
Abstract
Single-cell RNA sequencing (scRNA-seq) is emerging as an essential technique for studying the physiology of individual cells in populations. Although well-established and optimized for mammalian cells, research of microorganisms has been faced with major technical challenges for using scRNA-seq, because of their rigid cell wall, smaller cell size and overall lower total RNA content per cell. Here, we describe an easy-to-implement adaptation of the protocol for the yeast Saccharomyces cerevisiae using the 10× Genomics platform, originally optimized for mammalian cells. Introducing Zymolyase, a cell wall–digesting enzyme, to one of the initial steps of single-cell droplet formation allows efficient in-droplet lysis of yeast cells, without affecting the droplet emulsion and further sample processing. In addition, we also describe the downstream data analysis, which combines established scRNA-seq analysis protocols with specific adaptations for yeast, and R-scripts for further secondary analysis of the data.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
High-throughput single-cell RNA sequencing (scRNA-seq) has become a well-established technique to study cell-to-cell heterogeneity, rare phenotypes and their effect on how populations function. Various methods have been developed for scRNA-seq, with different single cell isolation and library preparation strategies depending on the specific organisms involved [1,2,3,4,5,6,7,8,9,10]. Cell isolation methods range from micromanipulation and index sorting to more recent microfluidics-based methods. While each method has its strengths and weaknesses, droplet-based methods, such as Drop-seq [11, 20] or the 10× Genomics’ microfluidic platform, are often considered the gold standard because they generally yield higher throughput while reducing cost and workload. On the flipside, these droplet-based methods do require specialized microfluidics equipment and have been shown to generally detect a lower number of genes per cell [12]. Currently, the droplet-based method from 10× Genomics has become one of the most commonly used commercial systems [4, 13,14,15,16]. The platform uses Gel Bead-In-Emulsions (GEMs), trapping single cells in separate emulsion droplets along with reverse-transcription reagents and a uniquely labeled primer hydrogel bead. This allows in-droplet barcoding and reverse transcription into cDNA before pooling and bulk-level sequencing.
The use of this technique to study microbial cells has, however, been limited because of practical hurdles, such as the existence of a rigid cell wall and lower total amount of mRNA per cell. Here, we provide an easy-to-implement adaptation to the existing 10× Genomics’ protocol that allows using this technology for scRNA-seq in Saccharomyces cerevisiae by including an in-droplet cell wall digesting step. In addition, the bioinformatic pipelines for further analysis of the data are also described in detail.
2 Materials
2.1 Growth Media and Sampling Buffers
-
1.
Desired growth medium: autoclaved and filter-sterilized (0.2 μm filter) (see Notes 1 and 2).
-
2.
50% glycerol: 1260 g/L glycerol. To make 500 mL, add 250 mL of dH2O to 250 mL (equivalent to 315 g) of glycerol and filter-sterilize (0.2 μm filter).
2.2 In-Droplet Spheroplasting and Cell Lysis
-
1.
100× Zymolyase stock solution: 100 mg/mL Zymolyase 100 T (Amsbio) dissolved into buffer of a reverse transcriptase (see Notes 3 and 4). Depending on the version of the kit, a different stock solution might be required. Filter-sterilize (low retention syringe filter—0.2 μm pores), keep on ice (alternatively, aliquot the Zymolyase and freeze. Thaw when ready to use and avoid multiple freeze-thaw cycles).
-
2.
PBS: Phosphate buffered saline, filter-sterilized, pH 7.2. Make a 10× PBS solution and dilute to 1× in MilliQ H2O. To make 1 L 10× PBS, add into 800 mL MilliQ water: 81 g NaCl, 2.0 g KCl, 14.4 g Na2HPO4, 2.4 g KH2PO4. Add a stirrer bar, adjust to pH 7.2 (with HCl solution) and volume up to 1 L with MilliQ water. Filter-sterilize (see Note 5).
-
3.
10× Genomics Chromium Single-Cell Reagent Kit protocol, can be found on the 10× Genomics’ support site, under
2.3 Software Installations
-
1.
R Statistical Programming Language
-
2.
R packages.
-
(a)
Seurat. More information on https://satijalab.org/seurat/.
-
(b)
ggplot2.
-
(c)
dplyr.
-
(a)
-
3.
Cell Ranger.
The software can be found on the 10× Genomics’ support site, https://support.10xgenomics.com/.
-
4.
bcl2fastq.
The software can be found on the Illumina support site, https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software/downloads.html.
-
5.
A BASH Shell.
Should be preinstalled for users on Linux/MacOS.
3 Methods
For sample and single-cell library preparation, follow all standard precautions usually taken to perform RNA- and single-cell work, for example, dedicated space, pipettes and reagents, and use of filter tips and nuclease-free reagents.
3.1 Sample Preparation
-
1.
Grow yeast in media of interest, until density of interest is reached (see Notes 1, 2, and 6).
-
2.
Freeze 0.5 mL of cell culture mixed with 0.5 mL of ice-cold 50% glycerol at −80 °C until droplet generation using the 10× Genomics Chromium device (see Notes 7 and 8).
3.2 Single-Cell Library Preparation
To accommodate in-droplet spheroplasting and lysis of the yeast cells, a Zymolyase solution is added to the reverse transcriptase mix before running the Chromium device in the 2.0 or 3.0 version of the 10× kit and protocol. For version 3.1, cells take up to 18 min at room temperature to be encapsulated with beads (rather than 6 to 8.5 min for v2.0–3.0), causing premature lysis of cells and high levels of background mRNA. Adding Zymolyase solution to the GEM beads therefore works better for v3.1. This approach may also work for v2.0 and v3.0, but we have not tested this, as older versions are being phased out by 10× and are not readily available.
-
1.
Thaw cell cultures on ice, measure cell count, and pellet the cells (900 × g, 3′, 4 °C) (see Note 9). Resuspend into ice-cold filter-sterilized PBS to reach the desired cell count, according to the 10× Genomics Chromium Single Cell protocol (see Note 10). In order to keep the proportion of hydrogel beads that contain more than one cell low, it is advisable to aim for 1000 to 2000 recovered cells.
-
2.
From here on, follow the well-documented protocol of the 10× Genomics Chromium device, except for adding the Zymolyase solution to the single-cell master mix (v2.0 and 3.0) or to the beads (v3.1). To add the Zymolyase solution to the single-cell master mix in v2.0 and 3.0 (which is essentially the reverse transcription master mix), replace 1 μL of water in the master mix with Zymolyase stock solution at the appropriate concentration. For v2.0, the total reaction volume will be 100 μL, and the Zymolyase stock solution should be at 100× (100 mg/mL). For v3.0, the total reaction volume is 80 μL, hence the Zymolyase stock solution should only be 80× (80 mg/mL). Then, prepare the microfluidic Chromium Single Cell Chip, add the cell suspension to the single-cell master mix and immediately transfer to the microfluidics chip. Add hydrogel beads and partitioning oil according to the protocol, and commence encapsulating the cells with hydrogel beads using the 10× Genomics Chromium device.
To add the Zymolyase solution to the beads (v3.1, possibly earlier versions), defrost 70 μL aliquots of hydrogel beads, remove 1 μL of beads and add 1 μL of 70× Zymolyase. Then vortex the beads for 30 s, spin down for 5 s and load 50 μL of beads to the appropriate well according to the Chromium v3.1 protocol. Add cell suspension and reverse transcription master mix to the chip according to the protocol, and commence encapsulating the cells with hydrogel beads.
-
3.
After emulsification, perform reverse transcription, cleanup, cDNA amplification and library construction (see Note 11) according to the protocol.
-
4.
After library construction and QC, sequence the prepared libraries (see Note 12).
3.3 Cell Ranger Pipeline
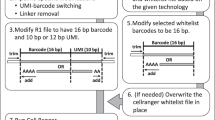
Cell Ranger consists of a set of analysis pipelines developed by 10× Genomics (downloadable from the 10× Genomics website) to process the single-cell RNA-sequencing output (Fig. 1). For some commands, it is useful to set limits on memory and CPU usage. We used a Linux 64 bit machine with 32 GB of RAM and Intel Core i7–4800 CPU to execute the following analysis pipeline.

Overview of the general bioinformatics pipeline to analyze 10× Genomics single-cell RNA-seq data. The first part of the analysis consists of a set of pipelines from the Cell Ranger software (10× Genomics) to process the scRNA-seq raw BCL files into output ready for secondary analysis. Further secondary data analysis mainly makes use of the Seurat R-package for data exploration and visualization
3.3.1 Demultiplex Raw Base Call (BCL) into FASTQ Files
-
1.
Create a sample sheet, as an input comma-separated (csv) file, containing the following three columns: the sequencer lane number (“Lane”), the sample name (“Sample”) and the index position (used in the library construction) from the GEM well of the 10× 96-well plate (“Index”) (see Note 13).
-
2.
Convert raw BCL files from the Illumina sequencer to FASTQ files, separated for each sample and created in the “outs” directory, using “cellranger mkfastq” (see Note 14). You can set the maximal GB the pipeline may request using the “localmem” option.
$ cellranger mkfastq --run=/path/to/folder_with_BCL_files/ \ --id=mkfastqOut \ --csv=/path/to/sample_sheet_mkfastq.csv \ --localmem=3
3.3.2 Mapping to Reference Genome and UMI Counting
-
1.
Build a custom yeast reference genome package using “cellranger mkref.” Following input files are needed: (1) fasta file containing the genomic DNA sequence for each chromosome, (2) gtf file annotating the feature loci on the reference genome (see Note 15).
$ cellranger mkref --genome=S288C \ -- fasta=/path/to/fastafile.fa \ --genes=/path/to/gtffile.gtf
-
2.
Make a separate directory per sample to generate single cell feature counts. Using ‘cellranger count’, map the reads to the reference genome, create bam files and carry out UMI (Unique Molecular Identifier, represents absolute number of observed transcripts) counting for each gene (see Notes 16, 17, 18, and 19).
$ cellranger count --id=unique_run_ID_name \ --fastqs=mkfastqOut/outs/fastq_path \ --sample=sample_name \ --transcriptome=S288C \ --expect-cells=2000 \ --localcores=15 \ --localmem=40 \ --chemistry=SC3Pv2
3.3.3 Aggregate Data from Multiple Samples
-
1.
Create an input csv file which specifies a list of the “cellranger count” output files; containing 1 row per sample, with a sample name (“library_id”) and the path to the output of the “cellranger count” command (“molecule_h5”) (see Note 20).
-
2.
Aggregate the counts from the different samples using “cellranger aggr” (see Note 21) to create multiple output files, among which the filtered feature-barcode matrices MEX file.
$ cellranger aggr --id=unique_run_ID_string \ --csv=aggregation_input.csv --normalization=none
3.4 Secondary Analysis in R
There are multiple tools for secondary data analysis, including Monocle, Seurat, Scanpy, and Cell Ranger R. We use the Seurat R package for secondary analysis (see Note 22), the input of which is the output from the Cell Ranger pipeline (Fig. 1).
3.4.1 Read-In of the Data, QC and Filtering, Normalization
-
1.
Read the output generated in the last step of the Cell Ranger pipeline (more specifically the filtered feature-barcode matrices MEX) into a matrix, using the ‘Read10x’ function of the Seurat package.
data_merge <- Read10X(data.dir='/path/to/filtered_feature_bc_matrix')
This creates a data matrix where each column corresponds to a single cell, denoted by a molecular barcode followed by a numerical index (indicating which of the samples this particular cell comes from), for example, “AAACCTGAGTGATCGG-1”. Each row in this matrix corresponds to a gene based on the exon annotation of the .gtf file used in mkref, for example, “COS7”.
-
2.
Create a Seurat object from this matrix and assign sample names (based on the numerical index) through a metadata column. We assigned sample names based on the time point of sampling. Therefore, we introduced “timepoint” in the metadata. This can be adapted to the specific setup of the experiment. When creating the Seurat object, it can be useful to prefilter out the barcodes (potential cells) with no reads and with data in less than, for example, 5 cells using “min.cells” and “min.features” respectively.
min_cells = 1 min_features = 5 data_merge_obj <- CreateSeuratObject(counts=data_merge, min.cells=min_cells, min.features=min_features) data_merge_obj@meta.data$timepoint <- 'na' data_merge_obj@meta.data[grepl('-1$',colnames(data_merge)),'timepoint'] <- 'sample_name_1' data_merge_obj@meta.data[grepl('-2$',colnames(data_merge)),'timepoint'] <- 'sample_name_2' data_merge_obj@meta.data[grepl('-3$',colnames(data_merge)),'timepoint'] <- 'sample_name_3' … data_merge_obj@meta.data[grepl('-x$',colnames(data_merge)),'timepoint'] <- 'sample_name_x'
-
3.
Plot and inspect major QC metrics (percentage of mitochondrial reads, possible doublets, etc.) through violin plots of distribution values. Seurat automatically calculates QC metrics (in the metadata: nCount_RNA is number of UMIs, nFeature_RNA is features per cell). The proportion of mitochondrial reads could be an important metric for downstream analyses and interpretation, but needs to be annotated and calculated manually. In S. cerevisiae, mitochondrial gene names start with ‘Q’, hence we first define all those genes starting with ‘Q’ as mitochondrial, and add this to the metadata (see Note 23).
data_merge_obj[['percent.mt']] <- PercentageFeatureSet(data_merge_obj, pattern = '^Q') plot1 <- VlnPlot(data_merge_obj, features = c('nFeature_RNA', 'nCount_RNA'), ncol = 2, group.by='timepoint')
-
4.
The thresholds for filtering out cells can be determined by looking at the violin plots. We provide an example based on previously published data [17], in which yeast undergoing a shift from glucose to maltose were studied. High numbers of detected genes (nFeature_RNA) could indicate possible doublets. Therefore, the threshold for maximum total number of genes detected was set to 2000 for sample 1 (Fig. 2) (see Note 24). These thresholds can, within reason, be determined per sample.

QC plots before and after quality control and filtering for possible doublets in 5 scRNA-seq samples from [17]. Violin plots for number of detected genes and number of counts (a) before and (b) after filtering
data_merge_obj <- data_merge_obj[,(grepl('-1$', colnames(data_merge_obj)) & data_merge_obj$nFeature_RNA < 2000) | (grepl('-2$', colnames(data_merge_obj)) & data_merge_obj$nFeature_RNA < 2000) | (grepl('-4$', colnames(data_merge_obj)) & data_merge_obj$nFeature_RNA < 1500) | (grepl('-3$', colnames(data_merge_obj)) & data_merge_obj$nFeature_RNA < 1500) | (grepl('-5$', colnames(data_merge_obj)) & data_merge_obj$nFeature_RNA < 2000)]
-
5.
Normalize the data using the default global-scaling “LogNormalize” option, to correct for differences in capture efficiency between cells and differences in read depth between samples (see Note 25).
data_merge_obj <- NormalizeData(data_merge_obj)
-
6.
Detect highly variable genes across the single cells using “FindVariableFeatures,” using the default selection method option “vst.” The selection method defines how to choose the top variable features. You can decide on “nfeatures” depending on how many variable genes you want to use to cluster the cells. The downstream analysis will focus on these genes which will help highlight biological signals in the data.
data_merge_obj <- FindVariableFeatures(data_merge_obj, selection.method = 'vst', nfeatures = 300)
-
7.
To improve downstream dimensionality reduction and clustering, we need to scale (linearly transform) the data, in order to prevent highly expressed genes from dominating the next steps of the analysis (see Note 26).
data_merge_obj <- ScaleData(data_merge_obj, verbose = FALSE)
3.4.2 Dimensional Reduction and Visual Exploration of Data
-
1.
Run linear dimensional reduction using “RunPCA” on the scaled data (see Note 27).
data_merge_obj <- RunPCA(data_merge_obj, npcs = 20, verbose = FALSE)
-
2.
Run UMAP on the dimensionally reduced data, and visualize using “DimPlot” (Fig. 3). You can also visualize the data with cells colored by a quantitative feature (such as expression levels of specific genes/growth markers) using “FeaturePlot” (Fig. 4).

Visualization of dimensionally reduced data, colored by sample using “DimPlot.” UMAP dimensions were created using 20 principal components

Visualization of dimensionally reduced data, with cells colored by expression level of the specific marker genes MAL11, MAL12, HXK1, and MAL33, using “FeaturePlot”
data_merge_obj <- RunUMAP(data_merge_obj, reduction = 'pca', dims = 1:20 , n.epochs = 1000) DimPlot(data_merge_obj, reduction = 'umap', group.by = 'timepoint', pt.size = 0.1) FeaturePlot(data_merge_obj, c('GENE_NAME1','GENE_NAME2','GENE_NAMEX'), pt.size = 0.5, cols = c('gray50','red'), reduction= 'umap')
3.4.3 Clustering and Differential Gene Expression
-
1.
Cluster the cells using a graph-based approach. This method first calculates the k-nearest neighbors, then constructs the shared nearest neighbors’ graph. The resolution parameter (see Note 28) determines the number of clusters (Fig. 5).

Clustering of cells based on k-nearest neighbors. The top graph (a) shows clustering using a resolution of 0.1, resulting in a lower number of clusters retrieved compared to the bottom graph (b) using a resolution of 0.5
data_merge_obj <- FindNeighbors(data_merge_obj, dims = 1:20) data_merge_obj <- FindClusters(data_merge_obj, resolution = 0.1) DimPlot(data_merge_obj, pt.size = 0.1)
-
2.
Find specific biomarkers that define clusters via differential expression. The parameter “logfc.threshold” is used to limit the testing to genes which show a specific fold-difference between the groups of cells. You can test for markers of a specific cluster (specified in ident.1) or you can find markers distinguishing clusters from each other (e.g., ident.1 = 2, ident.2 = 3 in this example will find markers differing between cluster 2 and cluster 3). You can further limit the list of markers by extracting only genes with specific average log-fold changes or adjusted p-values you define. Export as a csv-file.
diff_exp_res <- FindMarkers(data_merge_obj, ident.1 = 2, ident.2 = 3, logfc.threshold = 0.01) diff_exp_res <- diff_exp_res[abs(diff_exp_res$avg_logFC)>0.25 & diff_exp_res$p_val_adj<0.05, ] write.csv(diff_exp_res, 'diff_exp_res.csv', row.names = F)
-
3.
Upload this csv-file to a Gene Ontology webtool, such as Gorilla (http://cbl-gorilla.cs.technion.ac.il/) to identify and visualize enriched GO terms in your gene list.
4 Notes
-
1.
We never autoclave sugar stock solutions to avoid Maillard reactions and to obtain accurate sugar concentrations.
-
2.
To avoid possible clogging of the 10× Genomics device, it is recommended to filter-sterilize all growth media and buffers.
-
3.
Zymolyase cannot be dissolved in water at our target concentration. We specifically used the Quantiscript RT Buffer from the QuantiTect reverse transcription kit, and diluted the buffer from a 5× to a 1× concentration before use. Alternatively, dissolving in PBS should also work.
-
4.
After dissolving the Zymolyase, the solution should be dark but completely homogeneous with no visible solid particles. If filter-sterilizing does not work well and clogs the filter, mixing the concentrated Zymolyase well before adding to the master mix without filter-sterilizing seems to work as well.
-
5.
Alternatively, you can use PBS + 400 μg/mL BSA as recommended for sample preparation in the 10× Cell Preparation Guide. Both seem to work equally well.
-
6.
When setting up the experiment, make sure to calculate in advance the minimal cell concentration and volume you would need to sample to recover 1000–2000 cells (according to the Cell Suspension Volume Calculator Table in the 10× protocol).
-
7.
Freezing the cells using liquid nitrogen is possibly even better, but from our experience −80 °C when working with ice-cold buffers should be sufficient.
-
8.
Make sure to mix the sample well and extract an aliquot to obtain an accurate cell count before freezing.
-
9.
Limit time between thawing and the Chromium run as much as possible. If needed, wash once in PBS (+ BSA) buffer prior to counting and diluting. If you encounter clumps of cells during cell counting, dependent on your yeast strains and experimental conditions, sonicating for 40–60 s in a water bath sonicator can help resolve this. If time needed for thawing, resuspension and counting becomes quite long, it might be a good idea to use RNAlater (Qiagen) [4].
-
10.
If you have multiple samples you might want to bring all samples to a similar cell count so you can combine all samples with the reverse transcription master mix at the same time.
-
11.
Make sure you write down the 10× Sample Index name while constructing the library, this is important information for the downstream analysis, especially if you are running multiple samples.
-
12.
Take into consideration that all lanes on the sequencer should be 10× samples, since this sequencing run has specific parameters especially for these types of samples. In terms of read depth, we found that we got about 90% sequence saturation with 20,000 reads/cell.
-
13.
If one 10× library has been sequenced on different flow cells/sequencers, the “cellranger mkfastq” command will have to be run separately for each sequencing run using separate sample sheets.
-
14.
The “cellranger mkfastq” command is in essence a wrapper around Illumina’s “bcl2fastq” software. Therefore, before running this command, you should make sure you have installed the ‘bcl2fastq’ software (downloadable from the Illumina website).
-
15.
Both the fasta file and the gtf file can be downloaded from UCSC genome browser (fasta file from the ‘Sequence and Annotation Downloads’ section, the gtf file from the ‘Table Browser’ section). Note that Cell Ranger only uses the exon feature in the gtf for its calculations. In S. cerevisiae, the exon annotation does not contain the 3’UTR which results in reads, that fall entirely within the 3’UTR, not being assigned. Losing these reads can be avoided by including 3’UTR annotations in the exon feature of the .gtf file, similar to the approach used in [4].
-
16.
You can run the “cellranger count” command in parallel for different samples in separate terminal windows.
-
17.
Set the expected number of cells option to the expected value based on the calculation table in the protocol of the 10× Genomics Chromium device. If you need to combine multiple sequencing runs of the same GEM well, you need to input multiple fastq files, separated by a comma here.
-
18.
You can put limits on memory and cpu usage of this command. This is the most computationally intensive step of the analysis. On a laptop with a Core i7–4800 CPU, and 32GB of RAM, these commands of “cellranger count” took about 30 h to run. Of course, this will be dependent on your read depth, and possibility of using parallel processing.
-
19.
The “chemistry” option allows you to specify which assay configuration and reagent kit you used. In principle, it should be detected automatically.
-
20.
This path would be something like: unique_run_ID_name/outs/molecule_info.h5.
-
21.
In principle, you can normalize across samples to equalize the average read depth per cell between groups before merging. We did not use aggregation normalization between samples since we knew that the RNA content of cells drops during the time course of our specific sampling scheme, and were therefore able to verify that the total UMI count generally decreased during the time course as expected. We later normalize each cell by the total UMI count prior to clustering and calculating differential expression.
-
22.
ggplot2 and dplyr are two additional R packages that we use here. Most of the analysis using Seurat is based on the, well-explained, Seurat Guided Clustering Tutorial (https://satijalab.org/seurat/articles/pbmc3k_tutorial.html).
-
23.
High levels of mitochondrial reads are generally flagged in mammalian cells as a sign of apoptosis, and therefore a threshold for proportion of mitochondrial reads is usually set as an extra filter to remove dead cells [18]. However, in yeast, large variation in mitochondrial reads more often indicates biological variability, which is why we here do not filter but rather include the proportion of mitochondrial reads for further downstream analysis.
-
24.
Another, perhaps more intuitive, option would be filtering on the total number of counts instead. Having doublets would result in more mRNA in a droplet, higher counts and thus detection of more genes, making nCount_RNA and nFeature_RNA highly correlated. Here again, using a per sample filter, based on the distribution of nCount, would be best.
-
25.
If the total mRNA per sample changes in your experiment, then this normalization step will cause relative differences between samples for specific genes that may not have absolute changes. If needed, you could further remove technical noise with imputation [19].
-
26.
Here, the default method has been used. There is an option “vars.to.regress” to regress out specific features to remove unwanted sources of variation (such as mitochondrial gene expression, number of detected molecules), which might be valuable to use.
-
27.
We decided on the number of PCs (here 20) by visual inspection of the UMAP clustering generated from a few different options. We chose the minimal number that maintains the expected UMAP projection shape. The section on determining dimensionality of the dataset from the Seurat Guided Clustering Tutorial provides a discussion on other approaches including the use of Jack Straw plots (JackStrawPlot), Elbow plots (ElbowPlot), and analyzing dimensionality heatmaps (DimHeatmap).
-
28.
Based on results from previous experiments, we tweaked this parameter to have the 4 expected clusters. According to the Seurat Guided Clustering Tutorial, a range between 0.4 and 1.2 usually gives good clustering.
References
Gasch AP, Yu FB, Hose J et al (2017) Single-cell RNA sequencing reveals intrinsic and extrinsic regulatory heterogeneity in yeast responding to stress. PLoS Biol 15:e2004050. https://doi.org/10.1371/journal.pbio.2004050
Nadal-Ribelles M, Islam S, Wei W et al (2019) Sensitive high-throughput single-cell RNA-seq reveals within-clonal transcript correlations in yeast populations. Nat Microbiol 4:683–692. https://doi.org/10.1038/s41564-018-0346-9
Saint M, Bertaux F, Tang W et al (2019) Single-cell imaging and RNA sequencing reveal patterns of gene expression heterogeneity during fission yeast growth and adaptation. Nat Microbiol 4:480–491. https://doi.org/10.1038/s41564-018-0330-4
Jackson CA, Castro DM, Saldi G-A et al (2020) Gene regulatory network reconstruction using single-cell RNA sequencing of barcoded genotypes in diverse environments. Elife 9:e51254. https://doi.org/10.7554/eLife.51254
Islam S, Zeisel A, Joost S et al (2014) Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods 11:163–166. https://doi.org/10.1038/nmeth.2772
Hwang B, Lee JH, Bang D (2018) Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp Mol Med 50:96. https://doi.org/10.1038/s12276-018-0071-8
Yin Y, Jiang Y, Lam K-WG et al (2019) High-throughput single-cell sequencing with linear amplification. Mol Cell 76:676–690.e10. https://doi.org/10.1016/j.molcel.2019.08.002
Cao J, Packer JS, Ramani V et al (2017) Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357:661–667. https://doi.org/10.1126/science.aam8940
Imdahl F, Vafadarnejad E, Homberger C et al (2020) Single-cell RNA-sequencing reports growth-condition-specific global transcriptomes of individual bacteria. Nat Microbiol 5:1202–1206. https://doi.org/10.1038/s41564-020-0774-1
Bettauer V, Massahi S, Khurdia S et al (2020) Candida albicans exhibits distinct cytoprotective responses to anti-fungal drugs that facilitate the evolution of drug resistance. bioRxiv. https://doi.org/10.1101/2020.01.21.914549
Macosko EZ, Basu A, Satija R et al (2015) Highly parallel genome-wide expression profiling of individual cells using Nanoliter droplets. Cell 161:1202–1214. https://doi.org/10.1016/j.cell.2015.05.002
Skinnider MA, Squair JW, Foster LJ (2019) Evaluating measures of association for single-cell transcriptomics. Nat Methods 16:381–386. https://doi.org/10.1038/s41592-019-0372-4
Adamson B, Norman TM, Jost M et al (2016) A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167:1867–1882.e21. https://doi.org/10.1016/j.cell.2016.11.048
Kaufmann E, Sanz J, Dunn JL et al (2018) BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell 172:176–190.e19. https://doi.org/10.1016/j.cell.2017.12.031
Yan KS, Janda CY, Chang J et al (2017) Non-equivalence of Wnt and R-spondin ligands during Lgr5+ intestinal stem-cell self-renewal. Nature 545:238–242. https://doi.org/10.1038/nature22313
Zheng GXY, Terry JM, Belgrader P et al (2017) Massively parallel digital transcriptional profiling of single cells. Nat Commun 8:14049. https://doi.org/10.1038/ncomms14049
Jariani A, Vermeersch L, Cerulus B et al (2020) A new protocol for single-cell RNA-seq reveals stochastic gene expression during lag phase in budding yeast. Elife 9:e55320. https://doi.org/10.7554/eLife.55320
Osorio D, Cai JJ (2020) Systematic determination of the mitochondrial proportion in human and mice tissues for single-cell RNA-sequencing data quality control. Bioinformatics 37(7):963–967. https://doi.org/10.1093/bioinformatics/btaa751
Tang W, Bertaux F, Thomas P et al (2019) bayNorm: Bayesian gene expression recovery, imputation and normalization for single-cell RNA-sequencing data. Bioinformatics 36(4):1174–1181. https://doi.org/10.1093/bioinformatics/btz726
Urbonaite G, Lee JTH, Liu P et al (2021) A yeast-optimized single-cell transcriptomics platform elucidates how mycophenolic acid and guanine alter global mRNA levels. Communications Biology 4(1) https://doi.org/10.1038/s42003-021-02320-w
Acknowledgments
LV, AJ, and KJV would like to thank Thomas Van Brussel, Bernard Thienpont, and Diether Lambrechts at the VIB Center for Cancer Biology for their support in setting up the 10× sequencing protocol. LV, AJ, and KJV experiments were partly funded by the European Research Council (ERC CoG682009), FWO, KU Leuven, and VIB. LV acknowledges FWO SB Grant SB/1S07117N. JH acknowledges FWO SB Grant SB/1S17117N. BH would like to thank Markus Ralser at the Francis Crick Institute for helpful advice and for funding support from the Francis Crick Institute which receives its core funding from Cancer Research UK (FC001134), the UK Medical Research Council (FC001134), and the Wellcome Trust (FC001134), and specific funding from Wellcome Trust (IA 200829/Z/16/Z), to M. Ralser. BH would like to thank Ivan Andrew, Laurence Game, and Samuel Marguerat at the MRC London Institute of Medical Sciences for their support in setting up his 10× sequencing protocol. BH experiments were partly funded by the UK Medical Research Council (MC-A658-5TY20).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this protocol

Cite this protocol
Vermeersch, L., Jariani, A., Helsen, J., Heineike, B.M., Verstrepen, K.J. (2022). Single-Cell RNA Sequencing in Yeast Using the 10× Genomics Chromium Device. In: Devaux, F. (eds) Yeast Functional Genomics. Methods in Molecular Biology, vol 2477. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2257-5_1
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2257-5_1
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2256-8
Online ISBN: 978-1-0716-2257-5
eBook Packages: Springer Protocols