
Abstract
Mice (Mus musculus) and rats (Rattus norvegicus) have long served as model systems for biomedical research. However, they are also excellent models for studying the evolution of populations, subspecies, and species. Within the past million years, they have spread in various waves across large parts of the globe, with the most recent spread in the wake of human civilization. They have developed into commensal species, but have also been able to colonize extreme environments on islands free of human civilization. Given that ample genomic and genetic resources are available for these species, they have thus also become ideal mammalian systems for evolutionary studies on adaptation and speciation, particularly in the combination with the rapid developments in population genomics. The chapter provides an overview of the systems and their history, as well as of available resources.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
Population genomics can address very different biological questions related to speciation, divergence of closely related species, within species population structure or within population evolutionary processes that affect adaptation. In the era of next-generation sequencing (NGS) with increasing taxonomic sampling, the crucial factor to apply population genomics is not any longer the number of genetic markers (quantity) but it is quality and complexity of the massive amount of available information that needs to be integrated and interpreted.
In this chapter, we focus on studies of population genomics in rodents and in particular on the Murinae. Murinae as a subfamily of rodents comprises more than one hundred genera and it is among mammals one of the largest subfamilies with species native to most continents. Murinae includes the house mouse (Mus musculus) and the brown rat (Rattus norvegicus) of which laboratory strains have been used since decades for biomedical research, as well as to serve as models to study human diseases. Further, as human commensal species, both harbor also vectors for spreading infectious diseases that makes the wild living animals and populations of special interest. But also their evolutionary histories make them perfect models for studying general evolutionary processes, such as speciation, rapid adaptation and behavioral changes.
1.1 History of the House Mouse
A recent book, “Evolution of the House Mouse” [1], provides a broad overview on a variety of evolutionary aspects for the house mouse. Other general reviews can be found in [2, 3]. Here, we provide a short summary.
Mice consist of four major clades (Coelomys, Mus, Nannomys, and Pyromys), of which the subgenus Mus harbors the species Mus musculus, the house mouse. House mouse genetics began early in the twentieth century based on the first inbred strains from wild derived animals to study modes of inheritance [4, 5]. The world-wide distribution range of the house mouse is depicted in Fig. 1. It shows three main subspecies, the southeastern Asian house mouse (Mus musculus castaneus), the eastern European house mouse (Mus musculus musculus) and the western European house mouse (Mus musculus domesticus). Next to these main subspecies, there exist other subspecies (e.g., M. m. molossinus, a presumptive hybrid species between M. m. castaneus and M. m. musculus; [6], M. m. gentilulus [7, 8], M. m. homoulus [9], and further recently diverged ones like M. m. helgolandicus [10]). Most inbred strains and the reference genome sequence are derived from M. m. domesticus. The mouse genome was the first sequenced mammalian genome published in 2002 (Mouse Genome Sequencing Consortium, 2002) [8]. The genome consists of 19 autosomes and 2 sex chromosomes (X and Y) with a total length of 2.7 Gbp (currently with 22,612 coding and 15,402 noncoding genes annotated). The mouse ENCODE [11] consortium and genome assemblies of wild-derived inbred strains of the main subspecies have further enhanced the available genomic information [12,13,14], complemented by detailed recombination maps [15,16,17]. Genomic and transcriptomic data from wild derived populations of the subspecies and the sister species Mus spretus were reported in [14].

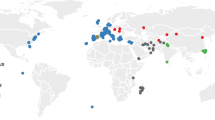
Sampling locations of mice for which public population scale WGS data exist. Population scale sampling locations of house mouse and close relatives
As one of the prominent human commensals, the dispersal and phylogenetic history of the house mouse were intensively studied. The ancestor of all subspecies within Mus musculus was initially thought to have lived in India [1, 18], but a broader sampling has shown that the Iranian plateau shows the highest diversity of lineages, including some as yet unnamed lineages [10]. The main subspecies started to diverge ~350–500 thousand years ago. As recently diverged species, one finds frequently phylogenetic discordance at different loci, whereby the statistical analysis of discordance patterns shows a strong deviation from a neutral model of pure lineage sorting [19]. Based on population data, it was shown that this is most likely due to secondary adaptive introgression, even across large geographic distances [20, 21]. The overall phylogenomic analysis suggests that M. m. musculus and M. m. castaneus are sister groups and that M. m. domesticus is more basal [12, 19].
The subspecies meet in several zones of secondary contact, where they form hybrid zones [2, 18, 22]. Fertility of offspring is impaired across these hybrid zones, and this serves as a general model to study the genetic basis of hybrid sterility as part of speciation processes (e.g., [22,23,24,25,26]).
Studies on house mouse phylogeography showed that the spread of the populations, especially those of M. m. domesticus, reflects human colonization and settlement history. For example, by looking into mtDNA haplotypes of worldwide distributed mouse samples, some historical human movements, such as following the seafarer routes of Vikings [27,28,29] or the colonization history of sub-Antarctic islands, could be reconstructed [30, 31].
Systematic population-level sampling of mouse populations has been introduced by Ihle et al. [32], where the sampling regime has taken care of the fact that mice tend to show inbreeding in family groups. Initial microsatellite based scans of populations that were sampled in this way suggested a high rate of positive selection between closely related populations [33]. The colonization history of Western European populations was traced by fossil evidence [34] and shown to be less than 3000 years ago. Nonetheless, these populations show clear genomic differentiation [20, 32, 33], differences in gene expression [35, 36], ultrasonic vocalization and mate choice [37, 38]. They harbor also a number of deme-specific MHC haplotypes [39].
Despite genomic resources, including a variant database of 17 laboratory inbred strains [12, 40], there was the need to derive laboratory strains that harbor most of the natural variation found in wild-derived populations [41, 42]. Genotype arrays were established that were constructed to maximize variant information at low sequencing costs [43]. The still commonly used genotyping arrays are MegaMUGA with a set of 77,808 SNP markers and GigaMUGA with a set of 143,259 SNP markers [44], which only represent a fraction of variants found between any sequenced inbred strain and the reference genome (~4 to 5 million SNPs; [12, 45]). However, researchers started to complement their analyses with NGS based datasets and genomic resources for wild populations of the house mouse are now common ground for subsequent analysis [14].
1.2 Brown Rat History
Mice and rats approximately diverged 7–12 million years ago [46]. Similar to house mice, brown rats (Rattus norvegicus) have been used for more than two centuries for biomedical studies to learn about the basis of human diseases and to deal with human pest management [47, 48]. The genome of the brown rat was published in 2004 [49] and consists of 20 autosomes and 2 sex chromosomes (X and Y) with a total length of 2.8 Gb (currently with 22,250 coding and 8934 noncoding genes annotated). The house mouse genome and the brown rat genome show a high number of shared syntenic homologous blocks with different levels of recombination [50]. Approximately 30% of the rat genome aligns only with the mouse genome, which might correspond to rodent-specific repeats [49]. A syntenic view of both genomes is given in Fig. 2 to illustrate the pairwise chromosome assignment obtained from the Synteny Portal (see Table 1 for web page URL link; [51]).

Syntenic blocks between house mouse and rat. House mouse (GRCm38/mm10) and brown rat (Rnor_6.0/rn6) chromosome-wide syntenic blocks obtained via the web-based Synteny Portal [51]
The origin of the laboratory brown rat (Rattus norvegicus) and the black rat (Rattus rattus) most likely lies in central Asia [52]. Spatial population genomics studies were conducted on brown rats living in New York City [53] and, like in mice studies, mtDNA haplotype data could disentangle the phylogeography of brown rats in the countries surrounding the South Atlantic Ocean [54]. While the phylogeography of black rats, like the phylogeography of house mice, reflects human colonization and settlement history [53, 55, 56, 57], brown rats did not appear in Europe until the sixteenth century. Their dispersal routes from Asia to Europe are still under debate [57]. For example, one route is thought to lead via northeast China and Siberia, while another route inferred on whole-genome sequencing may represent an expansion via a Southern East Asia route [58]. Figure 3 illustrates the sampling distribution of Rattus norvegicus from publicly available whole-genome data sets.

Sampling locations of rats for which public population scale WGS data exist. Population scale sampling locations of brown rats obtained from [58]
2 Population Genomics
As mammal species expand, they are faced with new abiotic and biotic factors, such as different climatic conditions, different food or new pathogens, prey and/or predators, which potentially lead to adaptation and contributes to shaping the genome over time. Evolutionary changes in the genome can result from mutation, gene flow, random genetic drift, recombination and selection. Genome-wide scans for deviation from modelled neutrality aim at revealing such evolutionary processes. Genome-wide scans can help to identify genotypic and phenotypic variation, and by taking demographic events into account, they can even detect genes under recent positive selection [59]. Negative selection leads to sequence conservation by removing disadvantageous alleles. Positive selection can yield to an excess of nonsynonymous fixed differences or lead to an altered allele-frequency spectrum (AFS). Multiple approaches exist to detect adaptation, each with its own caveats. For example, dN/dS ratios can be used in comparative studies to detect selection on genes. But this analysis is limited to species that represent a certain evolutionary distance to allow a sufficient number of substitutions to have occurred [60]. When samples are drawn from different populations of the same species, it is necessary to study frequency changes of polymorphisms instead of substitutions. As compared to studies with a limited number of neutral markers, population genomics uses high marker density to robustly infer genome-wide effects, usually as signals of departure from expectations of the neutral theory of molecular evolution (see Chapter 5 for a detailed description how to detect positive selection).
2.1 House Mouse Genetic Variation
Population genetic studies revealed a fairly large effective population size (Ne) for wild natural populations of mice in the order of Ne = 5 × 105 to 2 × 106 [61, 62] with two to three generations per year. Based on a genotyping array, the effective population sizes for the subspecies were estimated to range between Ne = 0.25 × 105 to 1.2 × 105 for M. m. musculus, Ne = 0.58 × 105 to 2 × 105 for M. m. domesticus and Ne = 2 × 105 to 7 × 105 for M. m. castaneus [63]. This assumption was validated recently by a population genomic study on nucleotide diversity within the subspecies of M. m. castaneus [64]. In the same study an excess of adaptive substitutions in protein-coding genes, UTRs and conserved noncoding elements (CNE) were observed [64]. A follow-up study based on the same data recently inferred the recombination landscape within the same subspecies and revealed that genetic diversity is positively correlated with the rate of recombination [17] (see ref. 13 for the recombination landscape in the collaborative cross [41] and see ref. 65 for mouse inbred strains). The frequency-weighted mean estimate of the recombination rate was inferred from a broad-scaled map to 4Ner/bp = 0.0092 for autosomes per bp and to 4Ner/bp = 0.0026 for the X chromosome [17].
One candidate gene that is known to influence recombination break points in mammals is PRDM9 [66,67,68,69]. PRDM9 is highly polymorphic in natural populations of the house mouse [70, 71] and it was recently shown that some alleles are preferred over others in hybrid mice [72]. What is remarkable in the study of Booker et al. [17] is the high level of variability of recombination hot spots within one population and between wild-derived and classical inbred strains, which is worth further consideration. For example, phasing approaches should depend on an accurate recombination map and the question arises whether global heterogeneous recombination rates provide sufficient information for fine-scaled phasing inference.
Researchers need to rely on high-quality genome information to perform reference-based whole-genome analysis to retain variant information for the populations under study. However, in some cases the sequence divergence of the analyzed population and the reference is high and might produce mapping artefacts [73]. To cope with such situations Sarver et al. [74] performed a pseudo-reference based approach using exome data to infer the phylogenetic relationship and gene tree incongruence of the Mus clade. While Sarver et al. [74] used the d-statistic [75] to detect introgression between M. m. musculus and M. m. domesticus, other methods have been recently applied to infer introgression signals [8, 20, 21, 76, 77].
In their genomic comparison, Harr et al. [14] incorporated the two other house mouse subspecies M. m. domesticus and M. m. musculus together with the M. m. castaneus samples. In total this study covers a divergence time of roughly two million years by complementing the data with samples from the sister species M. spretus and the recently diverged species M. m. helgolandicus [14]; see Fig. 1. In combination with the short generation time of mice, this constitutes a substantial molecular divergence, which is, for example, larger than the divergence between humans and Hominidae across the same time scale. Figure 4 represents the inferred population sizes for the subspecies M. m. domesticus and the diverged species M. m. helgolandicus, this data set was analyzed with the smc++ software setting the mutation rate to μ = 5 × 10−9 per base pair per generation [78].

Inferred population history for subspecies of the house mouse. Effective population size inference across populations of the house mouse subspecies M. m. domesticus and Mus musculus helgolandicus. SNP data from [14] was filtered to only retain intergenic regions without any feature annotation. For each population a separate smc++ [78] model was created setting the per generation mutation rate to 5 × 10−9 (see Note 1 for a detailed method description)
Population genetic variation in segmental duplications (copy number variation) was systematically studied by Pezer et al. [79]. They found among the most copy-number variable genes three highly conserved genes that encode the splicing factor CWC22, the spindle protein SFI1, and the Holliday junction recognition protein HJURP. These genes showed population-specific expansion patterns that suggested an involvement in local adaptations. Other variable genes were found to encode proteins that are relevant for environmental and behavioral interactions, such as vomeronasal and olfactory receptors, as well as major urinary proteins. In a follow-up study, it was suggested that duplications in the Androgen-binding protein gene region might specifically have contributed to species diversification [80].
Another study also identified the CWC22 region as a region which shows major segmental duplication in the house mouse. It received the genetic name R2d and it was shown that the structural mutation rate appears to depend on the diploid configuration at that locus [81]. By reconstructing the origin and history of copy-number variants (CNVs), the study of Morgan et al. [81] is a nice example how important refined analyses are to disentangle complex genome structures. This is particularly true for genomic regions that are duplicated and are absent from the reference genome, which the author termed the “missing genome” [81].
The sequence and structural diversity of Y chromosomes in natural populations was studied in [82]. The mouse Y chromosome is in comparison to other mammals larger and harbors more annotated genes. The authors could show that CNV on the long arm of both sex chromosomes is highly variable, but sequence diversity as compared to autosomes is low in nonrepetitive regions.
The autosomal AFS of neutral intergenic regions was used to infer demography of all subspecies with the software “∂a ∂i” [83]. All simple models applied predicted effective population sizes that fall inside the range mentioned above (M. m. domesticus: Ne = 1.6 × 105, M. m. musculus: Ne = 1.6 × 105, M. m. domesticus: Ne = 4.2 × 105; [82] but could not explain the reduction of sex chromosome diversity. Important findings are for instance that there is a moderately strong selective sweep on the Y chromosome in the M. m. domesticus population and that positive selection of genes expressed in the male germline might shape the sex chromosomes.
2.2 Brown Rat Genetic Variation
Rats and in particular the species Rattus norvegicus have an effective population size comparable to that of the mice subspecies M. m. domesticus and M. m. musculus. Denium et al. [84] estimated the effective population size to be Ne = 1.24 × 105, based on silent mutations of 12 wild-derived animals. The authors highlight a recent bottleneck in rats (20,000 years ago) based on a ‘PSMC’ [85] analysis (see Chapter 7 for a discussion of MSMC and MSMC2). This bottleneck might be the cause of negative estimates of the rate of adaptive evolution in proteins and noncoding elements. Compared to mice, rats show a larger proportion of mildly deleterious mutations and concordantly a lower rate of highly deleterious mutations [84]. However, the reduction in diversity around exons is comparable to values obtained for mice [64]. Considering the different Ne of mice and rats, Denium et al. [84] estimated linkage disequilibrium (LD) decay to be six to seven times faster in mice than in rats.
As for mice, researchers looked into speciation and introgression events using population genomics. Teng et al. [86] used the Himalayan field rat (Rattus nitidius) as an outgroup, which is geographically restricted to Southeast Asia, to investigate introgression in brown rats sampled in China. With whole-genome data from 44 individuals, the Ne for brown rats and Himalayan field rats was estimated to Ne brown rats = 2.53 × 105 and Ne Himalayan field rats = 5.18 × 105, which reflects a difference of similar order to that of the house mice subspecies M. m. musculus and M. m. castaneus. According to the “PSMC” analysis the sibling species R. norvegicus and R. nitidius diverged ~650 thousand years ago, that is, within a time frame where the mouse divergence is suggested to be at the level of subspecies. The proportion of admixed fragments was estimated to 1.59% with admixture block sizes from 100 kbp to 1.42 Mbp [86]. Among the 346 introgressed regions detected, 92 loci were classified as adaptive. The strongest candidate is located on chromosome 1 overlapping with the “vomeronasal 1 receptor cluster,” a chemical communication protein. As in mice [20], the regions were enriched in biological terms like “chemosensory perception” and “immune response.” Next to regions showing signals of introgression, 352 regions were identified as having undergone a selective sweep based on allele frequency differentiation between populations “XP-CLR” [87] and cross population extended haplotype homozygosity calculations “XP-EHH” [88] which, like introgressed regions, are enriched in proteins involved in immune-response and metabolism.
Zeng et al. [58] extended the publicly available whole-genome sample set of brown rats to a world-wide distribution. With more than 100 individuals the authors investigated the geographic origin and migration paths. In contrast to previous hypothesis that Rattus norvegicus dispersed from northern Asia to Europe, their data supports the southern East Asian dispersal route to Europe [58]. Similar to Teng et al. [86], Zeng et al. [58] consistently identified candidate genes with signatures of positive selection that are associated with the immune-response by comparing European and Chinese populations.
3 Examples of Genes Under Positive Selection
In this section, we discuss three of several examples of genes that have been shown to be involved in adaptation in mice and rats. One prominent example is the evolution of the resistance against warfarin, a rodent pest management poison.
3.1 Rodent Resistance to Anticoagulants: Vkorc1
As vectors for human diseases, rodents have been reduced over half a century by rodenticides. Common compounds of rodenticides target the blood coagulation (e.g., warfarin) and target the vitamin K reductase reaction [89]. Several mutations have been found in house mice and brown rats within the Vkorc1 gene that confer resistance against warfarin [90]. Song et al. [76] suggested that an allele introgressed from the Algerian mouse (Mus spretus) into M. m. domesticus led to anticoagulant resistance. Both species live today in sympatry in south-western Europe. Vkorc1 was subject to adaptive protein evolution in M. spretus since it separated from other Mus lineages and four introgressed polymorphisms could be linked to a strong resistance phenotype [76, 91]. Based on whole-genome data [14], this region shows negative Tajima’s D values within western European mouse populations in contrast to a population from Iran (see Fig. 5a), compatible with recent positive selection acting on it.

Views from the UCSC genome browser showing haplotypes, nucleotide diversity and Tajima’s D values for M. m. domesticus subpopulations. UCSC tracks are shown for (a) Vkorc1 region on chromosome 7 and (b) Xpr1 region on chromosome 1. Tracks were obtained from data published in [14] via a “public track hub” showing haplotypes from SNPs, nucleotide diversity (pi) and Tajima’s D values for the subpopulations from France (DOM-FRA), Germany (DOM-GER) and Iran (DOM-IRA). pi and Tajima’s D was calculated on 10 kbp windows (see Table 1 for web page URL link)
3.2 Pathogen Related Resistance: Xpr1
Next to artificial human-made selection pressure, there exists natural selection caused by pathogens. Hasenkamp et al. [92] have studied the gene Xpr1, coding for the receptor of murine leukemia virus (MLV) They found that the gene has been subject to a recent selective sweep in the population from Iran and that the selected haplotype has adaptively introgressed into a population from France, where it has mixed with existing haplotypes and thus creates a higher average population diversity than in the nonintrogressed population from Germany (see Fig. 5b). It seems that the Xpr1 gene itself is under frequent positive selection and that alleles coping with new virus variants can rather quickly spread into other subpopulations if these are actively dealing with infectious cycles of that virus variant [92].
3.3 Segmental Duplications and Selective Sweeps: R2D2
As mentioned above, R2d is a CNV region on chromosome 2 that was found to cause nonrandom segregation [93]. Didion et al. [93] showed that signatures of selective sweeps obtained via genome-wide scans can be mimicked by “selfish” alleles. Within the 127 kbp genome region of R2d there is one annotated gene, namely Cwc22, which is a spliceosomal protein. Based on haplotype sharing, analysis of almost 400 individuals sampled across Europe revealed that all individuals with an extreme excess of shared identity showed a high copy number of R2d. If only one subpopulation was analyzed, the haplotype sharing methods failed to detect this “selfish” sweep. However, if individuals from different geographically locations were included in the analysis, R2d was identified as a selective sweep. Morgan et al. [81] showed for the same locus that an initial duplication event ~3.5 million years ago led to R2d1 and R2d2 and, therefore, mouse strains containing a single copy must have lost the second one. The authors identified nonallelic gene conversion in R2d1, which were transferred from R2d2 and caused the appearance of deep coalescence among R2d1 sequences [81]. Given both the patterns of concerted evolution, as well as the evolutionary dynamics of the selfish alleles, this could be a case of evolution through “molecular drive” [94].
3.4 The t-Haplotype as Meiotic Drive Element
Meiotic drive elements, or segregation distorters, transmit themselves to over 50% of the progeny of heterozygous individuals. The mouse t-haplotype, located within several inversions on chromosome 17, is a classic example of such a meiotic drive element [1]. Despite a strong driving capacity, t-haplotypes remain at relatively low frequency in natural populations, since homozygous individuals have strongly reduced viability [95]. The population genomics of the t-haplotype was studied in [96] based on the data provided in [14]. They found evidence for an accumulation of nonsynonymous substitutions within the inversions, but also signatures of recombination events that appear to have regenerated coding sequences that had accumulated deleterious mutations. Based on the corresponding transcriptome data in [14] they could show that individuals carrying a t-haplotype display also a change in the testis expression of genes outside of the t-complex.
4 Conclusion
Per sample cost reduction for sequencing has led to an exponential increase in available whole-genome data for model and nonmodel organisms. Being among the longest studied mammals, both house mouse and brown rat have proven to serve as models for studying the processes that shape genome evolution in natural populations, including introgression and positive selection. However, while the public domain is steadily filled with population genomic usable datasets, there is still a gap between studies that predict candidates and studies that functionally validate them. As a consequence, functional studies to prove that genes have a direct impact on fitness in a certain species should be extended. The experimental set up to measure fitness will always depend on the species level and should be imbedded in an environmental context.
5 Note
-
1.
SMC++ [78] analysis is based on 24 Mus musculus domesticus and 3 Mus musculus helgolandicus individuals described earlier [14]. SMC++ version 1.12.1 was used to infer population history for Mus musculus domesticus subpopulations based on a Variant Call Format (VCF) file obtained via the following URL: http://wwwuser.gwdg.de/~evolbio/evolgen/wildmouse/vcf/AllMouse.vcf_90_recalibrated_snps_raw_indels_reheader_PopSorted.vcf.gz. First, bcftools version 1.3.1 [97] was used to filter SNP positions (bcftools filter) aside indel regions (--SnpGap 3), setting genotypes of failed samples to missing values (--set-GTs.) and excluding all sites with either low coverage or low genotype quality (FORMAT/DP<5 | FORMAT/GQ<30). Further, bcftools was used to retain only biallelic SNPs (bcftools view -m2 -M2 -v snps) and SMC++ was used to convert the VCF file to SMC++ format. Only subpopulations indicated above were retained from the input VCF file and only autosomes were extracted individually by additionally masking all exons, regulatory features, simple repeats, and missing sites from the reference mm10 (exons URL: ftp://ftp.ensembl.org/pub/release-90/gtf/mus_musculus/Mus_musculus.GRCm38.90.chr.gtf; regulatory features URL: ftp://ftp.ensembl.org/pub/release-90/regulation/mus_musculus/mus_musculus.GRCm38.Regulatory_Build.regulatory_features.20161111.gff;
simple repeats URL: http://hgdownload.soe.ucsc.edu/goldenPath/mm10/database/simpleRepeat.txt). The per generation mutation rate was set to 5 × 10−9 to fit a size history for each subpopulation based on the extracted autosome data (scm++ estimate 5e-9 chr∗.smc.gz) and plotted with SMC++ (smc++ plot) as shown in Fig. 4.
References
Macholán M, Baird SJ, Munclinger P (eds) (2012) Evolution of the house mouse. Cambridge University Press, Cambridge
Guénet J-L, Bonhomme F (2003) Wild mice: an ever-increasing contribution to a popular mammalian model. Trends Genet 19:24
Phifer-Rixey M, Nachman MW (2015) The Natural History of Model Organisms: insights into mammalian biology from the wild house mouse Mus musculus. eLife Sci 4:e05959
Russell ES (1978) Origins and history of mouse inbred strains: contributions of Clarence Cook Little. Academic, New York
Silver LM (1995) Mouse genetics: concepts and applications. Oxford University Press, Oxford
Yonekawa H, Moriwaki K, Gotoh O et al (1988) Hybrid origin of Japanese mice “Mus musculus molossinus”: evidence from restriction analysis of mitochondrial DNA. Mol Biol Evol 5:63
Prager EM, Orrego C, Sage RD (1998) Genetic variation and phylogeography of central Asian and other house mice, including a major new mitochondrial lineage in Yemen. Genetics 150:835
Yang H, Wang JR, Didion JP et al (2011) Subspecific origin and haplotype diversity in the laboratory mouse. Nat Genet 43:648
Terashima M, Suyanto A, Tsuchiya K et al (2003) Geographic variation of Mus caroli from East and Southeast Asia based on mitochondrial cytochrome b gene sequences. Mammal Study 28:67
Hardouin EA, Orth A, Teschke M et al (2015) Eurasian house mouse (Mus musculus L.) differentiation at microsatellite loci identifies the Iranian plateau as a phylogeographic hotspot. BMC Evol Biol 15:26
Yue F, Cheng Y, Breschi A et al (2014) A comparative encyclopedia of DNA elements in the mouse genome. Nature 515:355
Keane TM, Goodstadt L, Danecek P et al (2011) Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477:289
Morgan AP, Gatti DM, Najarian ML et al (2017) Structural variation shapes the landscape of recombination in mouse. Genetics 206:603
Harr B, Karakoc E, Neme R et al (2016) Genomic resources for wild populations of the house mouse, Mus musculus and its close relative Mus spretus. Sci Data 3:160075
Cox A, Ackert-Bicknell CL, Dumont BL et al (2009) A new standard genetic map for the laboratory mouse. Genetics 182:1335
Liu EY, Morgan AP, Chesler EJ et al (2014) High-resolution sex-specific linkage maps of the mouse reveal polarized distribution of crossovers in male germline. Genetics 197:91
Booker TR, Ness RW, Keightley PD (2017) The recombination landscape in wild house mice inferred using population genomic data. Genetics 207:297
Boursot P, Auffray J-C, Britton-Davidian J et al (1993) The evolution of house mice. Annu Rev Ecol Syst 24:119
White MA, Ané C, Dewey CN et al (2009) Fine-scale phylogenetic discordance across the house mouse genome. PLoS Gen 5:e1000729
Staubach F, Lorenc A, Messer PW et al (2012) Genome patterns of selection and introgression of haplotypes in natural populations of the house mouse (Mus musculus). PLoS Gen 8:e1002891
Liu KJ, Steinberg E, Yozzo A et al (2015) Interspecific introgressive origin of genomic diversity in the house mouse. PNAS 112:196
Teeter KC, Payseur BA, Harris LW et al (2008) Genome-wide patterns of gene flow across a house mouse hybrid zone. Gen Res 18:67
Forejt J (1996) Hybrid sterility in the mouse. Trends Genet 12:412
Turner LM, Schwahn DJ, Harr B (2012) Reduced male fertility is common but highly variable in form and severity in a natural house mouse hybrid zone. Evolution 66:443
Turner LM, Harr B (2014) Genome-wide mapping in a house mouse hybrid zone reveals hybrid sterility loci and Dobzhansky-Muller interactions. eLife Sci 3:e02504
Dumont BL (2017) X-chromosome control of genome-scale recombination rates in house mice. Genetics 205:1649
Searle JB, Jones CS, Gündüz İ et al (2009) Of mice and (Viking?) men: phylogeography of British and Irish house mice. Proc R Soc B 276:201
Jones EP, van der Kooij J, Solheim R et al (2010) Colonization and interactions of two subspecies of house mouse ( Mus musculus) in Norway. Mol Ecol 19:5252
Jones EP, Skirnisson K, McGovern TH et al (2012) Fellow travellers: a concordance of colonization patterns between mice and men in the North Atlantic region. BMC Evol Biol 12:35
Hardouin EA, Chapuis J-L, Stevens MI et al (2010) House mouse colonization patterns on the sub-Antarctic Kerguelen Archipelago suggest singular primary invasions and resilience against re-invasion. BMC Evol Biol 10:325
Gray MM, Wegmann D, Haasl RJ et al (2014) Demographic history of a recent invasion of house mice on the isolated Island of Gough. Mol Ecol 23:1923
Ihle S, Ravaoarimanana I, Thomas M et al (2006) An analysis of signatures of selective sweeps in natural populations of the house mouse. Mol Biol Evol 23:790
Teschke M, Mukabayire O, Wiehe T et al (2008) Identification of selective sweeps in closely related populations of the house mouse based on microsatellite scans. Genetics 180:1537
Cucchi T, Vigne J-D, Auffray J-C (2005) First occurrence of the house mouse (Mus musculus domesticus Schwarz & Schwarz, 1943) in the Western Mediterranean: a zooarchaeological revision of subfossil occurrences. Biol J Linnean Soc 84:429
Bryk J, Somel M, Lorenc A et al (2013) Early gene expression divergence between allopatric populations of the house mouse (Mus musculus domesticus). Ecol Evol 3:558
Lorenc A, Linnenbrink M, Montero I et al (2014) Genetic differentiation of hypothalamus parentally biased transcripts in populations of the house mouse implicate the Prader–Willi syndrome imprinted region as a possible source of behavioral divergence. Mol Biol Evol 31:3240
von Merten S, Hoier S, Pfeifle C et al (2014) A role for ultrasonic vocalisation in social communication and divergence of natural populations of the house mouse (Mus musculus domesticus). PLoS One 9:e97244
Montero I, Teschke M, Tautz D (2013) Paternal imprinting of mating preferences between natural populations of house mice (Mus musculus domesticus). Mol Ecol 22:2549
Linnenbrink M, Teschke M, Montero I et al (2018) Meta-populational demes constitute a reservoir for large MHC allele diversity in wild house mice (Mus musculus). Front Zool 15:15
Morgan AP, Didion JP, Doran AG et al (2016) Whole genome sequence of two wild-derived Mus musculus domesticus inbred strains, LEWES/EiJ and ZALENDE/EiJ, with different diploid numbers. G3 6:4211
Churchill GA, Airey DC, Allayee H et al (2004) The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet 36:1133
de Koning D-J, McIntyre LM (2017) Back to the future: multiparent populations provide the key to unlocking the genetic basis of complex traits. Genetics 206:527
Yang H, Ding Y, Hutchins LN et al (2009) A customized and versatile high-density genotyping array for the mouse. Nat Metods 6:663
Morgan AP, Fu C-P, Kao C-Y et al (2015) The mouse universal genotyping array: from substrains to subspecies. G3 6:263
Doran AG, Wong K, Flint J et al (2016) Deep genome sequencing and variation analysis of 13 inbred mouse strains defines candidate phenotypic alleles, private variation and homozygous truncating mutations. Gen Biol 17:167
Wu S, Wu W, Zhang F et al (2012) Molecular and paleontological evidence for a post-cretaceous origin of rodents. PLoS ONE 7 (10):e46445
Jacob HJ (1999) Functional genomics and rat models. Gen Res 9:1013
Aitman TJ, Critser JK, Cuppen E et al (2008) Progress and prospects in rat genetics: a community view. Nat Genet 40:516
Consortium RGSP (2004) Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428:493
Jensen-Seaman MI, Furey TS, Payseur BA et al (2004) Comparative recombination rates in the rat, mouse, and human genomes. Gen Res 14:528
Lee J, Hong W, Cho M et al (2016) Synteny portal: a web-based application portal for synteny block analysis. Nucleic Acids Res 44:W35
Aplin KP, Suzuki H, Chinen AA et al (2011) Multiple geographic origins of commensalism and complex dispersal history of black rats. PLoS One 6:e26357
Combs M, Puckett EE, Richardson J et al (2018) Spatial population genomics of the brown rat (Rattus norvegicus) in New York City. Mol Ecol 27:83
Hingston M, Poncet S, Passfield K et al (2016) Phylogeography of Rattus norvegicus in the South Atlantic Ocean. Diversity 8:32
Matisoo-Smith E, Robins JH (2004) Origins and dispersals of Pacific peoples: evidence from mtDNA phylogenies of the Pacific rat. PNAS 101:9167
Tollenaere C, Brouat C, Duplantier JM et al (2010) Phylogeography of the introduced species Rattus rattus in the western Indian Ocean, with special emphasis on the colonization history of Madagascar. J Biogeogr 37:398
Puckett EE, Park J, Combs M et al (2016) Global population divergence and admixture of the brown rat (Rattus norvegicus). Proc R Soc B 283:20161762
Zeng L, Ming C, Li Y et al (2018) Out of Southern East Asia of the brown rat revealed by large-scale genome sequencing. Mol Biol Evol 35:149
Ometto L, Glinka S, De Lorenzo D et al (2005) Inferring the effects of demography and selection on Drosophila melanogaster populations from a chromosome-wide scan of DNA variation. Mol Biol Evol 22:2119
Pond SK, Muse SV (2005) Site-to-site variation of synonymous substitution rates. Mol Biol Evol 22:2375
Baines JF, Harr B (2007) Reduced X-linked diversity in derived populations of house mice. Genetics 175:1911
Halligan DL, Oliver F, Eyre-Walker A et al (2010) Evidence for pervasive adaptive protein evolution in wild mice. PLoS Genet 6:e1000825
Phifer-Rixey M, Bonhomme F, Boursot P et al (2012) Adaptive evolution and effective population size in wild house mice. Mol Biol Evol 29:2949
Halligan DL, Kousathanas A, Ness RW et al (2013) Contributions of protein-coding and regulatory change to adaptive molecular evolution in murid rodents. PLoS Genet 9:e1003995
Simecek P, Forejt J, Williams RW et al (2017) High-resolution maps of mouse reference populations. G3 7:3427
Brunschwig H, Levi L, Ben-David E et al (2012) Fine-scale maps of recombination rates and hotspots in the mouse genome. Genetics 191:757
Parvanov ED, Petkov PM, Paigen K (2010) Prdm9 controls activation of mammalian recombination hotspots. Science 327:835
Baudat F, Buard J, Grey C et al (2010) PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327:836
Berg IL, Neumann R, Lam K-WG et al (2010) PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nat Genet 42:859
Buard J, Rivals E, de Segonzac DD et al (2014) Diversity of Prdm9 zinc finger array in wild mice unravels new facets of the evolutionary turnover of this coding minisatellite. PLoS One 9:e85021
Kono H, Tamura M, Osada N et al (2014) Prdm9 polymorphism unveils mouse evolutionary tracks. DNA Res 21:315
Smagulova F, Brick K, Pu Y et al (2016) The evolutionary turnover of recombination hot spots contributes to speciation in mice. Genes Dev 30:266
Liu Q, Guo Y, Li J et al (2012) Steps to ensure accuracy in genotype and SNP calling from Illumina sequencing data. BMC Genomics 13:S8
Sarver BAJ, Keeble S, Cosart T et al (2017) Phylogenomic insights into mouse evolution using a pseudoreference approach. Gen Biol Evol 9:726
Durand EY, Patterson N, Reich D et al (2011) Testing for ancient admixture between closely related populations. Mol Biol Evol 28:2239
Song Y, Endepols S, Klemann N et al (2011) Adaptive introgression of anticoagulant rodent poison resistance by hybridization between Old World mice. Curr Biol 21:1296
Rosenzweig BK, Pease JB, Besansky NJ et al (2016) Powerful methods for detecting introgressed regions from population genomic data. Mol Ecol 25:2387
Terhorst J, Kamm JA, Song YS (2017) Robust and scalable inference of population history from hundreds of unphased whole-genomes. Nat Genet 49:303
Pezer Ž, Harr B, Teschke M et al (2015) Divergence patterns of genic copy number variation in natural populations of the house mouse (Mus musculus domesticus) reveal three conserved genes with major population-specific expansions. Gen Res 25:1114
Pezer Ž, Chung AG, Karn RC et al (2017) Analysis of copy number variation in the Abp gene regions of two house mouse subspecies suggests divergence during the gene family expansions. Gen Biol Evol 9:1393
Morgan AP, Holt JM, McMullan RC et al (2016) The evolutionary fates of a large segmental duplication in mouse. Genetics 204:267
Morgan AP, Pardo-Manuel de Villena F (2017) Sequence and structural diversity of mouse Y chromosomes. Mol Biol Evol 34:3186
Gutenkunst RN, Hernandez RD, Williamson SH et al (2009) Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet 5:e1000695
Deinum EE, Halligan DL, Ness RW et al (2015) Recent evolution in Rattus norvegicus is shaped by declining effective population size. Mol Biol Evol 32:2547
Li H, Durbin R (2011) Inference of human population history from individual whole-genome sequences. Nature 475:493
Teng H, Zhang Y, Shi C et al (2017) Population genomics reveals speciation and introgression between brown norway rats and their sibling species. Mol Biol Evol 34:2214
Chen H, Patterson N, Reich D (2010) Population differentiation as a test for selective sweeps. Gen Res 20:393
Sabeti PC, Varilly P, Fry B et al (2007) Genome-wide detection and characterization of positive selection in human populations. Nature 449:913
Pelz H-J, Rost S, Hünerberg M et al (2005) The genetic basis of resistance to anticoagulants in rodents. Genetics 170:1839
Rost S, Pelz H-J, Menzel S et al (2009) Novel mutations in the VKORC1 gene of wild rats and mice—a response to 50 years of selection pressure by warfarin? BMC Genet 10:4
Goulois J, Hascoët C, Dorani K et al (2017) Study of the efficiency of anticoagulant rodenticides to control Mus musculus domesticus introgressed with Mus spretus Vkorc1. Pest Manag Sci 73:325
Hasenkamp N, Solomon T, Tautz D (2015) Selective sweeps versus introgression-population genetic dynamics of the murine leukemia virus receptor Xpr1 in wild populations of the house mouse (Mus musculus). BMC Evol Biol 15:248
Didion JP, Morgan AP, Yadgary L et al (2016) R2d2 drives selfish sweeps in the house mouse. Mol Biol Evol 33:1381
Dover G (1982) Molecular drive: a cohesive mode of species evolution. Nature 299:111
Lindholm AK, Dyer KA, Firman RC et al (2016) The ecology and evolutionary dynamics of meiotic drive. Trends Ecol Evol 31:315
Kelemen RK, Vicoso B (2018) Complex history and differentiation patterns of the t-haplotype, a mouse meiotic driver. Genetics 208:365
Li H, Handsaker B, Wysoker A et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this protocol

Cite this protocol
Ullrich, K.K., Tautz, D. (2020). Population Genomics of the House Mouse and the Brown Rat. In: Dutheil, J.Y. (eds) Statistical Population Genomics. Methods in Molecular Biology, vol 2090. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-0199-0_18
Download citation
DOI: https://doi.org/10.1007/978-1-0716-0199-0_18
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-0198-3
Online ISBN: 978-1-0716-0199-0
eBook Packages: Springer Protocols