
Abstract
The use of primary neuronal cultures generated from Drosophila tissue provides a powerful model for studies of transport mechanisms. Cultured fly neurons provide similarly detailed subcellular resolution and applicability of pharmacology or fluorescent dyes as mammalian primary neurons. As an experimental advantage for the mechanistic dissection of transport, fly primary neurons can be combined with the fast and highly efficient combinatorial genetics of Drosophila, and genetic tools for the manipulation of virtually every fly gene are readily available. This strategy can be performed in parallel to in vivo transport studies to address relevance of any findings. Here we will describe the generation of primary neuronal cultures from Drosophila embryos and larvae, the use of external fluorescent dyes and genetic tools to label cargo, and the key strategies for live imaging and subsequent analysis.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
- Live imaging
- Axonal transport
- Drosophila primary neurons
- Motors
- Organelles
- Mitochondria
- Kinesin
- Dynein
- Lysotracker
- Mitotracker
1 Introduction
Neurons extend axons to communicate and innervate all parts of the body. Axons are the longest cellular protrusions, with motor neurons reaching up to a meter in humans. Axons predominantly form output synapses which are in their majority located at the distant end of the axon . The extreme spatial separation between synaptic terminals and the neuronal cell body or soma poses a logistical problem in that soma-derived proteins, RNAs, lipids, and organelles crucial for the sustainability and function of axons and their synapses need to be transported over large distances [1,2,3].
Long-distance axonal transport involves the coordinated action of motor proteins binding directly or indirectly (through adaptors) to the transported cargoes while walking along microtubules [2, 4,5,6]. For this, axonal microtubules are arranged into loose parallel bundles that run all along the axon shaft [7, 8]. Motor proteins involved in long-distance axonal transport use ATP as energy source to step along microtubules; they include dynein/dynactin responsible for retrograde movements and certain members of the kinesin family for anterograde transport (kinesin-1, −2, −3, −4 [3, 9, 10]). Apart from antero- and retrograde cargo transport, this system must also ensure the controlled release, hence delivery, of cargoes at the appropriate target sites [11, 12].
It is not surprising that many neurodegenerative diseases are linked to genetic defects in this transport machinery, thus highlighting the importance of this process for neuronal function and viability [5, 13]. Decrease in axonal transport is also considered as a widespread aging hallmark [14]. It is therefore pivotal to understand the transport machinery, its regulatory mechanisms, and the consequence of its dysfunction.
Despite the recent advances in understanding the mechanism of fast axonal transport (e.g., [15,16,17,18]), there are numerous unresolved issues in relation to the regulation of speed, directionality, selectivity of transport, targeted unloading of cargoes, and adaptation of transport to the specific cellular demands. Such complex regulatory processes are likely to involve the specific compositions of motor protein complex (e.g., type of motor protein and cargo adaptors [19, 20]), the interaction between different complexes and with other classes of microtubule binding proteins [2, 21, 22], posttranslational modifications (PTMs) of these complexes or of their microtubule tracks mediated by the orchestrating action of signaling pathways [23].
A powerful model to study transport mechanisms in its complexity is the fruit fly Drosophila melanogaster , which has been used for this purpose for more than two decades (e.g., [24,25,26,27,28,29]). The fundamental function and structure of Drosophila and vertebrate neurons is comparable [30,31,32,33,34], including the organization and regulation of microtubules which follow similar organizational principles as in vertebrates, in spite of the fact that neurofilaments are absent [8]. Furthermore, most motor proteins have counterparts in Drosophila, but there is less redundancy: while around 40 genes encode members of the kinesin super-family in vertebrates [35], only 18 kinesin coding genes cover a similar range of motor protein classes in Drosophila [3]. For example, kinesin light chain comprises three mouse members, but only one in Drosophila [3]. In addition, an arsenal of genetic tools for the manipulation of virtually every fly gene is readily available or can be efficiently generated through genomic engineering, and they can be easily targeted to specific neurons or combined to generate loss or gain of function of different genes or tools in the same neurons or organisms [36,37,38].
In its majority, research in Drosophila neurons makes use of in vivo studies in the whole organism. However, also the use of cultured Drosophila primary neurons, first used half a century ago (e.g., [39, 40]) has proven a valuable complementary strategy. As is the case for mammalian primary neurons, also fly neurons give access to subcellular detail and robust and sensitive readouts and are accessible to external application of compounds such as fluorescent dyes and drugs [41,42,43,44,45]. Importantly, many of the observed subcellular features and dynamics are very similar to those of vertebrate neurons [46]. However, as an important advantage, primary fly neurons are accessible to the powerful combinatorial genetics mentioned above: as long as genetically manipulated embryos can generate neurons, these can be analyzed in culture. Accordingly, primary Drosophila neuronal cultures have been used to successfully study various different aspect of neuronal physiology [47, 48], including cell division [49], axon guidance [50,51,52], cytoskeletal dynamics [41, 42, 53,54,55,56,57,58,59], neuronal activity [60, 61], protein aggregation in neurodegenerative disease models [62], receptor signaling and trafficking [44, 52] and for the study of axonal transport [43, 63, 64]. In this method paper, we will explain primary neuronal cultures from Drosophila as cellular system where axonal transport can be studied efficiently.
2 Materials
-
1.
Drosophila specimens: primary neuronal cultures can be performed from Drosophila embryos [65], late instar L3 larva [53], and late pupa [66] (see Note 1).
-
2.
Sodium hypochlorite solution (or household bleach), diluted 1:1 with water to 50%.
-
3.
Ultrafine sieves and brushes for the collection and cleaning of embryos.
-
4.
Agar plates: dissolve 2–4% agar in water, boil, pour into empty plastic petri dishes (50 mm diameter), let cool down and harden.
-
5.
70% Ethanol in ultrapure (Milli-Q) autoclaved water.
-
6.
Supplemented Schneider’s medium pH 6.8–7.0: Combine Schneider’s Drosophila medium with 20% non-heat-inactivated FBS. Filter-sterilize and protect from light with aluminum foil. To inactivate the serum’s complement system, incubate for 3 days at 26 °C (alternatively use heat-inactivated FBS and skip the 3 days incubation at 26 °C). Add 2 μg/mL insulin (2 mg/mL insulin stock can be aliquoted and kept at −20 °C). Use a small aliquot of medium to test the pH and adjust to 6.8–7.0 with 1 N HCl or NaOH if needed. Aliquots of 1–2 mL medium can be stored at −80 °C for 6 months. After thawing aliquots for use, filter-sterile with a 0.22-μm syringe filter and freshly add 10 U Penicillin/Streptomycin per mL medium.
-
7.
HBSS buffer: 30 mL 10 × Hanks’ Balanced Salt Solutions (GIBCO, no calcium or magnesium), 3 mL Penicillin/Streptomycin (GIBCO, 10,000 U/mL), 167 mL distilled water, 0.01 g phenylthiourea; aid to dissolve by incubating at 37 °C for 30 min, sterile filter with 0.22-μm filter. This buffer can be stored in aliquots at 4 °C for 6 months.
-
8.
Dispersion or dissociation medium: dissolve 0.005 mg Dispase II and 0.001 mg Collagenase type V in 2 mL HBSS medium. Incubate at 37 °C for 30 min until dissolved. Filter through a 0.22-μm filter and store at 4 °C. It can be used for up to a week.
-
9.
Pestles: small autoclavable plastic pestles for 1.5-mL microcentrifuge tubes with a tight fit.
-
10.
Custom-made culture chambers or 35-mm glass bottom dishes (MatTek) to grow primary neurons: culture chambers can be assembled from one lead-free glass microscope slide (Menzel Gläser) with a 15-mm hole (custom-made at a workshop) and one intact slide glued together with aquarium silicone. Dry (up to 5 days) and scrape off any excess silicone. Clean with 70% ethanol followed by 100% acetone. The chambers can be reused after cleaning with acetone and autoclaving. Alternatively, use 35-mm glass bottom dishes (for example, from MatTek) with optical quality glass for microscopy. While incubation chambers and glass bottom plates can be used with inverted microscopes, incubation chambers are ideal for upright microscopes.
-
11.
4× concanavalin A stock solution: in a laminar flow hood, dissolve concanavalin A (conA) in ultrapure autoclaved water to a concentration of 20 μg/mL. This stock solution can be frozen in aliquots at −20 °C.
-
12.
Treated coverslips: Drosophila primary neurons can grow on acetone-treated or conA-coated glass coverslip (Menzel Gläser, 24 × 24 mm, VWR international MENZBB024024A123). For acetone treatment, dip coverslips in 100% acetone, let dry and autoclave. For conA treatment, incubate coverslips with 150 μL conA at a concentration of 5 μg/mL at 37 °C for 1.5 h. Remove conA solution, wash with 150 μL sterile water, remove and dry at ∼50 °C overnight. Note that the morphology of neurons changes when growing on conA [65]. When using glass bottom dishes for live imaging, treat the glass coverslip that is attached to the bottom of the dish.
-
13.
Vaseline for sealing chambers/dishes with coverslips.
-
14.
100 nM LysoTracker DND-99/50 nM MitoTracker Green FM/CMXRos in supplemented Schneider’s medium.
-
15.
Fixatives: We carry out standard fixations in 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.2). To prepare 100 mL fixative, dissolve 4 g paraformaldehyde in 36 mL 0.2 M NaH2PO4 (warm up slightly and stir), add 14 mL 0.2 M NaH2PO4, and fill up with water. Adjust pH if necessary.
-
16.
PBT: 0.3% Triton X-100 (v/v) in 1× PBS.
3 Methods
3.1 Drosophila Embryonic Primary Neuronal Cultures (Fig. 1a–d)
Drosophila primary neuronal cultures can be prepared from several developmental stages (embryo, larva, and pupa) [65]. In this chapter, we will discuss our protocol for primary neuronal cultures derived from whole embryos or third instar larval CNSs, in which our transport studies have been performed (Figs. 1 and 2).
-
1.
Collect embryos (Fig. 1A.1) in vials or cages for ∼15 h at 20–25 °C (the temperature can be adapted to increase the yield of stages 11–12 embryos [67], see Notes 1 and 2).
-
2.
Dechorionate embryos (Fig. 1A.2) with 50% bleach for 90 s. Wash thoroughly with water in a sieve. Note that extended incubations as well as the quality of the bleach can affect the viability of the cultures. Carefully collect embryos with a thin paint brush from the sieve and transfer them to an agar plate.
-
3.
Select stages 11–12 embryos (Fig. 1A.3) by autofluorescence pattern (for embryo stages see [67]; see Note 3 a–c). If mutant Drosophila stocks are used (with adequate GFP-balancers), mutant embryos can now be selected based in the absence of GFP, using a fluorescent dissection microscope. For cultures on acetone-treated glass coverslip select 10 embryos per culture (the number of embryos can be altered to achieve different densities of neurons in culture). For cultures on conA-coated glass coverslip select five embryos per culture.
-
4.
Transfer the embryos (Fig. 1b.4, 5) into a 1.5-mL tube with a 5 μL drop of supplemented Schneider’s medium using clean forceps (with 70% ethanol). From this point on the next steps will be performed in a laminar flow hood wearing gloves and with autoclaved materials whenever possible.
-
5.
Wash the embryos (Fig. 1b.4, 5) with 70% ethanol solution for 30 s by gently inverting the tube several times.
-
6.
Remove ethanol and wash embryos in 500 μL supplemented Schneider’s medium (Fig. 1b.6). Take care not to accidentally pipette the embryos. Pipetting against a black background can help to visualize the embryos.
-
7.
Replace Schneider’s medium with 100 μL HBSS-based dispersion medium at RT (Fig. 1b.7–9; see Note 4).
-
8.
Grind embryos in the tube with an autoclaved pestle to break down the embryos (Fig. 1b.7–7), use one pestle per genotype (see Note 5). At this step, there is the option to add a preculture step to reduce maternal contribution of proteins or allow for extra time for knockdowns to work (see Subheading 3.2).
-
9.
Incubate the tubes with the cell dispersion for 4–7 min at 37 °C (Fig. 1b.7–9). OPTION: This step can be omitted leading to neurons with more complex morphologies and less isolated single neurons.
-
10.
Stop the dispersion reaction by removing from 37 °C and adding 200 μL of supplemented Schneider’s medium (Fig. 1b.10).
-
11.
Sediment cells at 600–750 × g for 4 min (Fig. 1b.11, 12). Remember the orientation of the tube during centrifugation since it may be difficult to see the pellet.
-
12.
Aspirate and discard the medium (Fig. 1b.11, 12). Make sure this step is performed promptly to avoid cell pellets resuspending in the old medium. Add 31 μL of fresh supplemented Schneider’s medium per culture/chamber. Adjust volume depending on the final number of cultures (we usually prepare three cultures per condition at a time). Gently resuspend cells in Schneider’s medium without generating bubbles.
-
13.
Distribute 30 μL cell suspension either to glass bottom dishes (treated with acetone or coated with conA) and cover with a regular coverslip using Vaseline as sealant or to custom-made culture chambers and cover with a lead-free coverslips (treated with acetone or coated with conA) and seal with Vaseline (Fig. 1c.13). Note that the morphology of neurons changes when growing on conA [65].
-
14.
Let the cells settle onto the acetone or conA treated surfaces for 2 h at 26 °C (Fig. 1c.14).
-
15.
Flip over the incubation chambers/glass bottom dishes and grow primary neurons on top of the chamber as hanging drop cultures (Fig. 1c.15) until the desired time in vitro has been reached (see Note 6).

Workflow to generate Drosophila primary neuronal cultures for short- and long-term imaging. The numbers in the figure correspond to the protocol steps in Subheading 3.3. (a) Diagram showing the steps for embryo collection, removal or the chorion, and selection of embryos for primary neuronal cultures. (b) Diagram showing the main steps to generate a suspension of primary cells from Drosophila embryos. Note that the protocol can be modified to use late stage 3 larval brains as well (see Subheading 3.3 for more detail). (c) Diagram showing the hanging drop culturing technique. Drosophila primary cells are allowed to attach to coverslips before inverting the sealed culture chambers (upper row)/glass bottom dishes (lower row). Cells are then grown the desired time in vitro (hours to days). Cultures can optionally be incubated with drugs and/or dye-based organelle /vesicle markers before medium is exchanged for imaging. (d) For long-term imaging, cells can be allowed to either (a) directly attached to coated or uncoated glass bottom dishes or (b) grown as hanging drop cultures in glass bottom dishes, then unsealed and Vaseline removed (see c). Cells can then be treated with drugs and/or dye-based organelle /vesicle labels. The dish is then filled up with Schneider’s medium and sealed with a coverslip to prevent evaporation of the medium during long-term imaging

Transport of vesicles and organelles in Drosophila primary neuronal cultures. Single frame from time-lapse movies from Drosophila primary neuronal cultures showing different labeled cargo (cell bodies are at the bottom of the image), and kymograph plot showing the trajectory of cargoes. (a) Larval primary cultures treated with the dye-based fluorescent label LysoTracker DND-99. (b–d) Embryonic primary neuronal cultures (b) treated with the dye-based fluorescent label MitoTracker Green FM, (c) expressing Synaptotagmin-GFP using the tubulin-Gal4 driver, and (d) expressing mito-mCherry using the elav-Gal4 driver. Magenta dashed lines mark the axon . Panels on the right in (a–d) show corresponding kymographs. Lines with positive slopes in kymographs indicate anterograde transport, lines with negative slopes retrograde transport, and horizontal lines indicate stationary objects. Scale bar in overview images is 5 μm in length
3.2 Pre-culture Step for Drosophila Embryonic Primary Neuronal Cultures
Cells can be cultured in a 1.5-mL tube for several days before being dispersed again and grown on coverslips in culture chambers. This step is useful in order to deplete healthy gene product deposited by heterozygous mothers in the egg cells (which otherwise carry mutations in a specific gene). Maternally contributed gene products can persist for several days and mask the homozygous mutant phenotype of embryos and larvae [68]. This step is also effective in allowing extra time for conditions of knockdowns.
-
1.
Follow steps 1–8 of Subheading 3.1. For this protocol, increase the number of embryos to 15 per slide and use ConA-coated coverslip.
-
2.
After grinding the embryos, immediately add 200 μL of supplemented Schneider’s medium.
-
3.
Sediment cells at 600–750 × g for 4 min.
-
4.
Aspirate the medium. Add 100 μL of fresh supplemented Schneider’s medium, gently resuspend cells, and incubate in the tube for the desired time at 26 °C (1–7 days).
-
5.
After the incubation, sediment cells at 600–750 × g for 4 min.
-
6.
Aspirate Schneider’s medium and add 100 μL HBSS-based dispersion medium.
-
7.
Gently resuspend the cell pellet and incubate 4–7 min at 37 °C.
-
8.
Continue from step 10 of Subheading 3.1.
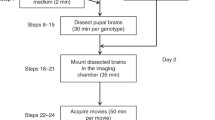
3.3 Third Instar Larval Brain Cultures
-
1.
Collect 2–3 late stage 3 larvae per culture.
-
2.
Wash larvae 3× in in 1.5 mL 70% ethanol.
-
3.
Wash larvae 3× in 1.5 mL sterile PBS.
-
4.
Transfer larvae into a sterile dissection dish with supplemented Schneider’s medium.
-
5.
Dissect out brains (use sterile forceps) and transfer to a 1.5-mL tube with supplemented Schneider’s medium.
-
6.
All following steps should be done in a laminar flow hood.
-
7.
Wash brains 3× in 500 μL supplemented Schneider’s medium.
-
8.
Remove medium and replace with 100 μL dispersion medium.
-
9.
Continue from step 8 of Subheading 3.1.
3.4 Labeling of Cargo
The use of primary neuronal cultures to study transport allows the labeling of cargo by (a) genetically encoded tools, mostly tagged proteins or localization sequences fused with GFP or other fluorophores and (b) fluorescent chemical compounds applied to the culture media such as LysoTracker, fluoro-dextranes, and mito-tracker. Here we describe the methods and tools we have been experimenting with. However, there is an arsenal of further tools that can be used to label specific cargo , such as dense-core vesicle labeled with atrial natriuretic factor-GFP (ANF-GFP), APP-positive vesicles labeled with App-YFP, Golgi vesicles labeled with GalT-RFP, endosomes labeled with Rab4-RFP, and ribonucleoprotein granules labeled with GFP-Imp to mention some [26, 29, 69].
3.4.1 LysoTracker Staining (See Note 7)
LysoTracker is a fluorescent probe which, due to its hydrophobic nature, diffuses into cells, and preferentially accumulates in vesicles with an acidic content. It is commonly used in vertebrate models to label late endosomes/lysosomes and to study their trafficking within cells including neurons [70,71,72,73,74]. LysoTracker has been used previously to label lysosomes/late endosomes in Drosophila primary neuronal cultures [75], Drosophila tissue, and cells lines [76]. Here we will describe its use in Drosophila primary neuronal culture (Fig. 2a, Supplementary Movie 1). Using this method, we find LysoTracker-positive vesicles move at ∼0.6–0.9 μm/s in either direction (compared to reported average speeds for lysotracker vesicles between 0.34 μm/s and 1.2 μm/s in vertebrate neurons) [77].
-
1.
Follow Subheadings 3.1–3.3. for the generation of Drosophila primary neuronal cultures.
-
2.
Prepare supplemented Schneider’s medium with 100 nM LysoTracker DND-99.
-
3.
Shortly before use, spin down solution to remove any precipitates that might have formed and could affect imaging.
-
4.
When cells have reached the desired maturity (see Note 8), detach the coverslips from the culture chambers or glass bottom dish and wash the cells (they will be attached to the coverslips if using culture chambers or at the bottom of the glass bottom dish) and culture chambers in supplemented Schneider’s medium with 100 nM LysoTracker DND-99.
-
5.
Remove all medium and add 50–60 μL supplemented Schneider’s medium with 100 nM LysoTracker DND-99 to the culture chambers/glass bottom dishes.
-
6.
Reseal the culture chambers/glass bottom dishes with the same coverslip, a new Vaseline seal may be required.
-
7.
Incubate for 15 min at the desired temperature.
-
8.
Replace labeling solution with 50–60 μL label-free supplemented Schneider’s medium.
-
9.
Let cells adjust to the condition in the imaging chamber for 15 min and proceed to image the cells.
3.4.2 MitoTracker Staining
Mitochondria can be labeled by both fluorescent mitochondrial dyes including MitoTracker and genetically encoded fluorescently tagged mitochondrial proteins or tagged mitochondria-targeting sequences (such as mito-GFP [78], mito-mCherry [69]). MitoTracker is a fluorescent dye which covalently binds free sulfhydryls. It is frequently used for transport studies in vertebrate neurons [73, 79, 80], and some MitoTracker variants have previously been used in Drosophila tissue and in Drosophila cell lines to label mitochondria [81]. We experienced that MitoTracker dyes in Drosophila primary neuronal cultures can diminish mitochondrial dynamics slightly, as has similarly been reported for rat hippocampal neurons [82]. Nonetheless, MitoTracker easily and reliably labels mitochondria in cultured neurons, and in our studies, it is instrumental in determining the number and distribution of mitochondria at the axon , cell body, and synaptic terminals in fixed samples [43]. Here we will describe how we use MitoTracker (Fig. 2b, Supplementary Movie 2), before explaining the use of genetically encoded fluorescently tagged mitochondrial proteins in the next section.
-
1.
Follow Subheadings 3.1–3.3. for the generation of Drosophila primary neuronal cultures.
-
2.
Prepare supplemented Schneider’s medium with 50 nM MitoTracker Green FM or CMXRos (note that not all MitoTracker variants will be retained after fixation).
-
3.
Shortly before use, spin down solution to remove any precipitates that might affect the imaging.
-
4.
Once primary neuronal cultures from the desired genotype have reached the preferred developmental stage, detach the coverslips from the culture chambers or glass bottom plates and wash the cells (they will be attached to the coverslips if using culture chambers or at the bottom of the glass bottom plates) and culture chambers in supplemented Schneider’s medium with 50 nM MitoTracker Green FM/CMXRos.
-
5.
Remove all medium and add again 50–60 μL supplemented Schneider’s medium with 50 nM MitoTracker Green FM/CMXRos. Be careful as the coverslips are prone to break.
-
6.
Replace Vaseline and reseal the culture chambers/glass bottom plates back.
-
7.
Incubate for 15 min.
-
8.
Remove medium and replace with regular supplemented Schneider’s medium.
-
9.
Proceed to imaging. OPTION: MitoTracker CMXRos-labeled cultures can be fixed by following the steps below.
-
10.
Prewarm the standard fixative to RT. With a black permanent pen, mark the region of the culture drop on the non-cell side of the coverslip. Remove the coverslip from culture chamber and position on a piece of plasticine or a rubber block with the cells facing up in a humid chamber. If glass bottom dishes are used, detach the coverslips from the culture chambers and proceed to step 8.
-
11.
Add 100–200 μL of 4% paraformaldehyde to the cells. Close the humid chamber and incubate for 30 min.
-
12.
Remove fixative and discard adequately, wash the cultures three times with PBT. MitoTracker can be combined with immunocytochemistry, using standard protocols [83].
3.4.3 Genetically Encoded Cargo Markers (e.g., Synaptotagmin-GFP and Mito-GFP/mCherry)
Genetically encoded fluorescent tools are particularly useful to examine the dynamics of motile organelles and transport vesicles in neurons and have been used to this end during decades in vivo and in vitro studies and in various organisms including rodents, zebrafish, Drosophila, and C. elegans [84,85,86,87]. Drosophila examples of such tools include synaptic vesicle proteins such as GFP-fused Synaptotagmin, Synaptobrevin, and Synaptogyrin to label synapses and synaptic transport, tagged Ras-associated binding GTPases, and Autophagy-related proteins to label endosomes and autophagosomes, respectively, and mitochondrial targeting sequences fused to fluorescent proteins (e.g., mito-GFP) to label mitochondria [43, 44, 64, 88, 89]. We have used primary cultures derived from transgenic Drosophila embryos expressing mito-GFP/mCherry, Synaptobrevin-GFP, and Synaptotagmin-GFP using the UAS/Gal4 system to study the impact mutations in microtubule regulators such as Tau and spectraplakins [43] and α1-tubulin84B have on axonal transport. Here we will describe how we use genetically encoded cargo markers (see Note 9), for example, see Fig. 2c, d; Supplementary Movies 3 and 4. Using this method, we find Synaptotagmin-positive vesicles move at a velocity of about 1.0–1.9 μm/s.
-
1.
Generate Drosophila primary neuronal cultures (Subheadings 3.1–3.3) from embryos or larvae expressing the genetically encoded cargo marker in the desired genetic background (see Note 10 for suitable drivers and transfection options).
-
2.
Once primary neuronal cultures have reached the adequate developmental stage, detach the coverslips from the culture chambers or glass bottom dishes, and wash the cells (they will be attached to the coverslips if using culture chambers or at the bottom of the glass bottom plates) and culture chambers in supplemented Schneider’s medium (∼60 μL). Be careful as the coverslips are prone to break. This step is important to remove debris from the cultures which may interfere with live imaging.
-
3.
Remove all medium without disturbing the cells and add 50–60 μL fresh supplemented Schneider’s medium to the chamber/dish.
-
4.
Re-apply new Vaseline if needed and place the coverslip back in place.
-
5.
Let the cells adjust to the imaging conditions for 30 min in the environmental chamber attached to the microscope.
3.5 Live Imaging of Neuronal Transport
Imaging neuronal transport is a delicate balancing act between sufficiently high resolution to visualize small vesicles, short exposure times to allow visualization of fast neuronal transport events and minimize exposure to avoid photo-cytotoxic effects and photo-bleaching. We make use of spinning disk microscopy as the systems have been specifically developed to reduce phototoxicity while providing high sensitivity.
-
1.
Image neurons using a spinning disk confocal (SDC) microscope with environmental chamber and at least 60× magnification objective (or equivalent equipment with good sensitivity and signal to noise ratio). Ideally, set the environmental chamber to the same conditions as the primary neurons were cultured at.
-
2.
Use a microscope with automated z-focus control (e.g., Definite Focus system or similar), which is highly advantageous as any focus drift could shift vesicles/organelles out of focus and prevent successful tracking of their movement (see Note 11).
-
3.
Use either eye piece widefield fluorescence, phase contrast, or SDC imaging to locate suitable cells.
-
4.
Make sure to take note of the position of the cell body and axon /neurite tips for all imaged cells/axons to determine antero- and retrograde directionality.
-
5.
Image with the appropriate timeframes and acquisition intervals, depending on the observed organelle /vesicle speeds, mutant conditions, labels used in the assay, and their intensity during imaging. For LysoTracker, Synaptobrevin-GFP, Synaptotagmin-GFP, and mito-GFP/mCherry, imaging timeframes in the range of 1–5 min at 0.25–1 s intervals have been used successfully. Due to the frequency of events and the slow transport component of mitochondria, imaging intervals might be expanded, and intervals prolonged for the analysis of mitochondrial transport.
-
6.
Adjust the imaging conditions to the moving, not the stationary objects.
-
7.
The number of cells to image depends on the frequency/number of visualized transport events. For the most part we have imaged 10–20 cells per each of the three coverslips, for statistical analysis of organelle /vesicle movements (e.g., Synaptotagmin-GFP, LysoTracker).
-
8.
Process the imaging time series for either direct object tracking or kymograph analyses.
3.6 Analyzing Axonal Transport
-
1.
Process the imaging time series for either direct object tracking (manual tracking plugin for ImageJ, TrackMate [90]) or kymograph generation and analyses (e.g., KymoResliceWide & Velocity measurement [91]; (http://dev.mri.cnrs.fr/projects/imagej-macros/wiki/Velocity_Measurement_Tool) or KymoAnalyzer plugins for ImageJ [92]).
-
2.
Suitable parameters to analyze are: the number of moving objects vs stationary objects, flux (number of moving particles through a defined length of the axon over a set time), directionality of movements (fraction of anterograde vs retrograde movement), average object track velocity (total speed over the whole kymograph track of a moving object including pauses, changes in speed and direction), transport segment velocity (speed calculated for each individual segment of a kymograph track of a moving object). A new segment within the track is defined each time there are changes in speed or direction of movement), transport run length (average length of a particle’s runs), number of pauses per transport track, number of direction switches.
4 Notes
-
1.
Any genetic combination can be used for primary cultures, these can include specimens with specific mutations, and/or expressing fluorescently tagged markers to visualize cargos. In the case of specimens carrying embryonic lethal mutations, the mutant stock will need to be balanced over a balancer chromosome which allows for the selection of mutant stage 11 embryos. For this we use the TwistGal4-UAS-GFP balancers (available in the Bloomington stock centre #6873, #6662, #6663).
-
2.
Collection times must be adapted when changing temperatures. For example, 15 h at 25 °C will favor stage 16 but hardly yield stage 11 embryos, while 15 h at 21–23 °C will be enriched with stage 11 embryos (for stages and precise timing see [67]).
-
3.
There are several factors to consider when selecting embryos:
-
(a)
Embryonic stages 11 would be ideal for primary neuronal cultures, at this stage most neurons in embryos are post-mitotic and are beginning axogenesis [65]. However, depending on the experimental setting, stage 15/16 might be useful, e.g., when the fluorescent signal from specific cargo is too week at younger stages (elav-Gal4-driven expression of synaptic markers is difficult to detect at stage 11).
-
(b)
When collecting for embryos expressing synaptic markers, select embryos with weak expression as this will facilitate imaging (strongly expressing cells will have more stationary objects that can negatively impact imaging). An inducible expression system and a short expression time might be desirable to reduce the number of labeled stationary objects.
-
(c)
When collecting mutant embryos against fluorescent balancers, make sure the balancer fluorescence is visible at the desired embryonic stage.
-
(a)
-
4.
When using more embryos/more larval brains, adjust all volumes proportionately after grinding and before incubating at 37 °C in HBSS-based dispersion medium at RT. Do not exceed 100 μL dispersion medium in a 1.5-mL tube for the grinding step as an increased volume can lead to spillage. We use approximately 100 μL dispersion medium per 15–30 embryos.
-
5.
The grinding step is a key point in the protocol. Make sure to have a tight fit between tube and pestle. Using too much force might adversely affect the cultures though. However, be sure to dissociate all embryos. Any intact embryos will survive, develop into larvae, and disrupt the rest of the cultured cells. An alternative for grinding is to harvest cells from the ventral region of the embryo using a glass capillary and a micromanipulator [65], this method is considerably more laborious but may lead to purer cultures.
-
6.
26 °C would be the standard incubation temperature for Drosophila primary neurons. However, Drosophila primary neurons can be cultured at a range of temperatures ranging from 12 to 29 °C. For instance, incubation at 29 °C can boost UAS-Gal4 expression. Make sure controls are incubated at the same temperature. If imaging of the initial outgrowth is required, the seeding and attachment time can be reduced to 30 min. However, to get the same cell density, a 2–3 times increased amounts of embryos will be required.
-
7.
The same protocol can be used to treat neurons with drugs and other live cell imaging fluorescent probes such as SiR-tubulin. We used 50–100 nM SiR-tubulin to label microtubules. Note that the higher end of SiR-tubulin concentrations can have effects on microtubule dynamics. A minimum of 30-min incubation will ensure microtubules are robustly labeled and can optionally be followed by a washing step with supplemented Schneider’s medium but is not required, longer incubation will yield stronger labeling. Microtubules can be visualized from 45–50 min onwards. SiR-tubulin labeling of Drosophila primary neurons does not require Verapamil treatment. We have successfully visualized neurite outgrowth and development from primary neurons as early as 45 min in vitro up to 22 h in vitro (for long-term imaging setup, see Fig. 1d). For this, the cell pellet is resuspended in supplemented Schneider’s medium that contains 50 nM SiR-tubulin. Other compounds (e.g., microtubule stabilizers or destabilizers, inhibitors of kinesins, kinases and phosphatases, oxidative stress inducers among others) can be added to the cell culture medium keeping in mind that each drug will need a different incubation time.
-
8.
Axonal transport of synaptic components in Drosophila primary neurons can be observed already at 6 h in vitro [43]; however, we frequently image neurons at 2–3 days in vitro, so that synaptic components are robustly established and long-range transport can be observed. Primary neurons cultured from larval brains allow imaging of lysotracker after 1 day in vitro.
-
9.
The same protocol can be used to visualize EB dynamics in neurons. Both expression of EB1-GFP or EB1-mCherry via elav-Gal4 or sca-Gal4 and transfection of neurons with pAc-EB1-mCherry have been successfully used in our laboratories [41, 42, 53, 54].
-
10.
For UAS-Gal4-based expression of fluorescently tagged proteins in primary neuronal cultures, suitable driver lines are: sca-Gal4, elav-Gal4, tubulin-Gal4, nSyb-Gal4. Note that sca-Gal4 expression is strong in young neurons (6HIV) but decreases over time and nSyb-Gal4 is weakly expressing in young neurons but increases over time and can be robustly detected after 2–3 days in vitro (Ines Hahn, personal communication). An alternative to UAS-Gal4-based expression is transfection of neuronal cultures. We find that the actin-promotor in the pAc5.1/V5 vector variants is suitable for the expression of constructs upon transfection of primary neurons (for protocol see [54]). Alternatively, Gal4 expressing neurons can be transfected with UAS-constructs.
-
11.
Be aware that due to the neurites/axons/dendrites being dynamic, they might move and leave the focal plane. ConA treatment of coverslips can be an advantage here since it increases the attachment axons.
References
Terenzio M, Schiavo G, Fainzilber M (2017) Compartmentalized signaling in neurons: from cell biology to neuroscience. Neuron 96(3):667–679. https://doi.org/10.1016/j.neuron.2017.10.015
Franker MA, Hoogenraad CC (2013) Microtubule-based transport - basic mechanisms, traffic rules and role in neurological pathogenesis. J Cell Sci 126(Pt 11):2319–2329. https://doi.org/10.1242/jcs.115030
Prokop A (2013) The intricate relationship between microtubules and their associated motor proteins during axon growth and maintenance. Neural Dev 8(1):17. https://doi.org/10.1186/1749-8104-8-17
Hirokawa N, Niwa S, Tanaka Y (2010) Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68(4):610–638. https://doi.org/10.1016/j.neuron.2010.09.039
Sleigh JN, Rossor AM, Fellows AD, Tosolini AP, Schiavo G (2019) Axonal transport and neurological disease. Nat Rev Neurol 15(12):691–703. https://doi.org/10.1038/s41582-019-0257-2
Guedes-Dias P, Holzbaur ELF (2019) Axonal transport: driving synaptic function. Science 366(6462):eaaw9997. https://doi.org/10.1126/science.aaw9997
Kevenaar JT, Hoogenraad CC (2015) The axonal cytoskeleton: from organization to function. Front Mol Neurosci 8:44. https://doi.org/10.3389/fnmol.2015.00044
Prokop A (2020) Cytoskeletal organization of axons in vertebrates and invertebrates. J Cell Biol 219(7). https://doi.org/10.1083/jcb.201912081
Hirokawa N, Noda Y, Tanaka Y, Niwa S (2009) Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol 10(10):682–696. https://doi.org/10.1038/nrm2774
Schiavo G, Greensmith L, Hafezparast M, Fisher EM (2013) Cytoplasmic dynein heavy chain: the servant of many masters. Trends Neurosci 36(11):641–651. https://doi.org/10.1016/j.tins.2013.08.001
Horiuchi D, Collins CA, Bhat P, Barkus RV, Diantonio A, Saxton WM (2007) Control of a kinesin-cargo linkage mechanism by JNK pathway kinases. Curr Biol 17(15):1313–1317. https://doi.org/10.1016/j.cub.2007.06.062
Guedes-Dias P, Nirschl JJ, Abreu N, Tokito MK, Janke C, Magiera MM, Holzbaur ELF (2019) Kinesin-3 responds to local microtubule dynamics to target synaptic cargo delivery to the Presynapse. Curr Biol 29(2):268–282.e268. https://doi.org/10.1016/j.cub.2018.11.065
Guo W, Stoklund Dittlau K, Van Den Bosch L (2020) Axonal transport defects and neurodegeneration: molecular mechanisms and therapeutic implications. Semin Cell Dev Biol 99:133–150. https://doi.org/10.1016/j.semcdb.2019.07.010
Mattedi F, Vagnoni A (2019) Temporal control of axonal transport: the extreme case of organismal ageing. Front Cell Neurosci 13:393. https://doi.org/10.3389/fncel.2019.00393
Hill SE, Colón-Ramos DA (2020) The journey of the synaptic autophagosome: a cell biological perspective. Neuron 105(6):961–973. https://doi.org/10.1016/j.neuron.2020.01.018
Corradi E, Dalla Costa I, Gavoci A, Iyer A, Roccuzzo M, Otto TA, Oliani E, Bridi S, Strohbuecker S, Santos-Rodriguez G, Valdembri D, Serini G, Abreu-Goodger C, Baudet ML (2020) Axonal precursor miRNAs hitchhike on endosomes and locally regulate the development of neural circuits. EMBO J 39(6):e102513. https://doi.org/10.15252/embj.2019102513
Cheng X-T, Sheng Z-H (2021) Developmental regulation of microtubule-based trafficking and anchoring of axonal mitochondria in health and diseases. Dev Neurobiol 81(3):284–299. https://doi.org/10.1002/dneu.22748
Zhao J, Fok AHK, Fan R, Kwan P-Y, Chan H-L, Lo LH-Y, Chan Y-S, Yung W-H, Huang J, Lai CSW, Lai K-O (2020) Specific depletion of the motor protein KIF5B leads to deficits in dendritic transport, synaptic plasticity and memory. eLife 9:e53456. https://doi.org/10.7554/eLife.53456
Maday S, Twelvetrees Alison E, Moughamian Armen J, Holzbaur Erika LF (2014) Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron 84(2):292–309. https://doi.org/10.1016/j.neuron.2014.10.019
Verhey KJ, Hammond JW (2009) Traffic control: regulation of kinesin motors. Nat Rev Mol Cell Biol 10(11):765–777. https://doi.org/10.1038/nrm2782
Kelliher MT, Saunders HAJ, Wildonger J (2019) Microtubule control of functional architecture in neurons. Curr Opin Neurobiol 57:39–45. https://doi.org/10.1016/j.conb.2019.01.003
Harterink M, Edwards SL, de Haan B, Yau KW, van den Heuvel S, Kapitein LC, Miller KG, Hoogenraad CC (2018) Local microtubule organization promotes cargo transport in C. elegans dendrites. J Cell Sci 131(20):jcs223107. https://doi.org/10.1242/jcs.223107
Gibbs KL, Greensmith L, Schiavo G (2015) Regulation of axonal transport by protein kinases. Trends Biochem Sci 40(10):597–610. https://doi.org/10.1016/j.tibs.2015.08.003
Gindhart JG Jr, Desai CJ, Beushausen S, Zinn K, Goldstein LSB (1998) Kinesin light chains are essential for axonal transport in drosophila. J Cell Biol 141(2):443–454. https://doi.org/10.1083/jcb.141.2.443
Reis GF, Yang G, Szpankowski L, Weaver C, Shah SB, Robinson JT, Hays TS, Danuser G, Goldstein LS (2012) Molecular motor function in axonal transport in vivo probed by genetic and computational analysis in drosophila. Mol Biol Cell 23(9):1700–1714. https://doi.org/10.1091/mbc.E11-11-0938
Weiss KR, Littleton JT (2016) Characterization of axonal transport defects in drosophila huntingtin mutants. J Neurogenet 30(3–4):212–221. https://doi.org/10.1080/01677063.2016.1202950
Vijayakumar J, Perrois C, Heim M, Bousset L, Alberti S, Besse F (2019) The prion-like domain of drosophila imp promotes axonal transport of RNP granules in vivo. Nat Commun 10(1):2593. https://doi.org/10.1038/s41467-019-10554-w
Duncan JE, Goldstein LSB (2006) The genetics of axonal transport and axonal transport disorders. PLoS Genet 2(9):e124. https://doi.org/10.1371/journal.pgen.0020124
Medioni C, Ephrussi A, Besse F (2015) Live imaging of axonal transport in drosophila pupal brain explants. Nat Protoc 10(4):574–584. https://doi.org/10.1038/nprot.2015.034
Sánchez-Soriano N, Bottenberg W, Fiala A, Haessler U, Kerassoviti A, Knust E, Löhr R, Prokop A (2005) Are dendrites in Drosophila homologous to vertebrate dendrites? Dev Biol 288(1):126–138. https://doi.org/10.1016/j.ydbio.2005.09.026
Bellen HJ, Tong C, Tsuda H (2010) 100 years of drosophila research and its impact on vertebrate neuroscience: a history lesson for the future. Nat Rev Neurosci 11(7):514–522. https://doi.org/10.1038/nrn2839
McGurk L, Berson A, Bonini NM (2015) Drosophila as an In Vivo model for human neurodegenerative disease. Genetics 201 (2):377–402. https://doi.org/10.1534/genetics.115.179457
Sánchez-Soriano N, Tear G, Whitington P, Prokop A (2007) Drosophilaas a genetic and cellular model for studies on axonal growth. Neural Dev 2(1):9. https://doi.org/10.1186/1749-8104-2-9
Sanes JR, Zipursky SL (2010) Design principles of insect and vertebrate visual systems. Neuron 66(1):15–36. https://doi.org/10.1016/j.neuron.2010.01.018
Hirokawa N, Tanaka Y (2015) Kinesin superfamily proteins (KIFs): various functions and their relevance for important phenomena in life and diseases. Exp Cell Res 334(1):16–25. https://doi.org/10.1016/j.yexcr.2015.02.016
Ejsmont RK, Hassan BA (2014) The little Fly that could: wizardry and artistry of drosophila genomics. Genes 5(2):385–414. https://doi.org/10.3390/genes5020385
Hahn I, Ronshaugen M, Sánchez-Soriano N, Prokop A (2016) Functional and genetic analysis of Spectraplakins in Drosophila. Methods Enzymol 569:373–405. https://doi.org/10.1016/bs.mie.2015.06.022
Bier E, Harrison MM, O’Connor-Giles KM, Wildonger J (2018) Advances in engineering the Fly genome with the CRISPR-Cas system. Genetics 208(1):1–18. https://doi.org/10.1534/genetics.117.1113
Donady JJ, Seecof RL (1972) Effect of the gene lethal (1) Myospheroid on drosophila embryonic cells in vitro. In Vitro 8(1):7–12
Seecof RL, Alléaume N, Teplitz RL, Gerson I (1971) Differentiation of neurons and myocytes in cell cultures made from drosophila gastrulae. Exp Cell Res 69(1):161–173. https://doi.org/10.1016/0014-4827(71)90321-1
Alves-Silva J, Sánchez-Soriano N, Beaven R, Klein M, Parkin J, Millard TH, Bellen HJ, Venken KJ, Ballestrem C, Kammerer RA, Prokop A (2012) Spectraplakins promote microtubule-mediated axonal growth by functioning as structural microtubule-associated proteins and EB1-dependent +TIPs (tip interacting proteins). J Neurosci 32(27):9143–9158. https://doi.org/10.1523/jneurosci.0416-12.2012
Szikora S, Földi I, Tóth K, Migh E, Vig A, Bugyi B, Maléth J, Hegyi P, Kaltenecker P, Sanchez-Soriano N, Mihály J (2017) The formin DAAM is required for coordination of the actin and microtubule cytoskeleton in axonal growth cones. J Cell Sci 130(15):2506–2519. https://doi.org/10.1242/jcs.203455
Voelzmann A, Okenve-Ramos P, Qu Y, Chojnowska-Monga M, Del Caño-Espinel M, Prokop A, Sanchez-Soriano N (2016) Tau and spectraplakins promote synapse formation and maintenance through Jun kinase and neuronal trafficking. Elife 5:e14694. https://doi.org/10.7554/eLife.14694
Zschätzsch M, Oliva C, Langen M, De Geest N, Ozel MN, Williamson WR, Lemon WC, Soldano A, Munck S, Hiesinger PR, Sanchez-Soriano N, Hassan BA (2014) Regulation of branching dynamics by axon-intrinsic asymmetries in tyrosine kinase receptor signaling. Elife 3:e01699. https://doi.org/10.7554/eLife.01699
Qu Y, Hahn I, Webb SED, Pearce SP, Prokop A (2017) Periodic actin structures in neuronal axons are required to maintain microtubules. Mol Biol Cell 28(2):296–308. https://doi.org/10.1091/mbc.E16-10-0727
Sánchez-Soriano N, Gonçalves-Pimentel C, Beaven R, Haessler U, Ofner-Ziegenfuss L, Ballestrem C, Prokop A (2010) Drosophila growth cones: a genetically tractable platform for the analysis of axonal growth dynamics. Dev Neurobiol 70(1):58–71. https://doi.org/10.1002/dneu.20762
Bai J, Sepp KJ, Perrimon N (2009) Culture of drosophila primary cells dissociated from gastrula embryos and their use in RNAi screening. Nat Protoc 4(10):1502–1512. https://doi.org/10.1038/nprot.2009.147
Küppers-Munther B, Letzkus JJ, Lüer K, Technau G, Schmidt H, Prokop A (2004) A new culturing strategy optimises drosophila primary cell cultures for structural and functional analyses. Dev Biol 269(2):459–478. https://doi.org/10.1016/j.ydbio.2004.01.038
Ceron J, Tejedor FJ, Moya F (2006) A primary cell culture of drosophila postembryonic larval neuroblasts to study cell cycle and asymmetric division. Eur J Cell Biol 85(6):567–575. https://doi.org/10.1016/j.ejcb.2006.02.006
Katsuki T, Ailani D, Hiramoto M, Hiromi Y (2009) Intra-axonal patterning: intrinsic compartmentalization of the axonal membrane in drosophila neurons. Neuron 64(2):188–199. https://doi.org/10.1016/j.neuron.2009.08.019
Slováková J, Speicher S, Sánchez-Soriano N, Prokop A, Carmena A (2012) The actin-binding protein canoe/AF-6 forms a complex with Robo and is required for slit-Robo signaling during axon pathfinding at the CNS midline. J Neurosci 32(29):10035–10044. https://doi.org/10.1523/jneurosci.6342-11.2012
Oliva C, Soldano A, Mora N, De Geest N, Claeys A, Erfurth ML, Sierralta J, Ramaekers A, Dascenco D, Ejsmont RK, Schmucker D, Sanchez-Soriano N, Hassan BA (2016) Regulation of drosophila brain wiring by neuropil interactions via a slit-Robo-RPTP signaling complex. Dev Cell 39(2):267–278. https://doi.org/10.1016/j.devcel.2016.09.028
Hahn I, Voelzmann A, Parkin J, Fuelle J, Slater PG, Lowery LA, Sanchez-Soriano N, Prokop A (2020) Tau, XMAP215/Msps and Eb1 jointly regulate microtubule polymerisation and bundle formation in axons. bioRxiv:2020.2008.2019.257808. https://doi.org/10.1101/2020.08.19.257808
Qu Y, Hahn I, Lees M, Parkin J, Voelzmann A, Dorey K, Rathbone A, Friel CT, Allan VJ, Okenve-Ramos P, Sanchez-Soriano N, Prokop A (2019) Efa6 protects axons and regulates their growth and branching by inhibiting microtubule polymerisation at the cortex. eLife 8:e50319. https://doi.org/10.7554/eLife.50319
Prokop A, Beaven R, Qu Y, Sánchez-Soriano N (2013) Using fly genetics to dissect the cytoskeletal machinery of neurons during axonal growth and maintenance. J Cell Sci 126(Pt 11):2331–2341. https://doi.org/10.1242/jcs.126912
Sanchez-Soriano N, Travis M, Dajas-Bailador F, Gonçalves-Pimentel C, Whitmarsh AJ, Prokop A (2009) Mouse ACF7 and drosophila short stop modulate filopodia formation and microtubule organisation during neuronal growth. J Cell Sci 122(Pt 14):2534–2542. https://doi.org/10.1242/jcs.046268
Stephan D, Sánchez-Soriano N, Loschek LF, Gerhards R, Gutmann S, Storchova Z, Prokop A, Kadow IC (2012) Drosophila Psidin regulates olfactory neuron number and axon targeting through two distinct molecular mechanisms. J Neurosci 32(46):16080–16094. https://doi.org/10.1523/jneurosci.3116-12.2012
Kraft R, Escobar MM, Narro ML, Kurtis JL, Efrat A, Barnard K, Restifo LL (2006) Phenotypes of Drosophila brain neurons in primary culture reveal a role for Fascin in neurite shape and trajectory. J Neurosci 26(34):8734–8747. https://doi.org/10.1523/jneurosci.2106-06.2006
del Castillo U, Müller H-AJ, Gelfand VI (2020) Kinetochore protein spindly controls microtubule polarity in Drosophila axons. Proc Natl Acad Sci 117(22):12155–12163. https://doi.org/10.1073/pnas.2005394117
Yao W-D, Rusch J, Poo M-M, Wu C-F (2000) Spontaneous acetylcholine secretion from developing growth cones of Drosophila central neurons in culture: effects of cAMP-pathway mutations. J Neurosci 20(7):2626–2637. https://doi.org/10.1523/jneurosci.20-07-02626.2000
Küppers B, Sánchez-Soriano N, Letzkus J, Technau GM, Prokop A (2003) In developing drosophila neurones the production of gamma-amino butyric acid is tightly regulated downstream of glutamate decarboxylase translation and can be influenced by calcium. J Neurochem 84(5):939–951. https://doi.org/10.1046/j.1471-4159.2003.01554.x
West RJH, Sharpe JL, Voelzmann A, Munro AL, Hahn I, Baines RA, Pickering-Brown S (2020) Co-expression of C9orf72 related dipeptide-repeats over 1000 repeat units reveals age- and combination-specific phenotypic profiles in drosophila. Acta Neuropathol Commun 8(1):158. https://doi.org/10.1186/s40478-020-01028-y
Narayanareddy BR, Vartiainen S, Hariri N, O’Dowd DK, Gross SP (2014) A biophysical analysis of mitochondrial movement: differences between transport in neuronal cell bodies versus processes. Traffic 15(7):762–771. https://doi.org/10.1111/tra.12171
Liao P-C, Tandarich LC, Hollenbeck PJ (2017) ROS regulation of axonal mitochondrial transport is mediated by Ca2+ and JNK in drosophila. PLoS One 12(5):e0178105. https://doi.org/10.1371/journal.pone.0178105
Prokop A, Küppers-Munther B, Sánchez-Soriano N (2012) Using primary neuron cultures of drosophila to analyze neuronal circuit formation and function. In: Hassan BA (ed) The making and un-making of neuronal circuits in drosophila. Humana Press, Totowa, NJ, pp 225–247. https://doi.org/10.1007/978-1-61779-830-6_10
Sicaeros B, Campusano JM, O’Dowd DK (2007) Primary neuronal cultures from the brains of late stage drosophila pupae. J Vis Exp 4:200–200. https://doi.org/10.3791/200
Campos-Ortega JA, Hartenstein V (1997) The embryonic development of Drosophila melanogaster. Springer Verlag, Berlin
Roote J, Prokop A (2013) How to design a genetic mating scheme: a basic training package for drosophila genetics. G3 (Bethesda, Md) 3(2):353–358. https://doi.org/10.1534/g3.112.004820
Vagnoni A, Bullock SL (2016) A simple method for imaging axonal transport in aging neurons using the adult drosophila wing. Nat Protoc 11(9):1711–1723. https://doi.org/10.1038/nprot.2016.112
Lee S, Sato Y, Nixon RA (2011) Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J Neurosci 31(21):7817–7830. https://doi.org/10.1523/jneurosci.6412-10.2011
Maday S, Wallace KE, Holzbaur ELF (2012) Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol 196(4):407–417. https://doi.org/10.1083/jcb.201106120
Moughamian AJ, Osborn GE, Lazarus JE, Maday S, Holzbaur EL (2013) Ordered recruitment of dynactin to the microtubule plus-end is required for efficient initiation of retrograde axonal transport. J Neurosci 33(32):13190–13203. https://doi.org/10.1523/jneurosci.0935-13.2013
Evans CS, Holzbaur ELF (2020) Degradation of engulfed mitochondria is rate-limiting in Optineurin-mediated mitophagy in neurons. eLife 9:e50260. https://doi.org/10.7554/eLife.50260
Perrot R, Julien JP (2009) Real-time imaging reveals defects of fast axonal transport induced by disorganization of intermediate filaments. FASEB J 23(9):3213–3225. https://doi.org/10.1096/fj.09-129585
Beaven R, Dzhindzhev NS, Qu Y, Hahn I, Dajas-Bailador F, Ohkura H, Prokop A (2015) Drosophila CLIP-190 and mammalian CLIP-170 display reduced microtubule plus end association in the nervous system. Mol Biol Cell 26(8):1491–1508. https://doi.org/10.1091/mbc.E14-06-1083
DeVorkin L, Gorski SM (2014) LysoTracker staining to aid in monitoring autophagy in drosophila. Cold Spring Harb Protoc 2014(9):951–958. https://doi.org/10.1101/pdb.prot080325
Zhao X, Chen XQ, Han E, Hu Y, Paik P, Ding Z, Overman J, Lau AL, Shahmoradian SH, Chiu W, Thompson LM, Wu C, Mobley WC (2016) TRiC subunits enhance BDNF axonal transport and rescue striatal atrophy in Huntington’s disease. Proc Natl Acad Sci U S A 113(38):E5655–E5664. https://doi.org/10.1073/pnas.1603020113
Pilling AD, Horiuchi D, Lively CM, Saxton WM (2006) Kinesin-1 and dynein are the primary motors for fast transport of mitochondria in drosophila motor axons. Mol Biol Cell 17(4):2057–2068. https://doi.org/10.1091/mbc.e05-06-0526
Moutaux E, Christaller W, Scaramuzzino C, Genoux A, Charlot B, Cazorla M, Saudou F (2018) Neuronal network maturation differently affects secretory vesicles and mitochondria transport in axons. Sci Rep 8(1):13429. https://doi.org/10.1038/s41598-018-31759-x
Sheng Z-H (2014) Mitochondrial trafficking and anchoring in neurons: new insight and implications. J Cell Biol 204(7):1087–1098. https://doi.org/10.1083/jcb.201312123
Da-Rè C, Franzolin E, Biscontin A, Piazzesi A, Pacchioni B, Gagliani MC, Mazzotta G, Tacchetti C, Zordan MA, Zeviani M, Bernardi P, Bianchi V, De Pittà C, Costa R (2014) Functional characterization of drim2, the Drosophila melanogaster homolog of the yeast mitochondrial deoxynucleotide transporter. J Biol Chem 289(11):7448–7459. https://doi.org/10.1074/jbc.M113.543926
Wang X, Schwarz TL (2009) Imaging axonal transport of mitochondria. Methods Enzymol 457:319–333. https://doi.org/10.1016/S0076-6879(09)05018-6
Budnik V, Gorczyca M, Prokop A (2006) Selected methods for the anatomical study of drosophila embryonic and larval neuromuscular junctions. Int Rev Neurobiol 75:323–365. https://doi.org/10.1016/s0074-7742(06)75015-2
Bercier V, Rosello M, Del Bene F, Revenu C (2019) Zebrafish as a model for the study of live in vivo Processive transport in neurons. Front Cell Dev Biol 7:17. https://doi.org/10.3389/fcell.2019.00017
Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS (2003) Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in drosophila. Neuron 40(1):25–40. https://doi.org/10.1016/s0896-6273(03)00594-4
Nonet ML (1999) Visualization of synaptic specializations in live C. elegans with synaptic vesicle protein-GFP fusions. J Neurosci Methods 89(1):33–40. https://doi.org/10.1016/s0165-0270(99)00031-x
Surana S, Villarroel-Campos D, Lazo OM, Moretto E, Tosolini AP, Rhymes ER, Richter S, Sleigh JN, Schiavo G (2020) The evolution of the axonal transport toolkit. Traffic (Copenhagen, Denmark) 21(1):13–33. https://doi.org/10.1111/tra.12710
Lu W, Del Castillo U, Gelfand VI (2013) Organelle transport in cultured drosophila cells: S2 cell line and primary neurons. J Vis Exp 81:e50838. https://doi.org/10.3791/50838
Dey S, Banker G, Ray K (2017) Anterograde transport of Rab4-associated vesicles regulates synapse Organization in Drosophila. Cell Rep 18(10):2452–2463. https://doi.org/10.1016/j.celrep.2017.02.034
Jean-Yves Tineveza NP, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, Eliceiri KW (2017) TrackMate: an open and extensible platform for single-particle tracking. Methods 115:80–90
Katrukha E (2020), KymoResliceWide plugin for ImageJ, v0.5, Zenodo https://doi.org/10.5281/zenodo.4281086. software download link: https://github.com/ekatrukha/KymoResliceWide
Neumann S, Chassefeyre R, Campbell GE, Encalada SE (2017) KymoAnalyzer: a software tool for the quantitative analysis of intracellular transport in neurons. Traffic (Copenhagen, Denmark) 18(1):71–88. https://doi.org/10.1111/tra.12456
Acknowledgments
This work was made possible through support by the BBSRC to N.S.S. (BB/M007456/1, BB/R018960/1), A.V. is funded by the BBSRC (BB/M007553/1, awarded to Andreas Prokop). The Bioimaging Facility microscopes (3i Spinning Disk Confocal/Photometrics Primer95B sCMOS camera; Andor Dragonfly Upright/Zyla 4.2 Plus sCMOS) used in this study were purchased with grants from BBSRC, Wellcome and the University of Manchester Strategic Fund. We also acknowledge the ITM Biomedical imaging facility at the University of Liverpool hosting the 3i Marianas spinning disk confocal system used for some at the studies. The Fly Facility has been supported by funds from The University of Manchester and the Wellcome Trust (087742/Z/08/Z). Special thanks go to Peter March and Steven Marsden for their help with the microscopy and to Andreas Prokop, Ryan J. H. West and Ines Hahn for helpful comments on the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
1 Electronics Supplementary Materials
Supplementary Movie 1
Live movie of larval Drosophila primary neurons grown for 1 day in vitro and incubated for 15 min with 100 nM LysoTracker DND-99. Scale bar represents 5 μm, time is given in seconds. (AVI 63747 kb)
Supplementary Movie 2
Live movie of embryonic Drosophila primary neurons precultured in a 1.5-mL tube, grown for 1 day in vitro, and incubated for 15 min with 50 nM MitoTracker Green FM. Scale bar represents 5 μm, time is given in seconds. (AVI 7783 kb)
Supplementary Movie 3
Live movie of Drosophila embryonic primary neurons expressing UAS-Synaptotagmin-GFP under control of the tubulin-Gal4 driver line. Cells were grown for 3 days in vitro. Scale bar represents 5 μm, time is given in seconds. (AVI 8609 kb)
Supplementary Movie 4
Live movie of embryonic Drosophila primary neurons expressing UAS-mito-mCherry under control of the elav-Gal4 driver line. Cells were grown for 1 day in vitro. Scale bar represents 5 μm, time is given in seconds. (AVI 7495 kb)
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this protocol

Cite this protocol
Voelzmann, A., Sanchez-Soriano, N. (2022). Drosophila Primary Neuronal Cultures as a Useful Cellular Model to Study and Image Axonal Transport. In: Vagnoni, A. (eds) Axonal Transport. Methods in Molecular Biology, vol 2431. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-1990-2_23
Download citation
DOI: https://doi.org/10.1007/978-1-0716-1990-2_23
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-1989-6
Online ISBN: 978-1-0716-1990-2
eBook Packages: Springer Protocols