
Abstract
Satellite cells are a population of adult muscle stem cells that play a key role in mediating muscle regeneration. Activation of these quiescent stem cells in response to muscle injury involves modulating expression of multiple developmentally regulated genes, including mediators of the muscle-specific transcription program: Pax7, Myf5, MyoD and myogenin. Here we present evidence suggesting an essential role for the antagonistic Polycomb group and Trithorax group proteins in the epigenetic marking of muscle-specific genes to ensure proper temporal and spatial expression during muscle regeneration. The importance of Polycomb group and Trithorax group proteins in establishing chromatin structure at muscle-specific genes suggests that therapeutic modulation of their activity in satellite cells could represent a viable approach for repairing damaged muscle in muscular dystrophy.
Similar content being viewed by others

Introduction
Skeletal muscle regeneration is mediated by myogenic cell populations that reside in the muscle and behave as adult stem cells [1–3]. In the present article we will focus on satellite cells, which represent the best characterized population of adult muscle stem cells. Satellite cells are a population of mononuclear cells that reside between the muscle fiber and the basal lamina [1, 4].
While satellite cells spend most of their lifetime in a quiescent state, upon muscle damage they can re-enter the cell cycle and either: undergo a symmetric cell division to self-renew and expand the satellite cell population; or undergo an asymmetric cell division that results in the cell on the basal lamina side maintaining the satellite cell identity, while the cell adjacent to the muscle fiber enters the myogenic differentiation program [5, 6]. Cell fate decisions undertaken by the satellite cells upon muscle damage are thought to be regulated through epigenetic mechanisms that modify the structure of chromatin without changing the DNA sequence. These epigenetic changes lead to altered gene expression profiles that contribute to defining cellular identity. Understanding the nature, origin and raison d'être of these epigenetic modifications in the regenerating muscle will be critical to determining how satellite cells can be maintained ex vivo such that this adult stem cell population can be amplified for therapeutic use to treat muscle-wasting diseases.
Polycomb group and Trithorax group proteins in muscle regeneration
Genetic screens for mutations that caused patterning defects in Drosophila led to the identification of Polycomb group (PcG) proteins, which act to repress developmentally regulated gene expression [7, 8]. Further screening to identify genes that rescued the Polycomb phenotype resulted in the identification of an antagonistic group of proteins, termed Trithorax group (TrxG) proteins, which act to establish high levels of transcription from these same developmentally regulated loci. Over the past 5 years, studies in human and mouse embryonic stem cells have suggested that PcG and TrxG families of epigenetic regulators modulate pluripotency and lineage restriction of a number of cell types [9].
While not all PcG and TrxG proteins have been extensively studied, the role of the PcG and TrxG histone methyltransferases in regulating gene expression is well characterized. These histone methyltransferases include the lysine methyltransferase family 6 (KMT6) enzymes Ezh1 and Ezh2 that act as the active subunit of the polycomb repressor complex 2 (PRC2), and the TrxG lysine methyltransferase family 2 (KMT2) members (that is, MLL1, MLL2, MLL3, MLL4, hSET1A, and hSET1B) that act as the active subunit of Ash2L-containing methyltransferase complexes. The KMT6 family of methyltransferases is involved in depositing the transcriptionally repressive mark trimethyl histone H3 at lysine 27 (H3K27me3) on developmentally regulated genes, whereas the transcriptionally permissive mark trimethylation of H3 at lysine 4 (H3K4me3) is mediated by the KMT2 family of methyltransferases. As the repressive H3K27me3 mark is heritably transmitted to daughter cells [10], and is dominant over H3K4me3 [11], the activation of transcription at developmentally regulated genes requires the activity of a third family of enzymatic proteins, which act as H3K27me3 demethylases - namely, lysine demethylase family 6 (KDM6) members UTX and JMJD3 [12–14]. The KMT6 family of enzymes thus establishes gene silencing at developmentally regulated loci, while the KDM6 and KMT2 families of enzymes work together to antagonize this repressive activity and permit gene expression in specific cell types. Reciprocally, KMT6-mediated methylation of histones is used to silence developmentally regulated genes as lineage restriction takes place [15].
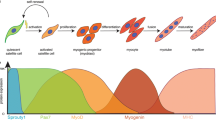
Several developmentally regulated, lineage-specific regulators have been defined in muscle regeneration. These include Pax7 in the quiescent and activated satellite cells, MyoD and Myf5 in the proliferating myoblasts, and myogenin (Myog) in the fusion-competent myocytes that repair the damaged fiber (see Figure 1). While the complete pathway of epigenetics that modulate the temporal and spatial expression of these lineage-specific regulators remains to be elucidated, strong evidence exists showing a role for PcG/TrxG antagonism in modulating the expression of these muscle-specific transcriptional regulators at the different stages of muscle regeneration.

Epigenetic regulation of developmentally regulated genes in satellite-cell-mediated muscle regeneration. Regulation of gene expression by Polycomb group (PcG) and Trithorax group (TrxG) methyltransferase complexes at developmentally regulated loci is depicted. Histone modifications or the presence of PcG/TrxG complexes on the gene highlighted in grey are predicted and have not been formally shown (please see text for rationale on the predictions). (a) Cells undergoing symmetrical cell division will express Pax7 and the genes involved in cell-cycle progression. These genes are predicted to be marked by TrxG-mediated H3K4me3, while the repressed MyoD/Myf5 and Myog genes would be marked by the repressive H3K27me3 mark. In the case of Myf5/MyoD, it will be interesting to determine whether these are bivalently marked genes poised for transcription. (b) During asymmetrical cell division, one of the two cells will go on to become a proliferating myoblast. The proliferating myoblast will express genes involved in cell-cycle progression, as well as Pax7, and Myf5/MyoD. These genes are known to be marked by H3K4me3 in proliferating myoblasts, and in the case of Myf5 it has been shown that this mark is established through the recruitment of TrxG proteins by Pax7. (c) In terminally differentiating cells that will fuse to the damaged fiber, Pax7 is silenced along with genes involved in cell-cycle progression. This repression involves PcG-mediated incorporation of H3K27me3 into the chromatin at these genes. At this time, the Myog gene becomes expressed as MyoD collaborates with Mef2d and Six4 to establish the transcriptionally permissive state of H3K4me3. MRF, muscle regulatory factor.
In quiescent satellite cells, the Pax7 gene is expressed while modulators of cell-cycle progression and muscle-specific transcriptional regulators remain silenced. To date, epigenetic analysis of quiescent satellite cells has been limited by technical challenges. Firstly, the current techniques for explanting muscle tissue and growing progenitors ex vivo are by themselves sufficient to trigger satellite cell differentiation, altering the epigenomic profile. Secondly, the limited number of quiescent satellite cells present on a muscle fiber [16] has to date precluded chromatin immunoprecipitation analysis to determine the role of PcG and TrxG proteins in establishing the epigenetic state of these cells. The existence of histone modifications at developmentally regulated genes during the later stages of myogenesis, however, implies a regulation through the antagonistic functions of PcG and TrxG proteins. For instance, a transition from a transcripionally permissive H3K4me3 mark to a repressive H3K27me3 mark induced by Ezh2 was observed on the Pax7 gene as proliferating myoblasts turn off this important marker of satellite cell identity, and prepare for differentiation [17]. Similarly, genes involved in cell-cycle progression are enriched for the permissive H3K4me3 mark in proliferation conditions [18], and then become enriched for the repressive H3K27me3 mark [19] through a process involving the E2F family of transcription factors and the retinoblastoma protein as the cells exit the cell cycle to terminally differentiate [20]. A role for PRC2-mediated repression at the Myf5 locus in quiescent satellite cells can also be inferred from the observation that this gene becomes marked by the antagonizing TrxG-mediated mark of H3K4me3 in proliferating myoblasts [21]. While these findings are strongly suggestive of a role for TrxG and PcG in maintaining the quiescent state, confirmation of this mechanism will require the use of more sensitive detection techniques such as chromatin immunoprecipitation sequencing for H3K4me3 and H3K27 on satellite cells obtained by laser-capture micro-dissection of fixed muscle tissue.
Upon muscle injury, satellite cells become activated and re-enter the cell cycle. These cells begin to express cell-cycle regulatory genes, which become marked by H3K4me3 [18]. Satellite cells that divide in the planar orientation along the fiber will undergo a symmetrical cell division and give rise to two satellite cells that can reenter the quiescent state [5]. In contrast, cells that divide in the apical-basal plane will undergo asymmetric cell division to give rise to one satellite cell (which returns to the quiescent state), and one proliferating myoblast [5]. The proliferating myoblast is characterized by the expression of Myf5/MyoD [5] as well as genes that regulate cell-cycle progression while the satellite cell marker Pax7 is progressively silenced. As described above, the activation of transcription at Myf5 and genes involved in cell-cycle progression coincides with enrichment of the transcriptionally permissive H3K4me3 mark within their chromatin [21]. In contrast, the Pax7 gene transitions from a transcriptionally permissive state of H3K4me3 to a repressive state of H3K27me3 as the cell proceeds through differentiation [17].
The formation of multinucleated myotubes requires the downregulation of Pax7, Myf5, and cell-cycle regulatory genes, and the activation of Myog. Expression of the Myog gene coincides with the removal of the repressive H3K27me3 mark [22, 23] and the appearance of the transcriptionally permissive H3K4me3 mark within the 5' end of the gene [23, 24]. Coincident with the terminal differentiation, myoblasts exit the cell cycle as regulators of this process are silenced through incorporation of the H3K27me3 modification into chromatin marking their respective genes [19]. While our knowledge of epigenetic marking of chromatin in proliferating and differentiating myoblasts is currently restricted to a limited number of genes, advances in high-throughput sequencing should soon provide the epigenetic status for the entire muscle transcriptome at different stages of muscle regeneration.
Targeting Polycomb group and Trithorax group proteins to muscle-specific genes
The H3K27me3 mark is established by proteins of the KMT6 (Ezh1 and Ezh2) family of PcG proteins. In 2004, Caretti and colleagues were the first to demonstrate the involvement of PcG proteins in myogenic gene silencing [25]. They showed that the expression of two terminal muscle differentiation genes, Myh10 (myosin, heavy polypeptide 10, nonmuscle) and Ckm (muscle creatine kinase), are silenced via PcG repression in proliferating myoblasts, and that this silencing is lifted upon differentiation. An interesting aspect of these findings is that the recruitment of Ezh2 (KMT6B), the catalytic subunit of PRC2, to its target genes in precursor cells is mediated by the sequence-specific transcription factor YY1. The mechanism that allows YY1-mediated targeting of Ezh2 to these muscle-specific genes is intriguing, as both proteins are ubiquitously expressed. Furthermore, as hundreds of genes are coordinately induced upon myogenic differentiation [26–29], it will be important to identify those genes that are silenced by PRC2 in precursor cells, and to determine whether YY1 mediates KMT6 recruitment in all such instances.
Studies of the Ezh2-mediated repression of Notch1 expression in TNFα-treated satellite cells did not examine the mechanism of PRC2 recruitment [30]. Studies of the Pax7 gene, however, showed that YY1 also mediates the recruitment of Ezh2 to the transcriptional regulatory region of this marker of satellite cell identity to silence its expression in proliferating myoblasts [17]. This recruitment of Ezh2 to the Pax7 gene is modulated through mitogen-activated protein kinase (MAPK) signaling, where Ezh2 is phosphorylated by p38 MAPK to permit its interaction with the enhancer bound YY1. In contrast, recruitment of Ezh2 by YY1 to the Myh10 gene is not modulated by p38 MAPK signaling [17]. Furthermore, Ezh2 departs the Myh10 and CKm genes when p38 MAPK signaling is activated upon terminal differentiation [25]. An important question raised by these studies, therefore, is how p38 MAPK can stimulate YY1 and Ezh2 recruitment to a given locus (Pax7, silenced in differentiated cells) but not to another (Myh10, silenced in proliferating cells). Possibly the composition of multiprotein complexes at the regulatory region of these genes is not entirely defined, and p38-mediated stimulation depends on additional unidentified factors that might be differentially recruited to these loci.
Evidence suggests a role for additional factors in targeting Ezh2 to specific loci since high-throughput studies in embryonic stem cells show that the genomic binding profiles of PRC2 and YY1 do not overlap [31]. In these pluripotent cells, the histone demethylase Jarid2 has been shown to mediate recruitment of PRC2 (Ezh2) to specific genes [32–34]. Jarid2 could also be involved in targeting Ezh2 to muscle-specific genes as it is expressed in satellite cells before being downregulated twofold during differentiation (FJD and AB, unpublished observations based on published datasets [35, 36]) Determining the relative role of these two pathways to the establishment of PRC2-mediated transcription repression during muscle regeneration will be of future interest, and will require satellite cell-specific knockout/knockdown of YY1 and/or Jarid2.
An important implication of the findings on gene repression mediated by PRC2 is that this silencing of muscle development genes must be lifted for differentiation to occur. Removal of the H3K27me3 mark is mediated by KDM6 family members UTX (KDM6A) and JMJD3 (KDM6B) [12–14]. Interestingly, the demethylase UTX can associate with TrxG proteins, which antagonize PcG function by marking genes for activation [37]. To date, the recruitment of KDM6 family members to muscle-specific genes has only been examined in terminally differentiating myoblasts. In these cells, UTX is first recruited to the promoter region of the Myog gene, where it then associates with the elongating RNA polymerase II to demethylate a region extending over the entire length of the gene [22, 23]. Interestingly, recruitment of the UTX demethylase to the Myog locus is mediated by the homeodomain transcription factor Six4 [22, 23]. Importantly, Six1 and Six4 factors are involved in upregulating the expression of Pax3, MyoD, Myf5 and Myog [38–40] and of fast-twitch muscle-function genes [41] during muscle development. Furthermore, Six1 and Six4 are essential for terminal differentiation in adult myoblasts [38, 42] and they function in part by cooperating with the muscle regulatory factors (MRFs) MyoD and Myog in activating target gene transcription [42]. Genome-wide profiling of Six1 binding in myoblasts revealed a strong correlation between Six binding and target gene activation during differentiation [42]. This observation suggests that Six factors may have a global function in recruiting UTX complexes to developmentally regulated genes during myoblast differentiation.
Following removal of the repressive H3K27me3 modification, developmentally regulated genes become enriched for the transcriptionally permissive H3K4me3 mark to establish high levels of gene expression. Indeed, studies in proliferating myoblasts have shown that Pax7 is responsible for recruitment of the TrxG complex Ash2L into the Myf5 locus to mark the gene by H3K4me3 [21]. In terminally differentiating myoblasts, targeting of the Ash2L complex to the Myog promoter is mediated by the MADS-domain transcription factor Mef2d [23, 24]. Several different transactivators can thus clearly recruit Ash2L complexes to developmentally regulated genes to mediate the marking of chromatin by H3K4me3 during muscle regeneration. Importantly, the recruitment of Ash2L to the Myog gene has been shown to be modulated by p38 MAPK signaling through a direct phosphorylation of Mef2d [23, 24]. This ability to modulate the recruitment of Ash2L to the Myog promoter through inhibition of p38 MAPK signaling suggests a possible mechanism to regulate gene expression therapeutically.
DNA methylation in muscle regeneration
In addition to the repressive H3K27me3 mark mediated by PRC2/Ezh2 [10], methylation of CpG dinucleotides (5-methylcytosine) within a gene regulatory region can be heritably transmitted to daughter cells to block transcription [43, 44]. The importance of this methylation of DNA in myogenesis has been established from early studies showing that treatment of fibroblast with the inhibitor of DNA methylation (5-azacytidine) caused cells to differentiate towards the muscle lineage [45]. Subsequently, the Weintraub group used a genomic library obtained from 5-azacytidine-treated fibroblasts to clone the master regulator of muscle gene expression, MyoD [46].
Reciprocally, more recent studies have shown that treatment of C2C12 cells with an inhibitor of DNA methylation (zebularine) caused the cells to differentiate into a smooth muscle lineage [47]. This observation provides strong evidence that DNA methylation plays an important role in repressing factors involved in establishing alternate cell fates. Interestingly, the two repressive marks of CpG methylation and H3K27me3 have been shown to co-exist at specific genes in cells of restricted/limited potency [9, 48]. Moreover, co-existence of methylated H3K27 and CpG dinucleotides within transcriptional regulatory regions is not coincidental because Ezh2 has been shown to target the de novo DNA methyltransferase enzymes DNMT3a and DNMT3b to specific genes [49]. This combination of epigenetic marks is proposed to provide a more stable repression of transcription at genes coding for either mediators of pluripotency or determination factors that are specific to alternate cell lineages [9, 48]. Not all Ezh2 target genes, however, are marked by methylated CpG dinucleotides. Instead, genes with nonmethylated CpG dinucleotides are repressed through a bivalent chromatin state of nucleosomes doubly marked by methylated H3K4 and H3K27 that remain poised for activation [50]. The presence of methylated H3K4 within the nucleosome is proposed to block the recruitment of DNTM3a/DNTM3b to chromatin [51, 52] and to maintain the ability of these PcG marked genes to be activated later in lineage commitment. As a general rule, therefore, genes no longer required for lineage progression would be targeted for stable repression by a combination of H3K27me3 and CpG methylation, while genes required for further lineage progression would be bivalently marked by H3K4me3 and H3K27me3.
The importance of bivalent chromatin domains in regulating expression of muscle-specific genes remains to be established. The finding that methyl-CpG binding proteins mediate reorganization of chromatin during terminal myogenesis, however, confirms an essential role for this epigenetic mark in muscle regeneration [53]. Recent studies have shown the involvement of Ezh2 and DNMT3b in establishing repression at the Notch-1 promoter during satellite cell activation [30]. Indeed, down-regulation of Notch-1 occurs in an Ezh2-dependent manner and results in accumulation of the repressive H3K27me3 mark as well as recruitment of DNMT3b to mediate DNA methylation within the promoter region of this gene [30]. It remains to be determined whether Ezh2 or DNMT3b plays a role in downregulating other mediators of satellite cell function such as Pax7 in proliferating myoblasts. Overlaying genome-wide DNA methylation (obtained using either bisulfite sequencing or MeDIP) and H3K27me3 patterns (obtained using chromatin immunoprecipitation) in satellite cells will permit a full appreciation of the extent to which these complementary epigenetic marks modulate the myogenic gene expression program.
The function of Pax7 in satellite cells
Mice deficient in Pax7 expression are characterized by low-weight, skeletal muscle of small caliber and by null or very low numbers of satellite cells [54]. Surprisingly, it was recently reported in adult mice that myogenic regeneration occurs in the absence of Pax7 (and/or Pax3), suggesting that the homeodomain transcription factor would only be essential for growth and regeneration during the juvenile period [55]. However, considering the role of Pax7 in establishing the H3K4me3 marks at muscle regulatory genes such as Myf5 [21], an important role for Pax7 in the epigenetic modification of histones in adult satellite cells is likely to exist.
In light of the fact that satellite cells can regenerate damaged muscle in the absence of Pax7, we propose that this transcriptional regulator could act prior to the onset of adulthood to establish stable epigenetic modification of chromatin whose influence on gene expression continues after its expression has been ablated. This idea of epigenetic marking of chromatin to maintain cellular memory is supported by studies in Myf5-Cre/ROSA26-YFP mice, where it was shown that YFP+ satellite cells (which had previously expressed Myf5 and represent 90% of the satellite cell population) turn on expression of the endogenous Myf5 gene with faster kinetics than YFP- satellite cells [5]. Consistent with this, we propose a model in which Pax7-dependent epigenetic marks set up during the juvenile growth phase would establish satellite cell identity permanently. As these epigenetic marks could persist over successive cycles of proliferation/quiescence in satellite cells, such a scenario would make Pax7 expression dispensable in adult cells. However, the identification of Pax7-dependent marks in juvenile satellite cells, and of Pax7-bound genomic loci, will be required to verify this hypothesis formally.
How might Pax7 act to epigenetically mark genes of the muscle transcriptome? Pax7 could participate in the establishment of a bivalent state at muscle genes (such as Myf5) in quiescent satellite cells where the H3K4me3 mark co-exists with the repressive H3K27me3 mark to poise them for activation [50]. In such a case, activation of the muscle genes would no longer require Pax7 in the adult satellite cells as the chromatin would already be marked by H3K4me3 in juvenile satellite cells. This mark would persist through rounds of proliferation/quiescence, but would be counteracted at specific genes (depending on cellular context) by the regulated removal of the H3K27me3 mark. Recruitment of a KDM6 family histone demethylase specific to the gene by an additional transcription factor such as Six4 would thus be sufficient to establish expression of muscle development genes.
Alternatively, Pax7 could epigenetically mark genes of the muscle transcriptome through the introduction of variant histones within its target genes. Previous studies have shown that Pax7 can interact with HIRA, a chaperone specific for the variant histone H3.3 [56]. Because nucleosomes enriched in histone H3.3 are generally found at the start sites of transcribed genes [57] and are involved in epigenetic memory [58], the Pax7-HIRA interaction could prevent the permanent silencing of its target genes by marking them with H3.3. Indeed, the MyoD gene is marked by H3.3 in proliferating myoblasts [59]. Interestingly, this mark is stable enough to permit expression of MyoD in Xenopus oocytes that have undergone nuclear transfer using a nucleus from a muscle donor cell [58]. These two scenarios, which are not mutually exclusive, could explain how Pax7 would establish the inheritance of an active chromatin state at important loci in juvenile satellite cells, prior to their transcriptional activation.
Modulating epigenetics as a therapeutic approach to muscular dystrophy
The importance of the epigenetic pathways in modulation of tissue-specific gene expression makes them excellent candidate targets for disease interventions. Several drugs that attempt to modify epigenetic mechanisms are currently undergoing clinical trial [60, 61]. These include histone deacetylase inhibitors [61], histone methyltransferase inhibitors [62], as well as the inhibitor of DNA methylation 5-azacytidine [63].
In the case of muscular dystrophy, histone deacetylase inhibitors are currently being examined using the mdx mouse model for their ability to improve the dystrophic phenotype [64]. Here it is believed that deacetylase inhibitors prevent the effects of disrupted nitric oxide signaling on acetylation at chromatin within the diseased muscle [65]. The effects of prolonged treatment with drugs that inhibit these ubiquitously required chromatin-modifying enzymes, however, are of potential concern. As an alternative or complement to this strategy, the identification of small molecules that promote or disrupt the specific protein-protein interactions required for targeting the epigenetic enzymes to determined loci within the genome could have a similar benefit without the side effect of modifying gene expression in other cell types. Along this line of thought, a cell-permeable small molecule that inhibits the protein-protein interaction between bromodomain-containing protein BRD4 and histones H3-acetylated at lysine 14 has recently been reported [66]. The broad-reaching effects of blocking this interaction, however, maintain the same caveats described above for blocking the enzymatic activity of ubiquitously expressed epigenetic proteins.
Future screens should be directed at disrupting the interactions between the PcG and TrxG proteins and the transcriptional regulators that target these enzymes to muscle-specific genes. As many of the PcG and TrxG activities are present in multiprotein complexes, the screening of molecules to disrupt this targeted recruitment to muscle-specific genes will first require the delineation of specific subunits that mediate direct interactions with the transcriptional regulator of interest. The use of small molecules to disrupt interactions between transcriptional regulators and PcG and Trx proteins will thus require extensive research before they can be developed to treat muscular dystrophy.
An alternative approach to targeting PcG and TrxG activities to specific genes is the use of artificial zinc-finger transcription factors [67]. This technique has recently been used to target the VP16 transactivation domain to a 9-base-pair sequence within the utrophin promoter, allowing for an upregulation of expression from the endogenous gene in the mdx mouse [68]. In this case, a three-zinc-finger array fused to VP16 was expressed in transgenic animals using the muscle-specific myosin light-chain promoter. While a 9-base-pair target sequence is not sufficient to ensure a single genomic targeting event, artificial activators have been generated containing six zinc fingers that permit the targeting of a transactivation domain to an 18-base-pair sequence of the γ-globin gene that is unique in the genome [69]. As an alternative to the VP16 fusion with the gene-specific zinc finger array, an enzyme such as Ezh2, UTX, or MLL1 could be fused to these artificial DNA binding domains. In this way, TrxG or PcG fusion proteins could be targeted to individual loci within the genome to mediate silencing or activation of specific genes.
While utrophin is a therapeutically important gene for treatment of muscular dystrophy, an alternative target has been suggested by the recent finding that the discrepancy between the mild dystrophic phenotype observed in mdx mice and the severe phenotype observed in humans can be explained through the inactivation of the telomerase in the latter [70]. An artificial transcriptional zinc-finger-mediated upregulation of telomerase activity through epigenetic mechanisms specifically in satellite cells could perhaps lead to increased self-renewal such that the stem cells do not become depleted as the need for repair continues over the lifetime of the patient. A similar approach has recently been explored to repress expression of telomerase in transformed cells using artificial zinc fingers fused to the transcriptional repressor domain of KRAB [71]. Epigenetic enzymes could thus represent a viable target for future gene therapies to permit muscle repair in muscular dystrophy patients. However, current limitations associated with gene therapy remain - we must ensure that these zinc finger proteins are targeted to muscle cells efficiently while also ensuring that they do not activate muscle genes in other cell types.
Conclusions
There is little doubt that the incredible ability of certain structural features of chromatin to be perpetuated over several cell divisions is at play in controlling the fate of adult muscle stem cells. The elucidation of epigenetic mechanisms regulating the function of satellite cell function is still just beginning, but significant progress is being made at an exponential pace, thanks in part to our increasing knowledge of how these molecular pathways are laid out in embryonic stem cells. Moreover, technical advances are constantly emerging, speeding up our study of the inner workings of the epigenetic control machinery and helping with the design of new therapeutic approaches based on this knowledge. While most muscular illnesses are not epigenetic diseases per se, we can envision a near future where epigenetic therapies will be part of a successful treatment regimen for dystrophic patients.
Note
This article is part of a review series on Epigenetics and regulation. Other articles in the series can be found online at http://stemcellres.com/series/epigenetics
Abbreviations
- KDM6:
-
lysine demethylase family 6
- KMT2:
-
lysine methyltransferase family 2
- KMT6:
-
lysine methyltransferase family 6
- MAPK:
-
mitogen-activated protein kinase
- MRF:
-
muscle regulatory factor
- Myog:
-
myogenin
- PcG:
-
Polycomb group
- PRC2:
-
polycomb repressor complex 2
- TNF:
-
tumor necrosis factor
- TrxG:
-
Trithorax group.
References
Mauro A: Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol. 1961, 9: 493-495. 10.1083/jcb.9.2.493.
Peault B, Rudnicki M, Torrente Y, Cossu G, Tremblay JP, Partridge T, Gussoni E, Kunkel LM, Huard J: Stem and progenitor cells in skeletal muscle development, maintenance, and therapy. Mol Ther. 2007, 15: 867-877. 10.1038/mt.sj.6300145.
Mitchell KJ, Pannerec A, Cadot B, Parlakian A, Besson V, Gomes ER, Marazzi G, Sassoon DA: Identification and characterization of a non-satellite cell muscle resident progenitor during postnatal development. Nat Cell Biol. 2010, 12: 257-266.
Charge SB, Rudnicki MA: Cellular and molecular regulation of muscle regeneration. Physiol Rev. 2004, 84: 209-238. 10.1152/physrev.00019.2003.
Kuang S, Kuroda K, Le Grand F, Rudnicki MA: Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 2007, 129: 999-1010. 10.1016/j.cell.2007.03.044.
Le Grand F, Jones AE, Seale V, Scime A, Rudnicki MA: Wnt7a activates the planar cell polarity pathway to drive the symmetric expansion of satellite stem cells. Cell Stem Cell. 2009, 4: 535-547. 10.1016/j.stem.2009.03.013.
Sawarkar R, Paro R: Interpretation of developmental signaling at chromatin: the Polycomb perspective. Dev Cell. 2010, 19: 651-661. 10.1016/j.devcel.2010.10.012.
Morey L, Helin K: Polycomb group protein-mediated repression of transcription. Trends Biochem Sci. 2010, 35: 323-332. 10.1016/j.tibs.2010.02.009.
Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, Bibel M, Schubeler D: Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell. 2008, 30: 755-766. 10.1016/j.molcel.2008.05.007.
Hansen KH, Bracken AP, Pasini D, Dietrich N, Gehani SS, Monrad A, Rappsilber J, Lerdrup M, Helin K: A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol. 2008, 10: 1291-1300. 10.1038/ncb1787.
Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K: High-resolution profiling of histone methylations in the human genome. Cell. 2007, 129: 823-837. 10.1016/j.cell.2007.05.009.
Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE, Helin K: UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007, 449: 731-734. 10.1038/nature06145.
De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G: The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007, 130: 1083-1094. 10.1016/j.cell.2007.08.019.
Lan F, Bayliss PE, Rinn JL, Whetstine JR, Wang JK, Chen S, Iwase S, Alpatov R, Issaeva I, Canaani E, Roberts TM, Chang HY, Shi Y: A histone H3 lysine 27 demethylase regulates animal posterior development. Nature. 2007, 449: 689-694. 10.1038/nature06192.
Schuettengruber B, Cavalli G: Recruitment of polycomb group complexes and their role in the dynamic regulation of cell fate choice. Development. 2009, 136: 3531-3542. 10.1242/dev.033902.
Reimann J, Brimah K, Schroder R, Wernig A, Beauchamp JR, Partridge TA: Pax7 distribution in human skeletal muscle biopsies and myogenic tissue cultures. Cell Tissue Res. 2004, 315: 233-242. 10.1007/s00441-003-0833-y.
Palacios D, Mozzetta C, Consalvi S, Caretti G, Saccone V, Proserpio V, Marquez VE, Valente S, Mai A, Forcales SV, Sartorelli V, Puri PL: TNF/p38α/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell. 2010, 7: 455-469. 10.1016/j.stem.2010.08.013.
Sebastian S, Sreenivas P, Sambasivan R, Cheedipudi S, Kandalla P, Pavlath GK, Dhawan J: MLL5, a trithorax homolog, indirectly regulates H3K4 methylation, represses cyclin A2 expression, and promotes myogenic differentiation. Proc Natl Acad Sci USA. 2009, 106: 4719-4724. 10.1073/pnas.0807136106.
Blais A, van Oevelen CJ, Margueron R, Acosta-Alvear D, Dynlacht BD: Retinoblastoma tumor suppressor protein-dependent methylation of histone H3 lysine 27 is associated with irreversible cell cycle exit. J Cell Biol. 2007, 179: 1399-1412. 10.1083/jcb.200705051.
Blais A, Dynlacht BD: E2F-associated chromatin modifiers and cell cycle control. Curr Opin Cell Biol. 2007, 19: 658-662. 10.1016/j.ceb.2007.10.003.
McKinnell IW, Ishibashi J, Le Grand F, Punch VG, Addicks GC, Greenblatt JF, Dilworth FJ, Rudnicki MA: Pax7 activates myogenic genes by recruitment of a histone methyltransferase complex. Nat Cell Biol. 2008, 10: 77-84. 10.1038/ncb1671.
Seenundun S, Rampalli S, Liu QC, Aziz A, Palii C, Hong S, Blais A, Brand M, Ge K, Dilworth FJ: UTX mediates demethylation of H3K27me3 at musclespecific genes during myogenesis. Embo J. 2010, 29: 1401-1411. 10.1038/emboj.2010.37.
Aziz A, Liu QC, Dilworth FJ: Regulating a master regulator: establishing tissue-specific gene expression in skeletal muscle. Epigenetics. 2010, 5: 691-695. 10.4161/epi.5.8.13045.
Rampalli S, Li L, Mak E, Ge K, Brand M, Tapscott SJ, Dilworth FJ: p38 MAPK signaling regulates recruitment of Ash2L-containing methyltransferase complexes to specific genes during differentiation. Nat Struct Mol Biol. 2007, 14: 1150-1156. 10.1038/nsmb1316.
Caretti G, Di Padova M, Micales B, Lyons GE, Sartorelli V: The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev. 2004, 18: 2627-2638. 10.1101/gad.1241904.
Moran JL, Li Y, Hill AA, Mounts WM, Miller CP: Gene expression changes during mouse skeletal myoblast differentiation revealed by transcriptional profiling. Physiol Genomics. 2002, 10: 103-111.
Blais A, Tsikitis M, Acosta-Alvear D, Sharan R, Kluger Y, Dynlacht BD: An initial blueprint for myogenic differentiation. Genes Dev. 2005, 19: 553-569. 10.1101/gad.1281105.
Cao Y, Yao Z, Sarkar D, Lawrence M, Sanchez GJ, Parker MH, MacQuarrie KL, Davison J, Morgan MT, Ruzzo WL, Gentleman RC, Tapscott SJ: Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev Cell. 2010, 18: 662-674. 10.1016/j.devcel.2010.02.014.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L: Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010, 28: 511-515. 10.1038/nbt.1621.
Acharyya S, Sharma SM, Cheng AS, Ladner KJ, He W, Kline W, Wang H, Ostrowski MC, Huang TH, Guttridge DC: TNF inhibits Notch-1 in skeletal muscle cells by Ezh2 and DNA methylation mediated repression: implications in duchenne muscular dystrophy. PLoS One. 2010, 5: e12479-10.1371/journal.pone.0012479.
Squazzo SL, O'Geen H, Komashko VM, Krig SR, Jin VX, Jang SW, Margueron R, Reinberg D, Green R, Farnham PJ: Suz12 binds to silenced regions of the genome in a cell-type-specific manner. Genome Res. 2006, 16: 890-900. 10.1101/gr.5306606.
Peng JC, Valouev A, Swigut T, Zhang J, Zhao Y, Sidow A, Wysocka J: Jarid2/Jumonji coordinates control of PRC2 enzymatic activity and target gene occupancy in pluripotent cells. Cell. 2009, 139: 1290-1302. 10.1016/j.cell.2009.12.002.
Pasini D, Cloos PA, Walfridsson J, Olsson L, Bukowski JP, Johansen JV, Bak M, Tommerup N, Rappsilber J, Helin K: JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature. 2010, 464: 306-310. 10.1038/nature08788.
Li G, Margueron R, Ku M, Chambon P, Bernstein BE, Reinberg D: Jarid2 and PRC2, partners in regulating gene expression. Genes Dev. 2010, 24: 368-380. 10.1101/gad.1886410.
Fukada S, Uezumi A, Ikemoto M, Masuda S, Segawa M, Tanimura N, Yamamoto H, Miyagoe-Suzuki Y, Takeda S: Molecular signature of quiescent satellite cells in adult skeletal muscle. Stem Cells. 2007, 25: 2448-2459. 10.1634/stemcells.2007-0019.
Pallafacchina G, Francois S, Regnault B, Czarny B, Dive V, Cumano A, Montarras D, Buckingham M: An adult tissue-specific stem cell in its niche: a gene profiling analysis of in vivo quiescent and activated muscle satellite cells. Stem Cell Res. 2010, 4: 77-91. 10.1016/j.scr.2009.10.003.
Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, Di Croce L, Shiekhattar R: Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007, 318: 447-450. 10.1126/science.1149042.
Grifone R, Demignon J, Houbron C, Souil E, Niro C, Seller MJ, Hamard G, Maire P: Six1 and Six4 homeoproteins are required for Pax3 and Mrf expression during myogenesis in the mouse embryo. Development. 2005, 132: 2235-2249. 10.1242/dev.01773.
Giordani J, Bajard L, Demignon J, Daubas P, Buckingham M, Maire P: Six proteins regulate the activation of Myf5 expression in embryonic mouse limbs. Proc Natl Acad Sci USA. 2007, 104: 11310-11315. 10.1073/pnas.0611299104.
Spitz F, Demignon J, Porteu A, Kahn A, Concordet JP, Daegelen D, Maire P: Expression of myogenin during embryogenesis is controlled by Six/sine oculis homeoproteins through a conserved MEF3 binding site. Proc Natl Acad Sci USA. 1998, 95: 14220-14225. 10.1073/pnas.95.24.14220.
Niro C, Demignon J, Vincent S, Liu Y, Giordani J, Sgarioto N, Favier M, Guillet-Deniau I, Blais A, Maire P: Six1 and Six4 gene expression is necessary to activate the fast-type muscle gene program in the mouse primary myotome. Dev Biol. 2010, 338: 168-182. 10.1016/j.ydbio.2009.11.031.
Liu Y, Chu A, Chakroun I, Islam U, Blais A: Cooperation between myogenic regulatory factors and SIX family transcription factors is important for myoblast differentiation. Nucleic Acids Res. 2010, 38: 6857-6871. 10.1093/nar/gkq585.
Suzuki MM, Bird A: DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008, 9: 465-476. 10.1038/nrg2341.
Cedar H, Bergman Y: Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009, 10: 295-304. 10.1038/nrg2540.
Taylor SM, Jones PA: Multiple new phenotypes induced in 10T1/2 and 3T3 cells treated with 5-azacytidine. Cell. 1979, 17: 771-779. 10.1016/0092-8674(79)90317-9.
Lassar AB, Paterson BM, Weintraub H: Transfection of a DNA locus that mediates the conversion of 10T1/2 fibroblasts to myoblasts. Cell. 1986, 47: 649-656. 10.1016/0092-8674(86)90507-6.
Lee WJ, Kim HJ: Inhibition of DNA methylation is involved in transdifferentiation of myoblasts into smooth muscle cells. Mol Cells. 2007, 24: 441-444.
Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, Gnirke A, Jaenisch R, Lander ES: Genomescale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008, 454: 766-770.
Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, Bollen M, Esteller M, Di Croce L, de Launoit Y, Fuks F: The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006, 439: 871-874. 10.1038/nature04431.
Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES: A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006, 125: 315-326. 10.1016/j.cell.2006.02.041.
Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, Cheng X, Bestor TH: DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007, 448: 714-717. 10.1038/nature05987.
Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X: Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature. 2007, 449: 248-251. 10.1038/nature06146.
Brero A, Easwaran HP, Nowak D, Grunewald I, Cremer T, Leonhardt H, Cardoso MC: Methyl CpG-binding proteins induce large-scale chromatin reorganization during terminal differentiation. J Cell Biol. 2005, 169: 733-743. 10.1083/jcb.200502062.
Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, Rudnicki MA: Pax7 is required for the specification of myogenic satellite cells. Cell. 2000, 102: 777-786. 10.1016/S0092-8674(00)00066-0.
Lepper C, Conway SJ, Fan CM: Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature. 2009, 460: 627-631. 10.1038/nature08209.
Magnaghi P, Roberts C, Lorain S, Lipinski M, Scambler PJ: HIRA, a mammalian homologue of Saccharomyces cerevisiae transcriptional co-repressors, interacts with Pax3. Nat Genet. 1998, 20: 74-77. 10.1038/1739.
Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, Wen D, Chapgier A, DeKelver RC, Miller JC, Lee YL, Boydston EA, Holmes MC, Gregory PD, Greally JM, Rafii S, Yang C, Scambler PJ, Garrick D, Gibbons RJ, Higgs DR, Cristea IM, Urnov FD, Zheng D, Allis CD: Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010, 140: 678-691. 10.1016/j.cell.2010.01.003.
Ng RK, Gurdon JB: Epigenetic memory of an active gene state depends on histone H3.3 incorporation into chromatin in the absence of transcription. Nat Cell Biol. 2008, 10: 102-109. 10.1038/ncb1674.
Yang JH, Song Y, Seol JH, Park JY, Yang YJ, Han JW, Youn HD, Cho EJ: Myogenic transcriptional activation of MyoD mediated by replication-independent histone deposition. Proc Natl Acad Sci USA. 2011, 108: 85-90. 10.1073/pnas.1009830108.
Jain N, Rossi A, Garcia-Manero G: Epigenetic therapy of leukemia: an update. Int J Biochem Cell Biol. 2009, 41: 72-80. 10.1016/j.biocel.2008.10.006.
Colussi C, Illi B, Rosati J, Spallotta F, Farsetti A, Grasselli A, Mai A, Capogrossi MC, Gaetano C: Histone deacetylase inhibitors: keeping momentum for neuromuscular and cardiovascular diseases treatment. Pharmacol Res. 2010, 62: 3-10. 10.1016/j.phrs.2010.02.014.
Biancotto C, Frige G, Minucci S: Histone modification therapy of cancer. Adv Genet. 2010, 70: 341-386. full_text.
Gotze K, Platzbecker U, Giagounidis A, Haase D, Lubbert M, Aul C, Ganser A, Germing U, Hofmann WK: Azacitidine for treatment of patients with myelodysplastic syndromes (MDS): practical recommendations of the German MDS Study Group. Ann Hematol. 2010, 89: 841-850. 10.1007/s00277-010-1015-0.
Minetti GC, Colussi C, Adami R, Serra C, Mozzetta C, Parente V, Fortuni S, Straino S, Sampaolesi M, Di Padova M, Illi B, Gallinari P, Steinkuhler C, Capogrossi MC, Sartorelli V, Bottinelli R, Gaetano C, Puri PL: Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat Med. 2006, 12: 1147-1150. 10.1038/nm1479.
Colussi C, Gurtner A, Rosati J, Illi B, Ragone G, Piaggio G, Moggio M, Lamperti C, D'Angelo G, Clementi E, Minetti G, Mozzetta C, Antonini A, Capogrossi MC, Puri PL, Gaetano C: Nitric oxide deficiency determines global chromatin changes in Duchenne muscular dystrophy. FASEB J. 2009, 23: 2131-2141. 10.1096/fj.08-115618.
Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE: Selective inhibition of BET bromodomains. Nature. 2010, 468: 1067-1073. 10.1038/nature09504.
Beltran AS, Sun X, Lizardi PM, Blancafort P: Reprogramming epigenetic silencing: artificial transcription factors synergize with chromatin remodeling drugs to reactivate the tumor suppressor mammary serine protease inhibitor. Mol Cancer Ther. 2008, 7: 1080-1090. 10.1158/1535-7163.MCT-07-0526.
Di Certo MG, Corbi N, Strimpakos G, Onori A, Luvisetto S, Severini C, Guglielmotti A, Batassa EM, Pisani C, Floridi A, Benassi B, Fanciulli M, Magrelli A, Mattei E, Passananti C: The artificial gene Jazz, a transcriptional regulator of utrophin, corrects the dystrophic pathology in mdx mice. Hum Mol Genet. 2010, 19: 752-760. 10.1093/hmg/ddp539.
Wilber A, Tschulena U, Hargrove PW, Kim YS, Persons DA, Barbas CF, Nienhuis AW: A zinc-finger transcriptional activator designed to interact with the gamma-globin gene promoters enhances fetal hemoglobin production in primary human adult erythroblasts. Blood. 2010, 115: 3033-3041. 10.1182/blood-2009-08-240556.
Sacco A, Mourkioti F, Tran RK, Choi J, Llewellyn M, Kraft P, Shkreli M, Delp S, Pomerantz JH, Artandi SE, Blau HM: Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell. 2010, 143: 1059-1071. 10.1016/j.cell.2010.11.039.
Sohn JH, Yeh BI, Choi JW, Yoon J, Namkung J, Park KK, Kim HW: Repression of human telomerase reverse transcriptase using artificial zinc finger transcription factors. Mol Cancer Res. 2010, 8: 246-253. 10.1158/1541-7786.MCR-09-0141.
Acknowledgements
The authors would like to thank Dr Marjorie Brand for helpful discussions, and critically reading the manuscript. Work from the Blais laboratory is funded from grants from NSERC (355714-2008) and MDA (MDA68947), while work in the Dilworth laboratory is funded by CIHR (FRN 93777 and 77778). FJD holds a Canadian Research Chair in Epigenetic Regulation of Gene Expression.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Dilworth, F., Blais, A. Epigenetic regulation of satellite cell activation during muscle regeneration. Stem Cell Res Ther 2, 18 (2011). https://doi.org/10.1186/scrt59
Published:
DOI: https://doi.org/10.1186/scrt59