen AE, Deisseroth K, Häusser M (2015) Symposium all-optical interrogation of neural circuits. J Neurosci 35(41):13917–13926
Knöpfel T (2012) Genetically encoded optical indicators for the analysis of neuronal circuits. Nat Rev Neurosci 13:687–700
St-Pierre F, Marshall JD, Yang Y, Gong Y, Schnitzer MJ, Lin MZ (2014) High-fidelity optical reporting of neuronal electrical activity with an ultrafast fluorescent voltage sensor. Nature Neuroscience 17(6):884–889
Asakawa K, Kawakami K (2008) Targeted gene expression by the Gal4-UAS system in zebrafish. Dev Growth Differ 50:391–399
Antinucci P, Dumitrescu A, Deleuze C, Morley HJ, Leung K, Hagley T, Kubo F, Baier H, Bianco IH, Wyart C (2020) A calibrated optogenetic toolbox of stable zebrafish opsin lines. eLife 9
Rost BR, Schneider-Warme F, Schmitz D, Hegemann P (2017) Optogenetic tools for subcellular applications in neuroscience. Neuron 96:572–603
Pantoja C, Hoagland A, Carroll EC, Karalis V, Conner A, Isacoff EY (2016) Neuromodulatory regulation of behavioral individuality in zebrafish. Neuron 91(3):587–601
Wan Y, McDole K, Keller PJ (2019) Light-sheet microscopy and its potential for understanding developmental processes. Annu Rev Cell Dev Biol 35:655–681
Power RM, Huisken J (2017) A guide to light-sheet fluorescence microscopy for multiscale imaging. Nature Methods 14(4):360–373
Huisken J, Swoger J, Del Bene F, Wittbrodt J, Stelzer EH (2004) Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science 305(5686):56–86
Verveer PJ, Swoger J, Pampaloni F, Greger K, Marcello M, Stelzer EH (2007) High-resolution three-dimensional imaging of large specimens with light sheet-based microscopy. Nature Methods 4(4):311–313
Keller PJ, Schmidt AD, Wittbrodt J, Stelzer EH (2008) Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science 322(5904):1065-9
Gao L (2015) Extend the field of view of selective plan illumination microscopy by tiling the excitation light sheet. Optics Express 23(5):6102–6111
Vladimirov N, Wang C, Höckendorf B, Pujala A, Tanimoto M, Mu Y, Yang CT, Wittenbach JD, Freeman J, Preibisch S, Koyama M, Keller PJ, Ahrens MB (2018) Brain-wide circuit interrogation at the cellular level guided by online analysis of neuronal function. Nature Methods 15:1117–1125
Chhetri RK, Amat F, Wan Y, Höckendorf B, Lemon WC, Keller PJ (2015) Whole-animal functional and developmental imaging with isotropic spatial resolution. Nature Methods 12:1171–1178
Royer LA, Lemon WC, Chhetri RK, Keller PJ (2018) A practical guide to adaptive light-sheet microscopy. Nature Protocols 13(11):2462–2500
Dunsby C (2008) Optically sectioned imaging by oblique plane microscopy. Optics Express 16(25):20306–20316
Bouchard MB, Voleti V, Mendes CS, Lacefield C, Grueber WB, Mann RS, Bruno RM, Hillman EM (2015) Swept confocally-aligned planar excitation (SCAPE) microscopy for high-speed volumetric imaging of behaving organisms. Nature Photonics 9:113–119
Hillman EM, Voleti V, Li W, Yu H (2019) Light-sheet microscopy in neuroscience. Annu Rev Neurosci 42:295–313
Strack R (2021) Single-objective light sheet microscopy. Nature Methods 18:28
Galland R, Grenci G, Aravind A, Viasnoff V, Studer V, Sibarita JB (2015) 3D high-and super-resolution imaging using single-objective SPIM. Nature Methods 12(7):641–644
Meddens MBM, Liu S, Finnegan PS, Edwards TL, James CD, Lidke KA (2016) Single objective light-sheet microscopy for high-speed whole-cell 3D super-resolution. Biomedical Optics Express 7(6):2219–2236
Tomer R, Lovett-Barron M, Kauvar I, Andalman A, Burns VM, Sankaran S, Grosenick L, Broxton M, Yang S, Deisseroth K (2015) SPED light sheet microscopy: fast mapping of biological system structure and function. Cell 163(7):1796–1806
Quirin S, Vladimirov N, Yang C-T, Peterka DS, Yuste R, Ahrens MB (2016) Calcium imaging of neural circuits with extended depth-of-field light-sheet microscopy. Optics Letters 41(5):855–858
Truong TV, Holland DB, Madaan S, Andreev A, Keomanee-Dizon K, Troll JV, Koo DE, McFall-Ngai MJ, Fraser SE (2020) High-contrast, synchronous volumetric imaging with selective volume illumination microscopy. Communications Biology 3(1):74
Sapoznik E, Chang BJ, Ju RJ, Welf ES, Broadbent D, Carisey AF, Stehbens SJ, Lee KM, Marín A, Hanker AB, Schmidt JC, Arteaga CL, Yang B, Kruithoff R, Shepherd DP, Millett-Sikking A, York AG, Dean KM, Fiolka R (2020) A single-objective light-sheet microscope with 200 nm-scale resolution. bioRxiv
Mertz J (2011) Optical sectioning microscopy with planar or structured illumination. Nature Methods 8:811–819
Jemielita M, Taormina MJ, Delaurier A, Kimmel CB, Parthasarathy R (2013) Comparing phototoxicity during the development of a zebrafish craniofacial bone using confocal and light sheet fluorescence microscopy techniques. J Biophoton 6:920–928
Planchon TA, Gao L, Milkie DE, Davidson MW, Galbraith JA, Galbraith CG, Betzig E (2011) Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination. Nature Methods 8(5):417–423
Gao L, Shao L, Higgins CD, Poulton JS, Peifer M, Davidson MW, Wu X, Goldstein B, Betzig E (2012) Noninvasive imaging beyond the diffraction limit of 3D dynamics in thickly fluorescent specimens. Cell 151(6):1370–1385
Fahrbach FO, Gurchenkov V, Alessandri K, Nassoy P, Rohrbach A (2013) Light-sheet microscopy in thick media using scanned Bessel beams and two-photon fluorescence excitation. Optics Express 21(11):13824–13839
Chen BC, Legant WR, Wang K, Shao L, Milkie DE, Davidson MW, Janetopoulos C, Wu XS, Hammer JA, Liu Z, English BP, Mimori-Kiyosue Y, Romero DP, Ritter AT, Lippincott-Schwartz J, Fritz-Laylin L, Mullins RD, Mitchell DM, Bembenek JN, Reymann AC, Böhme R, Grill SW, Wang JT, Seydoux G, Tulu US, Kiehart DP, Betzig E (2014) Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 346(6208). https://doi.org/10.1126/science.125799
Liu TL, Upadhyayula S, Milkie DE, Singh V, Wang K, Swinburne IA, Mosaliganti KR, Collins ZM, Hiscock TW, Shea J, Kohrman AQ, Medwig TN, Dambournet D, Forster R, Cunniff B, Ruan Y, Yashiro H, Scholpp S, Meyerowitz EM, Hockemeyer D, Drubin DG, Martin BL, Matus DQ, Koyama M, Megason SG, Kirchhausen T, Betzig E (2018) Observing the cell in its native state: Imaging subcellular dynamics in multicellular organisms. Science 360(6386). https://doi.org/10.1126/science.aaq13
Baumgart E, Kubitscheck U (2012) Scanned light sheet microscopy with confocal slit detection. Optics Express 20(19):21805–21814
dal Maschio M, Donovan JC, Helmbrecht TO, Baier H (2017) Linking Neurons to Network Function and Behavior by Two-Photon Holographic Optogenetics and Volumetric Imaging. Neuron 94:774–789
McRaven C, Tanese D, Zhang L, Yang C-T, Ahrens MB, Emiliani V, Koyama M, High-throughput cellular-resolution synaptic connectivity mapping in vivo with concurrent two-photon optogenetics and volumetric Ca2+ imaging. bioRxiv. https://doi.org/10.1101/2020.02.21.959650
Ko K, Nig E (2000) Multiphoton microscopy in life sciences. J Microscopy 200(2):83–104
Supatto W, Truong TV, Débarre D, Beaurepaire E (2011) Advances in multiphoton microscopy for imaging embryos. Curr Opin Genet Dev 21:538–548
Truong TV, Supatto W, Koos DS, Choi JM, Fraser SE (2011) Deep and fast live imaging with two-photon scanned light-sheet microscopy. Nature Methods 8:757–762
Wolf S, Supatto W, Debrégeas G, Mahou P, Kruglik SG, Sintes JM, Beaurepaire E, Candelier R (2015) Whole-brain functional imaging with two-photon light-sheet microscopy. Nature Methods 12:379–380
Alberto D, Mauro R (2000) Two-photon excitation of fluorescence for three-dimensional optical imaging of biological structures. J Photochem Photobiol B Biol 55(1):1–8
Picot A, Dominguez S, Liu C, Chen IW, Tanese D, Ronzitti E, Berto P, Papagiakoumou E, Oron D, Tessier G, Forget BC, Emiliani V (2018) Temperature rise under two-photon optogenetic brain stimulation. Cell Reports 24:1243–1253
dal Maschio M, Donovan JC, Helmbrecht TO, Baier H (2017) Linking neurons to network function and behavior by two-photon holographic optogenetics and volumetric imaging. Neuron 94:774–789
Papagiakoumou E, Ronzitti E, Chen IW, Gajowa M, Picot A, Emiliani V (2018) Two-photon optogenetics by computer-generated holography. In: Neuromethods, vol 133, pp 175–197. Humana Press Inc.
Martin Persson DE, Goksör M (2012) Reducing the effect of pixel crosstalk in phase only spatial light modulators. Optics Express 20(20):22334–22343
Kaufmann A, Mickoleit M, Weber M, Huisken J (2012) Multilayer mounting enables long-term imaging of zebrafish development in a light sheet microscope. Development 139(17):3242–3247
Giovannucci A, Friedrich J, Gunn P, Kalfon J, Brown BL, Koay SA, Taxidis J, Najafi F, Gauthier JL, Zhou P, Khakh BS, Tank DW, Chklovskii DB, Pnevmatikakis EA (2019) CaImAn an open source tool for scalable calcium imaging data analysis. eLife 8. https://doi.org/10.7554/eLife.38173
Zhang J , Pégard N, Zhong J, Adesnik H, Waller L (2017) 3D computer-generated holography by non-convex optimization. Optica 4(10):1306–1313
Gerchberg RW, Saxton WO (1972) A practical algorithm for the determination of phase from image and diffraction plane pictures. Optik 35(2):337–246
Di Leonardo R, Ianni F, Ruocco G (2007) Computer generation of optimal holograms for optical trap arrays. Optics Express 15(4):169–175
Pozzi P, Maddalena L, Ceffa N, Soloviev O, Vdovin G, Carroll E, Verhaegen M (2018) Fast calculation of computer generated holograms for 3D photostimulation through compressive-sensing Gerchberg–Saxton algorithm. Methods and Protocols 2:2
Hossein Eybposh M, Caira NW, Atisa M, Chakravarthula P, Pégard NC (2020) DeepCGH: 3D computer-generated holography using deep learning. Optics Express 28:26636
Oron D, Papagiakoumou E, Anselmi F, Emiliani V (2012) Two-photon optogenetics. Progress Brain Res 196:119–143
Shirley P, Ashikhmin M, Marschner S (2009) Fundamentals of computer graphics. CRC Press
Goodman JW (2005) Introduction to Fourier optics. Roberts & Company Publishers
Pozzi P, Mapelli J (2021) Real time generation of three dimensional patterns for multiphoton stimulation. Front Cell Neurosci 15:609505
Acknowledgements
This work was supported by funding from TU Delft and The Netherlands Organisation for Health Research and Development (ZonMw).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2023 The Author(s)
About this protocol

Cite this protocol
Maddalena, L., Pozzi, P., Ceffa, N.G., Hoeven, B.v.d., Carroll, E.C. (2023). Optogenetics and Light-Sheet Microscopy. In: Papagiakoumou, E. (eds) All-Optical Methods to Study Neuronal Function. Neuromethods, vol 191. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2764-8_8
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2764-8_8
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2763-1
Online ISBN: 978-1-0716-2764-8
eBook Packages: Springer Protocols