
Abstract
Genome-wide transposon mutagenesis followed by deep sequencing allows the genome-wide mapping of growth-affecting loci in a straightforward and time-efficient way.
SAturated Transposon Analysis in Yeast (SATAY) takes advantage of a modified maize transposon that is highly mobilizable in S. cerevisiae. SATAY allows not only the genome-wide mapping of genes required for growth in select conditions (such as genetic interactions or drug sensitivity/resistance), but also of protein sub-domains, as well as the creation of gain- and separation-of-function alleles. From strain preparation to the mapping of sequencing reads, we detail all the steps for the making and analysis of SATAY libraries in any S. cerevisiae lab, requiring only ordinary equipment and access to a Next-Gen sequencing platform.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
Genome-wide saturated mutagenesis can be achieved in many microbes by generating dense libraries of transposon insertion mutants and identifying the insertion sites by deep sequencing. The number of sequencing reads mapping to a defined genomic location can then serve as a proxy for the fitness of the strain bearing this particular mutation, in the conditions used [1]. Increased and decreased fitness translate as increased and decreased number of sequencing reads for these transposons. Therefore, in haploid genomes, essential genes are typically devoid of sequencing reads.
A screen involves (1) inducing transposition in a chosen organism and strain, (2) growing and selecting the transposon mutants in defined conditions, (3) harvesting the mutant library and extracting genomic DNA, (4) amplifying the transposon–genome junctions selectively by PCR, (5) identifying the transposon insertion sites by deep sequencing and mapping reads onto the reference genome.
Such an approach has been developed and successfully used in a variety of bacteria [1,2,3,4] and eukaryotic microbes (including S. pombe [5], S. cerevisiae [6,7,8], S. uvarum [9], C. albicans [10, 11], C. glabrata [12], R. toruloides [13], and P. pastoris [14]), using various combinations of transposons/transposase systems, transposon–genome junction amplification methods, and sequencing platforms (recently reviewed in ref. [15]).
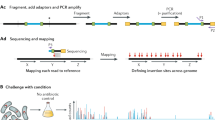
Here we describe a protocol for SAturated Transposon Analysis in Yeast (SATAY [6, 7]), which allows the rapid and effortless generation and sequencing of high-density transposon libraries in the budding yeast S. cerevisiae (Fig. 1). In SATAY, a modified non-autonomous Maize transposon (MiniDs) interrupts the ADE2 gene [16]. A galactose-induced hyperactive transposase (Ac) mobilizes the transposon. Homology-directed repair of ADE2 then confers adenine prototrophy to the transposed cells. A screen begins by transforming a plasmid (pBK549) encoding the galactose-induced transposase, the ADE2 gene interrupted by the MiniDs transposon and a URA3 selection marker, into an ade2- ura3- strain [7]. After some sanity checks, transformed clones are amplified and densely plated onto +galactose-adenine plates, allowing for (1) the induction of the transposition and (2) the growth of transposed Ade+ cells into colonies. These colonies are then scraped off the plate, re-grown in +glucose -adenine medium (plus any chosen environmental perturbation), before being processed for DNA extraction, amplification, sequencing, and mapping. Specific amplification of transposon–genome junction is achieved by circular PCR. Simultaneous sequencing of multiple libraries is achieved using the Illumina® system.

Principle of the SATAY method. (a) Principle of transposon insertion screening. (b) Outline of the steps involved in a SATAY screen. (c) Example data obtained by SATAY screens. This shows transposon insertion sites in a section of S. cerevisiae genetic map. Screens were performed in different conditions/genetic backgrounds. Data reveal essential genes (red dashed box), essential protein domains (magenta dashed box), and genes necessary for growth in defined conditions (green dashed box)
Optimized plating conditions yield 35,000–100,000 transposed colonies per 9.0 cm Ø petri dish, meaning that 30–70 petri dishes are sufficient to yield two million colonies. In our hands, this number allows mapping of transposons at ~250,000–400,000 independent genomic locations, averaging a resolution of one transposon every 50 bp [7].
Compared to synthetic genetic arrays and other high-throughput mutagenesis methods, SATAY possesses significant advantages. First, MiniDs targets the genome with little sequence specificity, minimizing insertion biases [16]. Second, its insertion within a gene does not preclude the expression of either the upstream or downstream sequences, allowing not only the mapping of genes required for growth in all or select conditions, but also of protein sub-domains, as well as the creation of gain- and separation-of-function alleles [6, 7, 17].
SATAY has been used to map genome-wide genetic interactions, as well as to identify genes involved in resistance and sensitivity to various chemicals, yielding insight into processes as diverse as signaling, organelle biology, lipid transport and metabolism, DNA repair, and transcriptional regulation [6, 7, 17,18,19,20,21].
2 Materials
All solutions are made with MilliQ (MQ) water.
2.1 Equipment
-
1.
UV-Vis Spectrophotometer.
-
2.
Stereomicroscope with transillumination.
-
3.
Bright field microscope with >10× long-distance objective.
-
4.
Shaking incubator (needs to accommodate up to 5 L Erlenmeyer flasks at 30 °C for yeast work, and culture tubes at 37 °C for bacteria work).
-
5.
30 °C static incubator (needs to accommodate at least 50 petri plates).
-
6.
37 °C static incubator for bacteria work.
-
7.
Horizontal gel electrophoresis apparatus.
-
8.
Repeater pipette, 5-mL sterile tips.
-
9.
Erlenmeyer flasks, sterile.
-
10.
Bunsen burner.
-
11.
Mini-Beadbeater (e.g., Disruptor Genie, Scientific Industrial).
-
12.
Thermomixer.
-
13.
Refrigerated microcentrifuge.
-
14.
Centrifuge (needs to accommodate 50-mL centrifuge tubes at 600 × g and 1600 × g, and 500-mL centrifuge tubes at 1600 × g).
-
15.
PCR thermocycler.
-
16.
Optional: Hemocytometer (Neubauer Improved Hemocytometer Counting Chamber).
2.2 Growth Media
-
1.
10× Amino Acid mix: 0.3 g/L l-Isoleucine, 3 g/L l-Valine, 0.4 g/L Adenine Hemisulfate salt, 0.2 g/L l-Arginine monohydrochloride, 0.2 g/L l-Histidine monohydrochloride monohydrate, 1 g/L l-Leucine, 0.3 g/L l-Lysine monohydrochloride, 0.2 g/L l-Methionine, 0.5 g/L l-Phenylalanine, 2 g/L l-Threonine, 0.4 g/L l-Tryptophan, 0.3 g/L l-Tyrosine, 0.2 g/L Uracil, 1 g/L l-Glutamic Acid monosodium salt hydrate, 1 g/L l-Aspartic Acid sodium salt monohydrate. Dissolve under constant agitation and gentle heat (~1 h). Autoclave for 15 min at 121 °C. Store at 4 °C.
-
2.
10× Drop Out mix: 10× Amino Acid mix in which the chosen component has been omitted.
-
3.
10× carbohydrate solution: 200 g/L of either d-(+)-glucose (Dextrose), d-(+)-Galactose, or d(+)-Raffinose. Aid dissolution by stirring on a heated (~40 °C) stirrer unit. Autoclave for 15 min at 121 °C. Store at room temperature.
-
4.
4× S solution: 6.8 g/L Yeast Nitrogen base (without Amino Acid and Ammonium Sulfate), 20 g/L Ammonium Sulfate. Autoclave for 15 min at 121 °C. Store at room temperature.
-
5.
SC plates: dissolve 1.7 g Yeast Nitrogen base (without Amino Acid and ammonium sulfate) with 5 g ammonium sulfate in 320 mL MQ water. In a separate flask, suspend 20 g granulated agar in 500 mL MQ water. Autoclave both the solutions for 15 min at 121 °C. Combine under constant stirring, allowing the mix to cool down to ~60 °C. Add 100 mL 10× carbohydrate solution and 100 mL 10× Drop Out mix (for instance, SC-D -ADE is obtained by adding 10× Dextrose solution and 10× -ADE Drop Out mix). Pour 25 mL in 9.0-cm Ø petri dishes.
-
6.
SC-Rd -URA liquid medium: 1× S solution, 1× -URA Drop Out mix, 1× carbohydrate solution (Raffinose), 0.1× carbohydrate solution (Dextrose). Store at 4 °C.
-
7.
SC-D -ADE liquid medium: 1× S solution, 1× -ADE Drop Out mix, 1× carbohydrate solution (Dextrose). Store at 4 °C.
-
8.
YPD: mix and autoclave 20 g BactoPeptone, 10 g Yeast Extract, 55 mg Adenine Hemisulfate salt in 900 mL. Add 100 mL 10× Dextrose solution. Store at room temperature.
2.3 DNA Work
-
1.
Filter pipette tips.
-
2.
Nonstick microfuge tubes (e.g., Ambion).
-
3.
Safe-Lock 1.5-mL microtubes.
-
4.
Glass beads, 0.5 mm, unwashed.
-
5.
PCR clean-up kit.
-
6.
PvuII.
-
7.
EcoRV.
-
8.
DpnII.
-
9.
NlaIII.
-
10.
5 Weiss Units/μL T4 ligase.
-
11.
5 U/μL Taq DNA Polymerase with ThermoPol Buffer (NEB).
-
12.
5× TBE: 0.45 M Tris–Base, 0.45 M Boric Acid, 0.01 M EDTA, pH 8.0.
-
13.
Cell Breaking Buffer: 2% Triton X-100, 1% SDS, 100 mM NaCl, 100 mM Tris–HCl, pH 8.0, 1 mM EDTA. All stock solutions autoclaved, except 100% Triton X-100.
-
14.
Ultrapure Phenol:Chloroform:Isoamylalcohol 25:24:1.
-
15.
25 mM dNTPs Mix. Store at −20 °C as 50 μL aliquots.
-
16.
TE: 10 mM Tris–HCl, pH 8.0, 1 mM EDTA.
-
17.
Ethanol 100% and 70%.
-
18.
3 M Sodium Acetate, pH 5.2.
-
19.
RNAse A:10 mg/mL RNase A, 10 mM Sodium Acetate, 0.1 M Tris–HCl, pH 7.4. Vortex RNAse in Sodium Acetate. Boil for 15 min. Adjust pH to 7.4 by adding 1 mL 1 M Tris–HCl pH 7.4. Aliquot in microtubes. Store at −20 °C.
-
20.
5 μg/μL Linear acrylamide.
-
21.
Agarose for electrophoresis.
2.4 Yeast Work
-
1.
9.0 cm Ø petri dishes.
-
2.
50-mL centrifuge tubes.
-
3.
Corning® 500-mL PP Centrifuge Tubes with Plug Seal Cap.
-
4.
Corning® 500-mL Polyetherimide Centrifuge Tube Cushions.
-
5.
Wood toothpicks, sterile.
-
6.
Glass pipettes, sterile.
-
7.
Glass beads 3 mm.
-
8.
Pasteur pipettes plain glass 230 mm.
-
9.
50% PEG 3350. Autoclave for 15 min at 121 °C. Store at room temperature. Use within a month.
-
10.
1 M LiOAc. Autoclave for 15 min at 121 °C. Store at room temperature.
-
11.
2 mg/mL salmon sperm DNA sodium salt. Dispense 1 mL aliquots in sterile 1.5-mL microtubes. Incubate at 95 °C for 5 min to denature the double stranded DNA. Immediately store at −20 °C. Thaw when needed, do not refreeze.
-
12.
Transformation mix: 40% PEG3350, 0.1 M LiOAc, 0.3 mg/mL salmon sperm DNA. Dispense 162 μL aliquots in 0.5-mL sterile microtubes. Store at −20 °C. Use one aliquot per transformation reaction. Thaw when needed, do not refreeze.
2.5 Sequencing
-
1.
MiSeq Reagent Kit v3 (150 Cycles) (Illumina®) or NextSeq 500/550 Mid Output Kit v2.5 (150 Cycles) (Illumina®) or NextSeq 500/550 High Output Kit v2.5 (75 Cycles) (Illumina®).
-
2.
PhiX Control v3 (Illumina®).
-
3.
1–200 μL Gel loading Tips, round (Exacta Cruz).
3 Methods
3.1 Generate Starting Yeast Clones
3.1.1 Verify pBK549 by Restriction Digest
-
1.
Miniprep 4–10 colonies of pBK549 using your favorite miniprep procedure.
-
2.
Digest 150 ng DNA with 20 units PvuII in a total volume of 20 μL.
-
3.
Digest 150 ng DNA with 20 units EcoRV in a total volume of 20 μL.
-
4.
Load 4 μL of each restriction digest on a 0.6% Agarose 1× TBE electrophoresis gel. Run the gel for 1.5 h at 80 V then image. Optional: run the gel for a further 30 min at 40 V.
-
5.
Identify and proceed with the DNA preparations that contain little or no recombined product (Fig. 2a–c, see Note 1).

The plasmid pBK549. (a) Map of the plasmid. Dashed lines represent homologies for repair of the plasmid after transposon (MiniDs) excision. Stars represent mutations that make the Ac transposase hyperactive. (b) Maps of recombinant species that appear spuriously in a transposase-independent manner, both upon bacterial plasmid expansion and yeast growth. (c) Diagnostic restriction digests of two pBK549 preparations, one with little recombinant species and perfectly usable (left), the other with more recombinant species (right). Fragment sizes for the diagnostic digest of the correct plasmid are, for EcoRV 5768, 3434 and 2284 bp, and for PvuII, 7036 and 4450 bp (marked with stars). Recombinant species 1 yields an EcoRV fragment of 6498 bp, and PvuII fragments of 4450 and 2048 bp. Recombinant species 2 yields an EcoRV fragment of 7140 bp, and PvuII fragments of 4450 and 2690 bp. (d) Streak of six independent pBK549-transformed yeast clones. The colonies appearing rarely on -ADE plates indicate that the clone bears an intact pBK549 (e.g., green sector) capable of generating Ade + progeny by spontaneous recombination. Clones that fail to produce any colonies on -ADE (red sector) might bear an already recombined species that is transposition-deficient. The clone in the green sector was then scraped off the -URA plate and processed further (see Subheading 3.2.1)
3.1.2 Transform pBK549 in Strains of Interest
-
1.
Inoculate the strain(s) of interest in 5 mL YPD. Grow at 30 °C, 140–200 rpm until saturation (see Notes 2 and 3).
-
2.
Measure the OD600 of each saturated culture. Dilute to OD600 0.2 in 20 mL fresh YPD. Incubate at 30 °C, 140–200 rpm until the culture has reached OD600 0.8.
-
3.
Spin each culture at 600 × g for 5 min at room temperature.
-
4.
Discard the supernatants. Resuspend the cells in 20 mL sterile MQ water and pellet the cells as above.
-
5.
Discard the supernatants. Resuspend the cells in 1 mL sterile MQ water, dispense 180 μL of cell suspension into a fresh 1.5-mL microtube and pellet the cells as above.
-
6.
Discard the supernatants. Add 16 μL pBK549 (7 ng/μL, 112 ng). Add 162 μL of transformation mix then mix well using the same pipette.
-
7.
Incubate at 30 °C for 30 min, no agitation.
-
8.
Incubate at 42 °C for 20 min, no agitation.
-
9.
Centrifuge at 600 × g for 5 min, room temperature.
-
10.
Discard the supernatants. Resuspend each cell pellet in 1 mL sterile MQ water. For each strain transformed, spread one SC-D -URA plate with 200 μL of the resuspended cells.
-
11.
Incubate at 30 °C for 2 days or until the colonies have reached the desired size. Store the plates at 4 °C (see Note 4).
3.1.3 Selection of Yeast Transformants
-
1.
Streak 12–18 independent transformants on 2–3 SC-D-URA and 2–3 SC-D -ADE plates (see Note 5).
-
2.
Pursue with clones that produce a low but detectable background of Ade+ colonies (Fig. 2d) (see Note 6).
3.2 Transposition
3.2.1 Day 0
-
1.
Scrape cells off selected sectors from the SC-D-URA plate and inoculate 40–60 mL SC-Rd -URA at OD600 0.2 (see Notes 7–9).
-
2.
Inoculate 3–5 pre-cultures from 3 to 5 independent sectors (see Notes 3 and 10).
-
3.
Grow at 30 °C for 16–20 h, with shaking at 160–200 rpm.
-
4.
Take the SC-Gal -ADE (transposition plates) and SC-D -ADE (control plates) plates from the cold room. Store these plates at room temperature overnight (see Notes 11 and 12).
3.2.2 Day 1
-
1.
Measure the OD600 of each pre-culture (see Note 13).
-
2.
Optional. Take a small aliquot of each overnight culture, dilute it 40,000× in sterile MQ water and plate 200 μL on one SC-D and one SC-D -URA plate (see Note 14).
-
3.
Transfer the cells to a 50-mL centrifuge tube and centrifuge for 5 min at 600 × g at room temperature.
-
4.
Calculate the concentration factor required to resuspend the cells at OD600 40 and remove the appropriate volume of supernatant using a pipette (see Notes 15 and 16).
-
5.
Resuspend the cell pellet using the same pipette.
-
6.
Arrange the SC-Gal -ADE plates in stacks of 10 plates. Dispense 200 μL of cell suspension on each plate of the first stack. Spread the cells with glass beads, keeping the plates horizontal and using a gentle circular motion. Ensure that no area of the agar surface has been missed in any of the plates (see Note 17).
-
7.
Repeat with the next set of 10 plates (see Note 18).
-
8.
The liquid spread in point 6 above should now be fully absorbed and the cells should form a uniform opaque film covering the agar surface entirely (see Note 19). Gently shake the stack of plates, this time using straight motions, two to three times in four directions, keeping the plates horizontal. Patterns on the agar surface become visible (see Notes 20 and 21).
-
9.
Remove the beads immediately. Do not allow the beads to remain on the agar surface.
-
10.
Proceed with the next stack. Repeat until all plates have been spread with the cells.
-
11.
As a negative control, dispense 200 μL of the same cell suspension onto one SC-D -ADE plate. Spread as above (see Note 22).
-
12.
Store the plates in plastic sleeves loosely sealed with tape, ensuring air circulation but preventing plates from drying out during the long incubation period. Incubate at 30 °C until ready to harvest (see Note 23).
3.2.3 Day 4
-
1.
Observe the plates after 3 days of incubation. Draw a square centimeter on the dish and count the number of clones contained within it using a stereomicroscope (Fig. 3a).

Growth and harvest of transposed clones. (a) Appearance of Ade+ colonies in galactose (inducing transposase) and glucose (repressing transposase) over time. Images are taken with a transilluminated stereomicroscope. Scale bar, 1 mm. (b) General aspect of transposed SC -ADE +Galactose plates after 17 days. Two genetic backgrounds (BY4741 and W303-1B) yield colonies of different sizes and number. (c) Manufacturing of harvesting rakes from long glass Pasteur pipettes. The opening of the pipette is first closed (1), then a first ~90° bend is generated ~0.5 cm from the tip (2). A second 45° bend is then generated ~4 cm from the tip in the same plane as the first bend (3)
3.2.4 Days 5–20
-
1.
Observe the plates every 3–4 days for the appearance of new colonies, for as long as 20 days (see Notes 24 and 25).
-
2.
When no new colonies appear, estimate the final number of transposed colonies and proceed to harvest (Fig. 3) (see Note 26).
3.3 Plate Harvest, Library Expansion, and Collection
3.3.1 Harvest the Plates
-
1.
Proceeding five plates at a time, dispense 2 mL of SC-D -ADE medium per plate using a glass pipette (see Note 27).
-
2.
Scrape the cells off the surface of the plate using a rake (Fig. 3c, see Note 28). Leave the plates tilted for the medium to collect on one side.
-
3.
Collect the cell suspensions using a pipette. Dispense into a sterile container (see Note 29).
-
4.
Proceed with the next five plates until all plates have been harvested (see Note 30).
3.3.2 Reseed
-
1.
Mix the harvested cell suspensions well.
-
2.
Measure the OD600 of each harvested cell suspension (see Note 31).
-
3.
Reseed the harvests in a total of 2 L SC-D -ADE at a density of OD600 0.2 (~2E+06 cells/mL) (see Note 32).
-
4.
Dilute an aliquot of the reseeded culture 1000×. Spread 200 μL on one SC-D and one SC-D -ADE plate using glass beads. Incubate for 3 days at 30 °C. Count the colonies to estimate the total number of cells, number of Ade + cells and number of cells per transposition event reseeded (see Note 33).
-
5.
Incubate at 30 °C in a shaking incubator for 16–24 h, 140–200 rpm.
-
6.
Optional. Determine the number of cells per transposition event after re-growth of the library (see Note 34).
-
7.
Optional. Dilute and re-grow the cultures to apply further treatments (e.g., exposure to drugs [6, 7, 17,18,19] (see Note 35).
-
8.
Optional. Freeze the excess harvest for further use (see Note 36).
3.3.3 Final Harvest
-
1.
Collect at least 500 mL of saturated culture (OD600 ~ 4), 5 min, 1600 × g, room temperature. Harvest the entire culture if centrifuge space is not limited (see Note 37).
-
2.
Resuspend each pellet in 150 mL MQ water. Centrifuge at room temperature for 5 min, 1600 × g.
-
3.
Weigh a 50-mL conical centrifuge tube.
-
4.
Resuspend each pellet in 40 mL MQ water and transfer the cells into the 50-mL tube.
-
5.
Repeat the centrifugation, remove all the supernatant and weigh the pellet by subtracting the tube’s weight from the total weight.
-
6.
Resuspend the pellet in an equal amount (weight/volume) of MQ water. Distribute 1 mL in 1.5-mL microtubes, so that each tube contains ~500 mg of cells in a dense suspension.
-
7.
Centrifuge at room temperature for 5 min at 1600 × g or for a few seconds at 16,000 × g. Remove all the supernatant using a P1000 followed by a P200 pipette, store the 500 mg cell pellets at −20 °C until further use.
3.4 DNA Template Preparation
3.4.1 Genomic DNA Extraction
The procedure below works with high efficiency and is easy, but generates phenol:chloroform waste. The method can be replaced by commercial DNA isolation kits.
-
1.
Resuspend 500 mg cell pellet in 500 μL of Cell Breaking Buffer.
-
2.
Distribute the cell suspension into 280 μL aliquots in 1.5-mL Safe-Lock microtubes.
-
3.
For each aliquot: add ~300 μL 0.5 mm unwashed glass beads, then 200 μL Phenol:Chloroform:Isoamylalcohol 25:24:1.
-
4.
Ensure that no glass bead is stuck between the wall of the tube and the cap, as this could prevent tight sealing and cause leakage.
-
5.
Vortex for 10 min at 4 °C using a Mini-Beadbeater.
-
6.
Add 200 μL 1× TE to the lysate. Vortex briefly then centrifuge for 5 min at 16,100 × g, 4 °C.
-
7.
Transfer the clear upper layer (~400 μL) to a fresh 1.5-mL tube, without disturbing the white interphase.
-
8.
Add 2.5 vol (1 mL) of 100% Ethanol and mix by inversion.
-
9.
Pellet the nucleic acids for 5 min at 16,100 × g, 20 °C (see Note 38).
-
10.
Pipette out as much supernatant as possible.
-
11.
Pulse centrifuge to ensure that all the liquid collects at the bottom of the tube.
-
12.
Remove the remaining supernatant by pipetting.
-
13.
Dilute 10 mg/mL RNase A to 250 μg/mL in MQ water. Prepare 220 μL of 250 μg/mL RNase A per nucleic acid pellet.
-
14.
Add 200 μL of 250 μg/mL RNase A on top of the pellet.
-
15.
Incubate for 15 min at 55 °C, 1000 rpm. Vortex briefly to completely resuspend the DNA pellet.
-
16.
Pulse centrifuge to ensure that all the liquid collects at the bottom of the tube.
-
17.
Add 2.5 vol (500 μL) 100% Ethanol and 0.1 vol (20 μL) 3 M Sodium Acetate, pH 5.2. Mix well by inversion or by pipetting.
-
18.
Centrifuge for 5 min at 16,100 × g, 20 °C to pellet the DNA.
-
19.
Pipette out as much supernatant as possible. Centrifuge briefly to ensure that all the liquid collects at the bottom of the tube. Pipette out the remaining supernatant.
-
20.
Add 1 mL 70% Ethanol. Place the tube in the opposite orientation to the previous centrifugation so that the pellet washes through the 70% Ethanol and collects on the opposite side of the tube. Centrifuge for 5 min at 16,100 × g, 20 °C.
-
21.
Remove most of the supernatant using a P1000 pipette. Centrifuge briefly to collect all the remaining 70% Ethanol to the bottom of the tube, then remove it entirely using a P20 pipette.
-
22.
With the microtube cap open, dry the pellet for 10 min at 37 °C.
-
23.
Distribute a total of 200 μL MQ water equally to all samples. Resuspend the pellets completely for 20–40 min at 55 °C on a thermomixer shaking at 700 rpm. Pool the resuspended genomic DNA aliquots.
3.4.2 Genomic DNA Quantification on Gel (See Note 39)
-
1.
Cast a 0.6% Agarose 1× TBE electrophoresis gel.
-
2.
Mix one volume of 6× home-made or commercial loading dye buffer with 3 volumes MQ water to make a working loading dye solution.
-
3.
Dilute 2 μL gDNA prep with 18 μL of loading buffer prepared above.
-
4.
Load various amounts of 1 kb GeneRuler™ and 5 μL of each diluted gDNA sample (Fig. 4a, see Note 40).
-
5.
Run the gel for 1.5 h at 80 V and image the gel avoiding saturation (see Note 41).
- 6.

DNA library generation. (a) Gel quantification of genomic DNA preps. Outer lanes contain indicated amounts of DNA ladder. Lower panel shows the quantification of indicated parts of the gel using Fiji/ImageJ. (b) Linear regression of the quantified standard DNA ladder bands and quantification of genomic preps 1–10
3.4.3 Genomic DNA Digestion with NlaIII and DpnII
-
1.
In parallel, digest 2 μg of genomic DNA with 50 units of DpnII, and 2 μg with 50 units of NlaIII. Both digestions take place overnight in 50 μL at 37 °C in Nonstick microfuge tubes.
-
2.
Heat-inactivate the reactions for 20 min at 65 °C.
-
3.
Run 2 μL (80 ng) of each reaction on a 1% Agarose 1× TBE electrophoresis gel (Fig. 5a, b).
-
4.
The digested DNA can be stored at −20 °C until further use.

Library sequencing. (a) Outline of the molecular biology steps. (b) Gel electrophoresis of genomic DNA before and after restriction digestion with indicated enzymes. (c) Gel electrophoresis of purified PCR products. The bands at 867 and 465 bp correspond to intact (untransposed) pBK549
3.4.4 Digested gDNA Circularization
-
1.
To the tube containing 48 μL digested DNA, add in the following order (see Note 42).
-
(a)
307 μL MQ water.
-
(b)
40 μL 10× T4 ligase buffer, mix well.
-
(c)
5 μL T4 ligase, mix gently.
-
(a)
-
2.
Incubate for 6 h at 22 °C.
-
3.
Add 40 μL 3 M Sodium Acetate pH 5.2, 1 μL (5 μg) linear acrylamide and mix well (see Note 43).
-
4.
Add 1 mL 100% Ethanol, mix well. Precipitate overnight or longer at −80 °C.
-
5.
Centrifuge for 20 min at 16,100 × g, 4 °C.
-
6.
Remove as much supernatant as possible without touching the pellet.
-
7.
Add 1 mL 70% Ethanol. Break the pellet using a pipette.
-
8.
Centrifuge for 20 min at 16,100 × g, 20 °C.
-
9.
Remove most of the supernatant using a P1000 pipette. Pulse centrifuge and remove the remaining supernatant completely using a P20 pipette.
-
10.
With the microtube cap open, dry the pellet for 10 min at 37 °C.
-
11.
Add 105 μL MQ water, incubate at 55 °C for 15–30 min with optional shaking at 1000 rpm.
-
12.
The ligated DNA can be stored at −20 °C until further use.
3.5 Library Amplification
3.5.1 Primer Design
-
1.
Each library is amplified using primer P5_MiniDs (common to all libraries) and two unique indexed primers (P7_(index)_MiniDs), one for the NlaIII sub-library and the other for the DpnII sub-library. The indexed primers are designed to specifically amplify the transposon-genome junctions and to generate products adapted to the Illumina® sequencing platforms (Fig. 5a, Table 1).
-
2.
Choose the best-suited sequencing platform according to availability, number and complexity (number of clones) of libraries, and budget (see Note 44).
-
3.
Choose combinations of indexes for the sequencing of pooled libraries according to Illumina®’s recommendations [23,24,25] (see Note 45).
-
4.
Resuspend all primers as 100 μM stock solutions in sterile MQ water on a Thermomixer at 55 °C shaking at 1000 rpm for 10 min. Store aliquots at −20 °C.
3.5.2 PCR and Purification
-
1.
Prepare one large reaction per sample by mixing the following reagents in the indicated order: 847.55 μL MQ water, 115 μL 10× ThermoPol Buffer, 9.2 μL 25 mM dNTPs, 11.5 μL Primer P5_MiniDs 100 μM, 11.5 μL Indexed Primer P7_(index)_MiniDs 100 μM, 40.25 μL (17.5 units) Taq DNA Polymerase. Omit the template at this stage.
-
2.
Dispense 90 μL of the PCR mix in a fresh PCR tube. Add 10 μL MQ water to make a no-template negative control.
-
3.
Add the remaining master mix into the thawed 105 μL aliquot of template DNA. Mix well using a P1000 pipette.
-
4.
Aliquot 100 μL of reaction into 10 PCR tubes (or 125 μL into a strip of 8 PCR tubes).
-
5.
Run the following PCR cycle: 95 °C 1 min [95 °C 30 s, 55 °C 30 s, 68 °C 3 min] × 35 cycles, 68 °C 10 min, 15 °C final hold.
-
6.
Pool the reactions corresponding to the NlaIII-digestion.
-
7.
Pool the reactions corresponding to the DpnII-digestion. Keep the NlaIII and DpnII pools separate.
-
8.
Run 2 μL of each pool and 2 μL of corresponding negative controls on a 1% agarose gel, 1× TBE (see Note 46).
-
9.
Purify 100 μL of each PCR pool using a PCR purification kit to generate the NlaIII and the DpnII sub-libraries (see Note 47).
-
10.
Run 2 μL of each sub-library on a 1% agarose gel, 1× TBE (Fig. 5c).
-
11.
Quantify the purified products using a UV-spectrophotometer (see Note 48).
3.6 Sequencing
Refer to Illumina® system guides for handling of reagents and run set up (Chap. 3 Sequencing in refs. [26, 27]).
SATAY-specific instructions are indicated below.
3.6.1 Combine Libraries to Be Sequenced Together
-
1.
Mix equal concentrations of the NlaIII and DpnII sub-libraries to generate a library (see Note 49).
-
2.
If mixing multiple libraries, adjust their amount in the pool so that the number of molecules per transposition event is kept constant (see Note 50).
-
3.
Quantify the pooled sample using a spectrophotometer (see Note 48). Confirm that the concentration is as expected.
-
4.
Estimate the molarity of the pooled sample using the following estimate for the average molecular weight of PCR products: 500 bp × 650 g/(mole × bp) = 325,000 g/mole.
3.6.2 Library Preparation
-
1.
Denature and dilute SATAY library and PhiX control to 20 pM in HT1 buffer according to manufacturer recommendations (Protocol A [27, 28]) (see Note 51).
-
2.
Combine the denatured SATAY library with 1% denatured PhiX control v3 to the following final volumes and concentrations in pre-chilled HT1 buffer (see Note 52). The library is ready to load.
-
MiSeq Kit v3 (150 cycles). 20 pM denatured DNA in 650 μL.
-
NextSeq 500/550 High Output Kit v2.5 (75 cycles). 2.6–3 pM denatured DNA in 1350 μL.
-
NextSeq 500/550 Mid Output Kit v2.5 (150 cycles). 2.5 pM denatured DNA in 1350 μL.
-
3.6.3 Loading of Library
-
1.
Miseq Kit v3 (150 cycles). Load 600 μL into Load Samples well [28].
-
2.
NextSeq 500/550 High Output Kit v2.5 (75 cycles). Load 1.3 mL into well 10 [29].
-
3.
NextSeq 500/550 Mid Output Kit v2.5 (150 cycles). Load 1.3 mL into well 10 [29].
3.6.4 Loading of Sequencing Primer (Sequencing of the Transposon–Genome Junctions)
-
1.
MiSeq Kit v3 (150 cycles). Load 3.4 μL of 100 μM Primer 688_minidsSEQ1210 into well 12 [30] (see Note 53).
-
2.
NextSeq 500/550 High Output Kit v2.5 (75 cycles). Load 5.16 μL of 100 μM Primer 688_minidsSEQ1210 into well 20 [30].
-
3.
NextSeq 500/550 Mid Output Kit v2.5 (150 cycles). Load 4.02 μL of 100 μM Primer 688_minidsSEQ1210 into well 20 [30].
3.6.5 Loading of Custom Index Primer (Sequencing of Indexes)
-
1.
MiSeq v3 (150 cycles). Load 600 μL of 0.5 μM Primer Custom_index1 diluted in pre-chilled HT1 buffer, into well 19 (section Preparing and Loading Custom Primers in ref. [31]).
-
2.
NextSeq 500/550 High Output Kit v2.5 (75 cycles). Load 2 mL of 0.3 μM Primer Custom_index1 diluted in pre-chilled HT1 buffer, into well 9 (section Prepare and Add Custom Primers in ref. [32]).
-
3.
NextSeq 500/550 Mid Output Kit v2.5 (150 cycles). Load 2 mL of 0.3 μM Primer Custom_index1 diluted in pre-chilled HT1 buffer, into well 9 (section Prepare and Add Custom Primers in ref. [32]).
3.6.6 Instrument Setup
-
1.
Generate a sample sheet following standard procedures (for instance, using Illumina® Experiment Manager, IEM), with the following parameters:
Miseq:
-
(a)
Index Reads: 1.
-
(b)
Read Type: Single Read.
-
(c)
Cycles Read 1: 76 (see Note 54).
-
(d)
Workflow: FastQ only.
-
(e)
FastQ Only Workflow-Specific Settings: √ Custom Primer for Index.
NextSeq:
-
(a)
Index Reads: 1.
-
(b)
Read Type: Single Read.
-
(c)
Read Length: Read 1 = 76, Read 2 = 0, Index 1 = 8, Index 2 = 0.
-
(d)
Custom Primers: √ Index 1.
-
(e)
Workflow: FastQ only.
-
(a)
-
2.
Upload the Sample Sheet to the instrument.
-
3.
Run the sequencing reaction.
3.7 Analysis
The analysis is performed using R and Bioconductor packages ShortRead [33], Rsamtools [34], and Rbowtie [35, 36].
The sequencing run generates one or more FASTQ file [37] per indexed sub-library (depending on the instrument). The analysis is conducted on raw FASTQ files directly.
You can either run an R script (Subheading 3.7.1, see Note 55) or use a web server (Subheading 3.7.2).
3.7.1 Reads Mapping Using R Script
-
1.
Install a working copy of Rstudio (https://www.rstudio.com/) or any other R working environment.
-
2.
Download the Fastq.gz files from the sequencing machine or service and store them into a dedicated folder locally (fastqFolder) (see Note 56).
-
3.
Download the analysis script (Supplement File 1), or if you want to be guided through the analysis, download the corresponding R notebook (Supplement File 2).
-
4.
Download a copy of the genome annotation (saccharomyces_cerevisiae_R64–2-1_20150113.gff) from the https://yeastgenome.org website and save it in a dedicated folder (genomepath). The indexed genome sequence used by the script will also be stored in this folder.
-
5.
Edit the script under the section “Declare variables” so as to point to your fastqFolder and genomepath.
-
6.
Run the script.
3.7.2 Reads Mapping Using Web Server
-
1.
Download the Fastq.gz files and store them into a folder locally.
-
2.
Generate *.zip or *.tar.gz archive files, one file per sample.
-
3.
Upload each archive individually on the web server at https://kornmann.bioch.ox.ac.uk/satay/submission.html.
-
4.
Download the resulting *.bed, *.wig, and *pergene.txt files. The *pergene.txt contains the number of transposon and reads mapping to all ORFs (see Note 57).
3.7.3 Visualizing Transposon Maps
-
1.
Open a browser and head to http://genome-euro.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=benjou&hgS_otherUserSessionName=23bDePuYrk [38].
-
2.
Click “Manage Custom Tracks” the “add custom track.”
-
3.
Click “Browse” and select the *.bed or *.wig file from the previous analysis, then click submit.
-
4.
Click on “go” and adjust zoom level in the genome browser.
-
5.
Adjust displayed tracks and settings as per genome-browser’s documentation.
4 Notes
-
1.
pBK549 recombines occasionally in a transposase-independent manner, both in bacteria (including recA-strains) and in yeast. The resulting species are transposition-deficient. The presence of some amount of recombined species in a pBK549 DNA prep is not an issue. Yeast clones that inherit them by transformation will be eliminated before starting the screen (Subheading 3.1.3). The highest the amount of recombined species relative to intact pBK549, the more yeast transformants you will need to check. Plasmid preps containing a mixture of species can be purified by retransformation in E. coli. To keep the amount of recombined pBK549 species as low as possible, we recommend to:
-
(a)
Systematically verify new plasmid DNA preps.
-
(b)
Extract plasmid DNA from isolated bacteria colonies only.
-
(c)
Inoculate the bacteria culture from a colony, not a pre-culture (even for MidiPrep).
-
(d)
Freeze an aliquot of bacteria culture in 30% glycerol, immediately after taking it out of the incubator. Use stocks corresponding to DNA preps that satisfy quality control to produce more DNA preps.
-
(a)
-
2.
The strains must be ura- ade-. Strains such as W303 are ade2- and can be used as is. Deletion of ADE2 (56 nt before ATG to 62 nt after the STOP) can be achieved using primers Ade2PriFwd and Ade2PriRev (Table 1) on any template from ref. [39].
-
3.
SATAY will pick up genetic interactions with secondary mutations spuriously present in a transposed strain. We recommend generating independent strains of the genotype of interest, if possible. Thanks to barcoding, each library can be divided into sub-libraries, providing biological replicates. The important and most labor-intensive step is to obtain a total of 2–4 million independent clones per condition/genotype (see Notes 16 and 26).
-
4.
Transformation plates can be used for up to 1 month, after which we recommend freshly re-transforming the vector.
-
5.
Here the goal is not to obtain isolated colonies but to propagate the transformants and check for spontaneous recombination. A small percentage of transformed cells will receive recombined species 1 or 2 and grow into clones that are transposition-deficient (Fig. 2b). These clones can be distinguished from clones harboring intact plasmids by streaking on -ADE medium. Clones bearing intact plasmids will yield a few (1–20) Ade+ colonies by spontaneous (transposase-independent) homologous recombination, while those bearing recombined species 1 and 2 will fail to yield any Ade + progeny. The ability to produce a few Ade+ colonies is a means to recognize transposition-proficient clones than can be used further. Use a flat toothpick to pick an isolated colony from the transformation plate, make a thick streak on SC-D-ADE first, then on SC-D-URA. Proceed to spread on the SC-D-URA plate with the same toothpick. Use a fresh toothpick to spread on the SC-D-ADE plate.
-
6.
To minimize any further appearance of Ade + spontaneous recombinants, store the streaked SC-D-URA plates at 4 °C and use within a week. Beyond a week, repeat the verification procedure directly from the transformation plate.
-
7.
Scrape the cells using a tip mounted on a P1000 pipette. Inoculate in a 1.5-mL microtube pre-filled with 1 mL sterile MQ water. Pipette the cells up and down to mix them well. Measure the OD600 of a 50× dilution of this cell suspension. Calculate the volume of cell suspension required to inoculate 40–60 mL SC-Rd-URA medium at OD600 0.2.
-
8.
One OD600 unit for cells scraped from a plate corresponds to ~1.6E+07 cells. One 1/6-plate sector is enough to inoculate 40–60 mL at OD600 0.2.
-
9.
The 0.2% glucose allows for a smooth diauxic transition from glucose to raffinose. The pre-culture in raffinose primes cells for subsequent galactose induction by minimizing glucose repression.
-
10.
2 mL of saturated (OD600 ~ 4) pre-culture per transposition plate or 100 mL for 50 plates.
-
11.
The goal is to ensure that the plates are dry before plating. Plating on moist plates will result in a long drying time and inhomogeneous plating.
-
12.
The number of SC-Gal -ADE (transposition) plates required depends on the transposition efficiency, which may vary with the genetic background. Aim to obtain 2–4 million transposed clones per condition/genotype. One SC-D -ADE (control) plate is required per pre-culture.
-
13.
The pre-cultures should have reached saturation (OD600 ~ 4 in our hands). Before measuring the OD600, dilute the culture to ensure the readout value is within the linear range of the spectrophotometer (usually 0.1–1).
-
14.
This step checks that most cells are viable and contain the plasmid. As a guideline, we find ~450 (By4741 background, from 2.5E+07 cells/OD as measured with a hemocytometer) and ~250 (W303 background, from 1E+07 cells/OD as measured with a hemocytometer) colonies on SC-D plates. Expect that up to 20% of the cells have become ura-, which is not an issue.
-
15.
For example, if the overnight pre-culture is at OD600 4, then you need to concentrate the cells 40/4 = 10-fold, therefore remove 90% of the initial volume. It might be necessary to perform two consecutive rounds of centrifugation to harvest the required amount of cells. In this case, remove the supernatant from the first centrifugation entirely, add the remaining culture and repeat the centrifugation. Remove the appropriate volume of the supernatant to attain the desired cell concentration.
-
16.
The procedures described here were optimized for our laboratory conditions (spectrophotometer, growth media, strains). We recommend testing several plating concentrations at small scale to determine which one leads to the highest number of transposed colonies. Plating at OD600 35–40 was optimal in our hands. Plating at OD600 70 decreased the yield, firstly because it became difficult to plate homogeneously and secondly because the cell density on the plate was so high that it prevented the growth of transposed clones. Plating below OD600 35 decreased the yield of transposed clones proportionally.
-
17.
Use a repeater pipette. Use a different tip for each pre-culture. Keep the tip in a sterile beaker while not in use.
-
18.
The number of plates to process at once depends on how fast the cell suspension is absorbed on the surface of the plate. The dryer the plates, the faster the liquid will be absorbed, therefore the fewer the plates that should be processed at once.
-
19.
Do not proceed further if the plates are still wet. Set the wet plates aside until they are dry while processing the remaining plates.
-
20.
Homogeneous plating is key. We determined that spreading 200 μL using 10–15 glass beads per plate was optimal. Fewer beads will result in inhomogeneous plating. More beads decrease the number of cells seeded on the plate. Dispense the beads on all the plates before plating. Store the plates upside down such that the beads remain in the lid until needed. Leaving the beads on the agar will produce dents, resulting in inhomogeneous plating and inhomogeneous size and distribution of the transposed colonies.
-
21.
Check the plating using a microscope equipped with a long-distance objective (e.g., a tetrad dissection microscope). Aim for a monolayer of cells covering 90% of the plate. Interspersed mounds of cells are perfectly normal.
-
22.
Perform one control plating on SC-D -ADE for each independent pre-culture. This control is necessary to estimate the number of Ade + cells present prior to transposition induction. Ade + cells can result from spontaneous transposase-independent recombination of pBK549 (see Note 5). Cells containing such recombination products will form colonies on both the SC-D -ADE and SC-Gal -ADE plates within the first 2–3 days after plating, while genuinely transposed clones will develop over the course of 10–15 days.
-
23.
In very dry incubators, drying of the plates can be prevented by incubating the sleeves in a humid chamber, for example, a plastic box containing moist towel paper.
-
24.
The incubation time might vary with the strain and media composition. We recommend performing a small-scale time course experiment to monitor the appearance of transposed clones. The number of clones as a function of the incubation time is reproducible and generally plateaus after 10 days. SC-Gal -ADE plates can be kept at 4 °C for at least 3 months before plating, without alteration of transposition efficiency.
-
25.
The clones might be difficult to differentiate from the background lawn of cells after only 3 days of incubation but will become more distinct rapidly. Unlike mounds of cells resulting from plating, colonies are characterized by their round shape, defined edges, and a distinctive refraction. Mark the colonies using permanent pens of different colors or take photos of the same area to follow the appearance of new ones. Colonies overlap above a density of ~500 per cm2, therefore estimate the number of overlapping colonies within a mound.
-
26.
ByK352 (By4741 ade2::HIS3) consistently produces 1200–1700 colonies/cm2 (66,000–93,500 colonies/plate) and ByK832 (W303-1B ade2::NatMX6), 350–500 colonies/cm2 (19,250–27,500 colonies/plate). Transposition-independent Ade + background colonies represent 2–10% of the final number of Ade + clones.
-
27.
Process a few plates at a time to avoid the liquid being absorbed into the agar before the cells have been harvested.
-
28.
The best rakes are made by bending a long Pasteur pipette with a Bunsen burner (Fig. 3c). Commercial glass rakes used for spreading are sometimes too thick to scrape the cells efficiently.
-
29.
To reduce the volume of medium needed, it is also possible to reuse the collected cell suspension to harvest the next set of plates.
-
30.
Because liquid absorbs in the agar, about 50 mL are collected from 50 harvested plates. The harvested material is thick and composed of ~70% Ade + cells. The red coloration is due to the presence of untransposed ade- cells that accumulated a red pigment on the SC-Gal -ADE transposition plates.
-
31.
The OD600 of the harvested material is ~50. Dilute to read within the linear range of the spectrophotometer.
-
32.
This step serves to expand the library and to dilute the ade- cells. The expanded library is then either processed for sequencing or exposed to various treatments and conditions prior to sequencing. It is possible to skip the expansion phase and to apply treatments in -ADE medium directly.
-
33.
2 L of reseed medium for a library of 2–4 million harvested colonies corresponds to reseeding ~700–1400 cells per transposition event. It is important that the number of cells per transposition event remains high to avoid losing complexity. This number is estimated as follows:
If the Ade + cells represent 70% of the total number of cells and OD600 0.2 represents 2E+06 cells/mL, then
[2000 mL × 2E+06 cells/mL × 70%] / 2–4E+06 clones = 700 – 1400 cells/transposition event.
This calculation can be refined once the SC-D and SC-D -ADE plates have been counted.
-
34.
Spread 200 μL of a 40,000-fold dilution of the saturated culture on one SC-D and one SC-D -ADE plate using glass beads. Incubate the plates for 3 days at 30 °C. Count the colonies to determine the number of cells per mL.
-
35.
To avoid decreasing the complexity of the library, it is important that each transposition event remains represented by a fair number of cells (see Note 33). We recommend more than 100 cells per event as a safe guideline.
-
36.
The harvest is in vast excess and can be frozen in 30% glycerol. Since viability upon freezing thawing is decreased, we recommend freezing small aliquots to measure cell viability post freezing, in addition to larger aliquots (to re-seed the library accordingly).
-
37.
1 L of culture at OD600 4 produces ~5 g of pellet.
-
38.
The dark red coloring of the nucleic acid pellet is due to the pigment produced by the ade- cells. This pigment is insoluble in ethanol but soluble in water. It does not alter the subsequent steps of library preparation and sequencing.
-
39.
The genomic DNA preparation contains RNAse and therefore cannot be quantified accurately using a spectrophotometer. Moreover, we surmise that the presence of the red pigment extracted from the ade- cells might alter DNA quantification depending on the method. Gel electrophoresis combined with staining with a DNA intercalating agent is insensitive to either of these issues.
-
40.
Any other quantified ladder as well as previously quantified genomic DNA preparations can serve as standards.
-
41.
Homogeneous staining is best achieved by staining the gel post running, especially when using charged intercalating agents like ethidium bromide.
-
42.
To ensure no genomic material is lost, all the following steps, including the assembly of the PCRs, are performed in the same tube as the digestion reaction.
-
43.
Linear acrylamide serves as a carrier during precipitation and is inert in subsequent reactions.
-
44.
We recommend sequencing a maximum of:
-
(a)
2 libraries per MiSeq (Reagent Kit v3, 150 cycles) run.
-
(b)
5 libraries per NextSeq (Mid Output Kit v2.5, 150 cycles) run.
-
(c)
20 libraries per NextSeq (High Output Kit v2.5, 75 cycles) run.
-
(a)
-
45.
It is important that the indexes differ by at least three consecutive nucleotides and are chosen with an appropriate balance of dyes for sequencing. The MiSeq platform uses four channels, while two are used by the NextSeq platform. During the first three cycles of index sequencing, no channels must be omitted. Information on selecting indexes with optimized color balance can be found in the Illumina® documentation (section Color Balance in ref. [23]).
-
46.
The NlaIII and DpnII sub-library products appear as smears centered around 500 bp, running from 250 bp up to 10 kb or beyond. Both the sub-libraries form discrete characteristic bands that correspond to the amplification of untransposed pBK549 (NlaIII, 867 bp; DpnII, 465 bp, Fig. 5c). The intensity of these untransposed bands relative to the rest of the smear diminishes with the length of the regrowth in SC-D -ADE (see Subheading 3.3.2). The negative control lanes should be clear except for possible primer dimers around 70 bp.
-
47.
Making 1 mL PCR and purifying only 100 μL appear to be a waste but the rationale is the following. To avoid losing complexity in the library, it is important that each transposon is represented by a fair number of molecules in the PCR. 2 μg DNA contains ~108 genomes. Therefore, each of the 2–4 million clones of a typical library is present in ~20–40 copies. Thus 2 μg DNA appears like a minimal amount, and 1 mL a minimal PCR volume for this amount of DNA. After amplification, the PCR product consists exclusively of transposon–genome junctions; therefore, much less is necessary downstream.
-
48.
The following instructions are based on quantification of the PCR products using a NanoDrop spectrophotometer. Other methods of quantification might lead to different values. It might be necessary to adjust the concentration of library on the sequencing cartridge.
-
49.
For example: if the concentration of the NlaIII sub-library is 300 ng/μL and that of the DpnII sub-library is 150 ng/μL, then mix 1 and 2 volumes of both the sub-libraries, respectively. The final concentration will be (300 + 2 × 150) / 3 = 200 ng/μL.
-
50.
For example: if the complexity of library A is twice that of library B, double the amount (in ng) of library A over that of B.
-
51.
The PhiX standard DNA is optional but serves as quality control and may allow Illumina® technical support to diagnose problematic runs.
-
52.
These are optimal DNA concentrations for SATAY libraries on Illumina® platforms in our hands.
-
53.
Use a thin gel loading tip to ensure the primer is loaded in the solution contained in well 12. Mix by moving the tip sideways.
-
54.
Set the “read length” on the instrument to 76 bp to actually read 75 bp.
-
55.
Other options are available from third parties (e.g., https://github.com/leilaicruz/LaanLab-SATAY-DataAnalysis).
-
56.
Depending on whether using the MiSeq or the NextSeq platform, the machine will output one or four *fastq.gz files per sample, respectively. This is because the MiSeq v3 flowcell harbors one sequencing lane versus four for the NexSeq v2.5 flowcell. Make sure that the files corresponding to the NlaIII- and DpnII-digested libraries contain “NlaIII” and “DpnII” in their file name, respectively, as this pattern will be used by the script to identify them.
-
57.
If submitting multiple samples, the output for each sample will be concatenated in the *.bed, *.wig, and *pergene.txt files.
References
Cain AK, Barquist L, Goodman AL et al (2020) A decade of advances in transposon-insertion sequencing. Nat Rev Genet 21:526–540. https://doi.org/10.1038/s41576-020-0244-x
van Opijnen T, Bodi KL, Camilli A (2009) Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods 6:767–772. https://doi.org/10.1038/nmeth.1377
Christen B, Abeliuk E, Collier JM et al (2014) The essential genome of a bacterium. Mol Syst Biol 7:528–528. https://doi.org/10.1038/msb.2011.58
Girgis HS, Liu Y, Ryu WS, Tavazoie S (2007) A comprehensive genetic characterization of bacterial motility. PLoS Genet 3:e154. https://doi.org/10.1371/journal.pgen.0030154
Guo Y, Park JM, Cui B et al (2013) Integration profiling of gene function with dense maps of transposon integration. Genetics 195:599–609. https://doi.org/10.1534/genetics.113.152744
Michel AH, Hatakeyama R, Kimmig P et al (2017) Functional mapping of yeast genomes by saturated transposition. eLife 6:e23570. https://doi.org/10.7554/eLife.23570
Michel AH, van Schie S, Mosbach A et al (2019) Exploiting homologous recombination increases SATAY efficiency for loss- and gain-of-function screening. In: bioRxiv. p 866483. https://doi.org/10.1101/866483
Edskes HK, Mukhamedova M, Edskes BK, Wickner RB (2018) Hermes transposon mutagenesis shows [URE3] prion pathology prevented by a ubiquitin-targeting protein: evidence for carbon/nitrogen assimilation cross talk and a second function for Ure2p in Saccharomyces cerevisiae. Genetics 209:789–800. https://doi.org/10.1534/genetics.118.300981
Sanchez MR, Payen C, Cheong F et al (2019) Transposon insertional mutagenesis in Saccharomyces uvarum reveals trans-acting effects influencing species-dependent essential genes. Genome Res 29:396–406. https://doi.org/10.1101/gr.232330.117
Segal ES, Gritsenko V, Levitan A et al (2018) Gene essentiality analyzed by in vivo transposon mutagenesis and machine learning in a stable haploid isolate of Candida albicans. mBio 9:e02048–e02118. https://doi.org/10.1128/mBio.02048-18
Gao J, Wang H, Li Z et al (2018) Candida albicans gains azole resistance by altering sphingolipid composition. Nat Commun 9:4495. https://doi.org/10.1038/s41467-018-06944-1
Gale AN, Sakhawala RM, Levitan A et al (2020) Identification of essential genes and fluconazole susceptibility genes in Candida glabrata by profiling Hermes transposon insertions. G3 (Bethesda) 10:3859–3870. https://doi.org/10.1534/g3.120.401595
Coradetti ST, Pinel D, Geiselman GM et al (2018) Functional genomics of lipid metabolism in the oleaginous yeast Rhodosporidium toruloides. eLife 7:e32110. https://doi.org/10.7554/eLife.32110
Zhu J, Gong R, Zhu Q et al (2018) Genome-wide determination of gene essentiality by transposon insertion sequencing in yeast Pichia pastoris. Sci Rep 8:10223. https://doi.org/10.1038/s41598-018-28217-z
Levitan A, Gale AN, Dallon EK et al (2020) Comparing the utility of in vivo transposon mutagenesis approaches in yeast species to infer gene essentiality. Curr Genet 66:1117–1134. https://doi.org/10.1007/s00294-020-01096-6
Lazarow K, Du M-L, Weimer R, Kunze R (2012) A hyperactive transposase of the maize transposable element activator (ac). Genetics 191:747–756. https://doi.org/10.1534/genetics.112.139642
Serbyn N, Noireterre A, Bagdiul I et al (2020) The aspartic protease Ddi1 contributes to DNA-protein crosslink repair in yeast. Mol Cell 77(5):1066–1079. https://doi.org/10.1016/j.molcel.2019.12.007
Haribowo AG, Hannich JT, Michel AH et al (2019) Cytotoxicity of 1-deoxysphingolipid unraveled by genome-wide genetic screens and lipidomics in Saccharomyces cerevisiae. MBoC 30:2814–2826. https://doi.org/10.1091/mbc.E19-07-0364
Nicastro R, Raucci S, Michel AH et al (2021) Indole-3-acetic acid is a physiological inhibitor of TORC1 in yeast. PLoS Genet 17:e1009414. https://doi.org/10.1371/journal.pgen.1009414
Serbyn N, Bagdiul I, Michel AH et al (2020) SUMO orchestrates multiple alternative DNA-protein crosslink repair pathways. Cell Rep 37(8):110034. SSRN: https://ssrn.com/abstract=3625817. https://doi.org/10.2139/ssrn.3625817
Jorgensen JR, Tei R, Baskin JM et al (2020) ESCRT-III and ER–PM contacts maintain lipid homeostasis. MBoC 31:1302–1313. https://doi.org/10.1091/mbc.E20-01-0061
Schindelin J, Arganda-Carreras I, Frise E et al (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. https://doi.org/10.1038/nmeth.2019
Illumina® (2018) Index Adapters Pooling Guide (1000000041074). https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/experiment-design/index-adapters-pooling-guide-1000000041074-05.pdf
Illumina® (2021) Indexed sequencing on Illumina Systems (15057455). https://support-docs.illumina.com/SHARE/IndexedSeq/indexed-sequencing.pdf
Illumina® (2016) Library pooling guidelines for the NextSeq 500/550 and MiniSeq systems. https://emea.support.illumina.com/bulletins/2016/10/library-pooling-guidelines-for-the-nextseq-and-miniseq-systems.html
Illumina® (2018) MiSeq System Guide (1000000061014). Available at https://support.illumina.com/content/dam/illumina-support/documents/documentation/system_documentation/miseq/miseq-system-guide-for-miseq-reporter-1000000061014-00.pdf
Illumina® (2019) NextSeq 550 System Guide (15069765). https://support.illumina.com/content/dam/illumina-support/documents/documentation/system_documentation/nextseq/nextseq-550-system-guide-15069765-06.pdf
Illumina® (2019) MiSeq System Denature and Dilute Libraries Guide (15039740). Available at https://support.illumina.com/content/dam/illumina-support/documents/documentation/system_documentation/miseq/miseq-denature-dilute-libraries-guide-15039740-10.pdf
Illumina® (2018) NextSeq System Denature and Dilute Libraries Guide (15048776). Available at https://support.illumina.com/content/dam/illumina-support/documents/documentation/system_documentation/nextseq/nextseq-denature-dilute-libraries-guide-15048776-09.pdf
Illumina® (2020) Spiking Custom Primers into the Illumina Sequencing Primers. Available at https://emea.support.illumina.com/bulletins/2016/04/spiking-custom-primers-into-the-illumina-sequencing-primers-.html
Illumina® (2016) MiSeq System Custom Primers Guide (15041638). Available at https://support.illumina.com/content/dam/illumina-support/documents/documentation/system_documentation/miseq/miseq-system-custom-primers-guide-15041638-01.pdf
Illumina® (2020) NextSeq 500 and NextSeq 550 Sequencing Systems Custom Primers Guide (15057456). Available at https://emea.support.illumina.com/content/dam/illumina-support/documents/documentation/system_documentation/nextseq/nextseq-500-550-custom-primers-guide-15057456-03.pdf
Morgan M, Anders S, Lawrence M et al (2009) ShortRead: a bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics 25:2607–2608. https://doi.org/10.1093/bioinformatics/btp450
Li H, Handsaker B, Wysoker A et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Wei Z, Zhang W, Fang H et al (2018) esATAC: an easy-to-use systematic pipeline for ATAC-seq data analysis. Bioinformatics 34:2664–2665. https://doi.org/10.1093/bioinformatics/bty141
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with bowtie 2. Nat Methods 9:357–359. https://doi.org/10.1038/nmeth.1923
Cock PJA, Fields CJ, Goto N et al (2010) The sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res 38:1767–1771. https://doi.org/10.1093/nar/gkp1137
Kent WJ, Sugnet CW, Furey TS et al (2002) The human genome browser at UCSC. Genome Res 12:996–1006. https://doi.org/10.1101/gr.229102
Longtine MS, McKenzie A, Demarini DJ et al (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast (Chichester, England) 14:953–961. https://doi.org/10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U
Acknowledgments
This work was supported by Wellcome Trust Investigator Award grant 214291/Z/18/Z.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
1 Electronic Supplementary Material
Supplementary File 1
(R 11 kb)
Supplementary File 2
(DOCX 86 kb)
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this protocol

Cite this protocol
Michel, A.H., Kornmann, B. (2022). SAturated Transposon Analysis in Yeast (SATAY) for Deep Functional Mapping of Yeast Genomes. In: Devaux, F. (eds) Yeast Functional Genomics. Methods in Molecular Biology, vol 2477. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2257-5_20
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2257-5_20
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2256-8
Online ISBN: 978-1-0716-2257-5
eBook Packages: Springer Protocols