
Abstract
Compromised bone-regenerating capability following a long bone fracture is often the result of reduced host bone marrow (BM) progenitor cell numbers and efficacy. Without surgical intervention, these malunions result in mobility restrictions, deformities, and disability. The clinical application of BM-derived mesenchymal stem cells (MSCs) is a feasible, minimally invasive therapeutic option to treat non-union fractures. This review focuses on novel, newly identified cell surface markers in both the mouse and human enabling the isolation and purification of osteogenic progenitor cells as well as their direct and indirect contributions to fracture repair upon administration. Furthermore, clinical success to date is summarized with commentary on autologous versus allogeneic cell sources and the methodology of cell administration. Given our clinical success to date in combination with recent advances in the identification, isolation, and mechanism of action of MSCs, there is a significant opportunity to develop improved technologies for defining therapeutic MSCs and potential to critically inform future clinical strategies for MSC-based bone regeneration.
Similar content being viewed by others
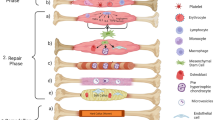

Introduction
Of the millions of long bone fractures incurred annually, approximately 10% result in impaired healing or non-union. Non-union fractures form as the result of significant trauma or a metabolic disease in which injury to the bone becomes atrophic rather than regenerating. This compromised bone-regenerating capability is a complication associated with reduced bone marrow (BM) progenitor cell numbers compounded by suppressed progenitor cell proliferation as a result of the disease itself [1]. Without surgical intervention, malunions result in severe lifestyle impairment, immobilization, and disability. Standard clinical methods for encouraging bone repair include distraction osteogenesis, autologous bone grafting, allograft demineralized bone matrix (DBM), and bone graft substitutes. However, these interventions are invasive and laden with hardware and result in donor site morbidity.
BM-derived mesenchymal stem cells (MSCs) represent an attractive therapeutic to stimulate the repair of a long bone fracture with their expansion potential, osteogenic capability, and potential to home to a site of injury [2]. Systemically administered MSCs localize in the lungs and liver within 24 hours of administration [3] and in the spleen, brain, or skin for the following 1 to 2 months [4], but when delivered in the presence of a long bone fracture, the circulating MSCs home to the fracture site [3] and integrate into the host marrow, bone, and cartilage [4].
MSC contribution to bone repair has been well established in trauma and disease-based pre-clinical models. Bruder and colleagues [5] first demonstrated that the implantation of a scaffold supplemented with BM MSCs supported osteogenesis over an empty scaffold, including the formation of a reparative callus that was absent in defects treated with scaffold alone. Furthermore, implanting pre-differentiated osteogenic MSC-scaffold constructs resulted in superior healing over scaffold alone [6]. Perhaps more importantly, the administration of MSCs with DBM into a clinically relevant model of diabetes resulted in augmentation of fracture healing over those that did not receive MSCs [7]. For additional information on the delivery of MSCs in combination with matrices for orthopedic repair, including the viability and retention of the transplanted cell, please refer to Bulman and colleagues [8].
Contrasting results demonstrate that autografting is more beneficial than defects treated with scaffold alone or cell-seeded scaffolds. Berner and colleagues [9] demonstrated comparable results in autologous MSC-seeded scaffolds, allogeneic MSC-seeded scaffolds, and empty scaffolds in terms of biomechanics; however, a greater bone volume and torsional stiffness were identified in autograft-treated defects. Bensaïd and colleagues [10] similarly observed superiority of autograft in healing critical-sized tibial defects over MSC-seeded hydroxyapatite (HA) constructs, whereas Cipitria and colleagues [11] reported a similar biomechanical stability and mineral density in MSC-seeded scaffolds as compared with autograft; however, there was reduced bone volume in the autograft-treated groups. Regardless, these studies clearly identify BM-derived MSCs as a viable, minimally invasive therapeutic option to treat fractures without the added complication of donor site morbidity associated with autograft.
Mesenchymal stem cells: recent advances in fundamental stromal cell biology
The concept of MSCs as progenitor cells was initially based on the identification of clonogenic fibroblastic cells within the BM that could differentiate into adipocytes, osteoblasts, and chondrocytes in vitro[12]. In the mid-1990s, BM-derived MSC preparations rapidly entered clinical testing to either replace cells in damaged skeletal tissues and, more recently, to provide paracrine signals to repair vascular injury or modulate pathological immune responses [13]. Progress has been made in elucidating the identity, location, heterogeneity, and physiological functions of the stromal cell populations in BM that expand in culture to generate MSCs. However, the mechanisms by which MSCs contribute to the homeostasis of tissues remains poorly understood.
The hypothesis that MSCs represent bona fide skeletal progenitor cells that generate skeletal tissues in vivo has been validated by several recent studies. First, with the serial transplantation of clonal CD45−CD146+ MSCs from human BM, Sacchetti and colleagues [14] demonstrated that cloned human MSCs retain the capacity to generate an ectopic bone and marrow compartment (hematopoietic microenvironment; HME) upon serial transplantation in vivo. Similar MSCs can be prospectively isolated from mouse marrow and enzyme-dissociated bone by using antibodies to platelet-derived growth factor receptor alpha (PDGFRα; CD140a) and stem cell antigen Sca-1 [15]. As with the CD146+ MSC isolated by Sacchetti and colleagues [14], the PDGFRα+Sca-1+ MSCs are capable of re-populating the HME when transferred to a donor.
Using the CXCL12 promoter to drive diphtheria toxin receptor (DTR), Omatsu and colleagues [16] used diphtheria toxin to ablate CXCL12-expressing stromal cells in vivo by demonstrating that mouse perivascular marrow stromal cells expressing CXCL12 were necessary for hematopoietic stem cell (HSC) proliferation and served as progenitors for osteoblasts and adipocytes. At the same time, the Frenette laboratory [17] reported that similar perivascular marrow stromal cells express the intermediate filament, nestin. Using a nestin-enhanced green fluorescent protein (eGFP) transgenic, they showed that nestin-eGFP+ CD45− cells express CXCL12, localize near HSCs, and retain the majority of the colony-forming unit-fibroblasts content of mouse BM. Lineage tracing of CD45−nestin+ BM cells reveal contribution of the MSCs to the long bone and growth plate cartilage [17]. Greenbaum and colleagues [18] further selectively deleted CXCL12 expression from distinct stromal cell populations, including osteoblasts, endothelial cells, and primitive stromal cells. This study indicated the presence of a distinct MSC population expressing the transcription factor PRX1 (Prrx1), supporting different aspects of hematopoiesis. The identified PRX1+CXCL12+ MSCs did not express nestin or leptin receptor, suggesting that PRX1+ MSCs may represent a precursor to cell populations previously identified.
The Scadden laboratory identified marrow stromal cells responsible for bone regeneration by using myxovirus resistance-1 (Mx1) promoter-specific lineage tracing [19]. Unlike the nestin+ and CXCL12+ MSCs, Mx1+ contributed to the osteogenic lineages but not adipogenic or chondrocytic lineages. They demonstrated that an ‘MSC-like’ cell population expressing the transcription factor Mx1 serves as a progenitor for osteoblasts following fracture and demonstrates characteristics of MSCs and bona fide stem cells in serial transplantation experiments.
Finally, two groups took different approaches to ablate fibroblast activation protein-alpha (FAP+) stromal cells from the tumor microenvironment. The Fearon laboratory used the same DTR/DTX system driven by the fap promoter to ablate FAP+ stromal cells from colon and pancreatic tumors. Notably, the ablation of FAP+ cells in healthy animals caused muscle wasting and anemia in the marrow, associated with deregulated hematopoiesis [20]. At the same time, the Rosenberg laboratory used chimeric antigen receptor (CAR) T-lymphocytes design to target FAP+ stroma with several murine tumors. Consistent with the Fearon report, Tran and colleagues [21] observed cachexia and osteopenia in mice treated with the FAP-targeting CAR T cells and found that these phenotypes were associated with a loss of CD45−PDGFRα+Sca1+ stromal cells in the BM.
Although there are still outstanding questions regarding the overlap of cells expressing FAP, nestin, CXCL12, Mx1, PRX1, and leptin receptor, these studies have yielded new knowledge regarding the identification in vivo role of MSCs, in particular their functional roles in maintaining bone regeneration, marrow, and muscle homeostasis. This recent evidence indicates that cells which serve as precursors to culture-expanded MSCs arise from a spectrum of bona fide progenitor cells that can be prospectively identified by their expression of novel functional proteins.
Equally important work has been conducted in human cell and tissue samples. Several groups have recently reported results of fluorescence-activated cell sorting-based purification of stromal cell populations from human BM by using multiple cell surface markers, including CD271, CD56, Stro-3, MSCA-1, and CD146 [22]. These studies confirm the presence of distinct stromal cell populations which differ in their differentiation capacities as well as in their relative abundance during fetal development and aging. Tormin and colleagues [23] demonstrated that these functional differences may correlate with differences in anatomical location as CD271+CD146+ cells were found in perivascular regions and CD271+CD146− cells at endosteal surfaces. In addition, Crisan and colleagues [24] demonstrated that stromal cells expressing the pericyte markers CD146 and NG2 are confined to perivascular regions and can be identified by cytometry from several human tissues, including skeletal muscle, pancreas, fat, placenta, and BM. Although both human and murine studies highlight the heterogeneity of stromal cell populations, they also highlight the lack of specific markers that enable prospective isolation of equivalent MSCs from experimental murine species and humans. Defined homogeneous cells may elicit greater therapeutic activity than heterogeneous MSC populations. In 2007, Sacchetti and colleagues [14] demonstrated that creation of the HME could be achieved only by adoptive transfer of defined CD146+ human MSCs; this HME was not produced by heterogeneous MSCs. Similarly, adoptive co-transfer of either mouse ALCAM+/Sca1− MSCs [25] or human CD146+ MSCs [26] enhance the engraftment and activity of co-transplanted HSCs when compared with heterogeneous MSCs. With regard to clinical development, these advances in fundamental stromal cell biology provide a better understanding of stromal/perivascular cell heterogeneity and function within the BM and other tissues. This knowledge will inevitably lead to improved technologies for defining therapeutic MSCs at the point of isolation and has the potential to critically inform future clinical strategies for MSC-based bone regeneration.
Mesenchymal stem cell mechanism of action
The method by which MSCs home to the site of injury is widely debated, but chemoattractant molecules released at the site of bone injury draw MSCs to participate in the healing process. At least 19 chemokine receptors are expressed by MSCs with varying significance to the chemotactic process [27]. Expression of stromal cell-derived factor 1 (SDF-1) by cells of the stromal niche is a well-documented attractant for circulating CXCR-4-expressing HSCs [28]. Mounting evidence suggested that the SDF-1/CXCR4 complex played a similar role in the attraction of MSCs to the site of trauma via an upregulation of CXCR4 by MSCs of the injured host [29]. Kitaori and colleagues [30] proposed SDF-1 as the primary chemoattractant present in the periosteum of a murine bone grafting model promoting host-progenitor cell recruitment and activation, whereby administration of an anti-SDF-1 neutralizing antibody significantly inhibited new bone formation. A range of other factors have been documented as chemotactic for MSCs, including RANTES, MIP-1α, MCP-1, CCL25, and CXCL16, but the mechanism of systemically administered MSCs homing to an injury is likely attractant/receptor-based [31].
Direct mesenchymal stem cell differentiation
In healthy repair, host progenitors contribute to fracture repair by migrating to the repair callus, where they contribute directly to remodeling by differentiation into osteoblasts [32]. In a poorly repairing fracture due to disease, administered MSCs must compensate for the malfunctioning host progenitors. Pereira and colleagues [33] showed that MSCs expanded in culture and injected into irradiated mice act as precursors for bone and cartilage for at least 5 months. Subsequently, BM administration to a non-ablated mouse model demonstrated the engraftment of MSCs, including their osteocytic differentiation [34]. Therefore, transplanted MSCs have the potential to contribute directly to repair by differentiation.
In mice, systemically administered bioluminescent MSCs migrate to the site of a fracture and integrate into the callus, contributing to repair by secreting bone morphogenetic protein (BMP)-2. Transplanted MSCs localized in the margins of the woven bone and differentiated into osteoblasts, as confirmed by their expression of osteocalcin [35]. In humans, the osteogenic capacity of transplanted BM, and therefore MSCs, has been demonstrated in a small-scale clinical study in children with osteogenesis imperfecta. Functional engraftment of these systemically administered allogeneic MSCs was observed 100 days after administration with associated clinical improvements [36]. Systemic administration can therefore result in homing of MSCs and their direct contribution to orthopedic repair.
The addition of osteo- or angiogenic growth factors to engineer tissue repair in situ has gained popularity. MSC differentiation by the addition of growth factors such as BMP-2 or -7 results in superior osteoblastic differentiation and increased mechanical integrity when applied in vivo[3][37, 38]. Furthermore, the combination of vascular endothelial growth factor and BMPs simulates both osteogenesis and angiogenesis, resulting in robust bone growth and increased mechanical strength [3]. However, Tortelli and colleagues [39] and Scotti and colleagues [40] have demonstrated that fracture repair is dependent on the maturation state of the implanted cell. Transplanted osteoblastic progenitors stimulate host cells to repair the fracture via intramembranous ossification while undifferentiated or chondrogenically primed MSCs instruct the host to repair via endochondral ossification (EO), a process reminiscent of embryonic skeletal development. The presence of the chondrogenic MSC-recruited host CD31+ endothelial cells increased vascularity and consequently host osteoprogenitor cell recruitment, enabling the development of high-quality reparative tissue through EO. Scotti and colleagues [41] have further presented promising results by using ‘developmental engineering’ to recapitulate EO with human MSC-seeded collagen scaffolds implanted in a mouse, resulting in complete formation of a bone organ.
Indirect paracrine influence
The administration of MSCs to a site of injury or in a disease state will expose the cells to an environment not conductive to cell survival. However, recent findings of von Bahr and colleagues [42] have demonstrated that the therapeutic efficacy of these cells did not depend on their sustained viability, but instead on their indirect paracrine influence. Fractures trigger the secretion of a variety of proinflammatory cytokines, including IL-1α, IL-1β, IL-6, and IL-18 and TNF-α [43]. This controlled inflammatory reaction establishes the angiogenic and osteogenic environment that facilitates repair. Prolonged or chronic inflammation, as found in disease states, is inhibitory to bone repair and contributes to non-union formation. In healthy repair, TNF-α recruits osteoclasts and MSCs to the fracture site while causing apoptosis of hypertrophic chondrocytes in support of EO in the early inflammatory response [44]; however, prolonged elevated TNF-α levels inhibit repair by accelerating cartilage removal and elevating osteoclast numbers [45].
The immune conditioning properties of MSCs offer a unique opportunity to regulate the extreme immune response during abnormal fracture repair. As demonstrated in human clinical trials, MSCs administered for the treatment of graft-versus-host disease result in dramatically improved survival rates [46]. Granero-Moltó and colleagues [35] further demonstrated MSC modulation of the local inflammatory environment at a fracture site and this led to lower IL-1β, IL-6, and TNF-α expression in the repair callus and thus to increased biomechanical stability. Therefore, systemically administered MSCs are efficacious in regulating exaggerated immune responses and thereby can contribute indirectly to fracture repair.
Transplanted MSCs also instruct the resident host progenitors, ensuring their optimal contribution to repair in addition to modulating the immune milieu at the fracture site. Xenotransplantation studies demonstrated the instructive capability of donor MSCs on host progenitors when quail progenitors were implanted subcutaneously into mice. In the first month of repair, donor progenitors were responsible for the neogenic osseous tissue; however, with time, the host cells were predominately responsible for the heterogeneous donor-host bone [47]. More recently, MSC-seeded ceramic scaffolds were shown to be depleted of the transplanted MSCs and replaced by the host’s own circulating osteoprogenitors in vivo. A wave of infiltrating host endothelial cells was observed to respond to the donor MSCs, followed by an influx of CD146+ pericytes. Together, the transplanted MSCs condition the local environment to stimulate host angiogenesis and therefore osseous repair [48]. Still, the contribution made by exogenously implanted MSCs in fracture repair remains unclear, but the potential for MSCs as an orthopedic cell therapy strategy is promising.
Clinical application of mesenchymal stem cells
In 1995, the first MSC clinical study used autologous, culture-expanded MSCs in patients with hematological malignancies, demonstrating safety with no reports of adverse events [49]. Since this first trial, over 3,000 patients enrolled in over 100 clinical trials have met safety endpoints with no serious adverse events reported to date.
In 2001, a correspondence to the New England Journal of Medicine[50] presented the first preliminary clinical data (a pilot, unblended, uncontrolled, unrandomized study) on three patients who received cell-based treatment for long bone non-unions due to trauma. Here, autologous BM was harvested and the MSCs culture expanded before implantation in combination with an HA scaffold. At 15 to 27 months, all patients had promising callus formation and the absence of complications [50]. After 6 to 7 years, complete fusion was observed, including integration with the healthy host bone, thereby demonstrating the therapeutic potential of MSCs on an osteoconductive scaffold [51]. Furthermore, osteogenically pre-differentiated patient-specific autologous MSCs clinically implanted on HA demonstrated ‘healing potential’ supported by incorporation with the host bone, increased radiographic density, and no loosening of the implants upon follow-up [52]. More recently, Liebergall and colleagues [53] clinically demonstrated that administration of MSCs in combination with DBM directly administered to tibial fractures resulted in more rapid fracture union, whereas Giannotti and colleagues [54] have demonstrated complete healing of all patients through the application of autologous cultured MSCs, which were osteogenically differentiated and then implanted into the fracture in autologous fibrin scaffolds. Together, these preliminary studies demonstrate the safety and potential efficacy of autologous MSCs in orthopedic repair.
Although recent pre-clinical reports have raised the concern that implanted BM-derived MSCs may stimulate harmful ectopic bone formation by the host [55], the published clinical reports discussed above have fully examined the site of MSC administration radiographically and no evidence of ectopic bone formation has been described. Furthermore, von Bahr and colleagues [42], who examined autopsy material of 18 patients who received MSC transplantations, have found no evidence of sustained retention of the transplanted cells or their involvement in forming ectopic tissue.
Future clinical application of MSCs to support long bone fracture healing will mature from the elementary (the implantation of heterogeneous autologous BM combined with a scaffold) to the advanced systemic application of a selected allogenic osteoprogenitor. The transition from autologous therapy to allogeneic treatment is a prerequisite, as those who require progenitor cell therapy to heal a fracture often have reduced progenitor cell number themselves, the initial reason for their development of a non-union [56]. Unfortunately, all current clinical investigations, summarized in Table 1, are fully focused on autologous heterogenic cell-based therapy. Most interestingly, though, is the significant transition of nearly half of the ongoing studies to the minimally invasive, local, or systemic administration of MSCs without the addition of a carrier.
Conclusions
Compounded with recent advancements in the identification of osteogenic precursors, the good manufacturing practice-grade isolation of clinically efficacious cells is an attractive possibility, enabling the delivery of a smaller dose of cells to the patient. The challenge for the upcoming decade of orthopedic research is therefore one of translation: to render, from the outstanding recent advancements in progenitor cell identification and our improved understanding of the therapeutic mechanism of action, clinically relevant therapies to advance the treatment of long bone non-union.
Abbreviations
- BM:
-
Bone marrow
- BMP:
-
Bone morphogenetic protein
- CAR:
-
Chimeric antigen receptor
- DBM:
-
Demineralized bone matrix
- DTR:
-
Diptheria toxin receptor
- eGFP:
-
Enhanced green fluorescent protein
- EO:
-
Endochondral ossification
- FAP:
-
Fibroblast activation protein alpha
- HA:
-
Hydroxyapatite
- HME:
-
Hematopoietic microenvironment
- HSC:
-
Hematopoietic stem cell
- IL:
-
Interleukin
- MSC:
-
Mesenchymal stem cell
- Mx1:
-
Myxovirus resistance-1
- PDGFRα:
-
Platelet-derived growth factor receptor alpha
- Sca:
-
Stem cell antigen
- SDF-1:
-
Stromal cell-derived factor-1
- TNF-α:
-
Tumor necrosis factor-alpha.
References
Mathieu M, Rigutto S, Ingels A, Spruyt D, Stricwant N, Kharroubi I, Albarani V, Jayankura M, Rasschaert J, Bastianelli E, Gangji V: Decreased pool of mesenchymal stem cells is associated with altered chemokines serum levels in atrophic nonunion fractures. Bone. 2013, 53: 391-398. 10.1016/j.bone.2013.01.005.
Ren G, Chen X, Dong F, Li W, Ren X, Zhang Y, Shi Y: Concise review: mesenchymal stem cells and translational medicine: emerging issues. Stem Cells Transl Med. 2012, 1: 51-58. 10.5966/sctm.2011-0019.
Kumar S, Wan C, Ramaswamy G, Clemens TL, Ponnazhagan S: Mesenchymal stem cells expressing osteogenic and angiogenic factors synergistically enhance bone formation in a mouse model of segmental bone defect. Mol Ther. 2010, 18: 1026-1034. 10.1038/mt.2009.315.
Pereira RF, O’Hara MD, Laptev AV, Halford KW, Pollard MD, Class R, Simon D, Livezey K, Prockop DJ: Marrow stromal cells as a source of progenitor cells for nonhematopoietic tissues in transgenic mice with a phenotype of osteogenesis imperfecta. Proc Natl Acad Sci U S A. 1998, 95: 1142-1147. 10.1073/pnas.95.3.1142.
Bruder SP, Kraus KH, Goldberg VM, Kadiyala S: The effect of implants loaded with autologous mesenchymal stem cells on the healing of canine segmental bone defects. J Bone Joint Surg Am. 1998, 80: 985-996.
Petite H, Viateau V, Bensaïd W, Meunier A, de Pollak C, Bourguignon M, Oudina K, Sedel L, Guillemin G: Tissue-engineered bone regeneration. Nat Biotechnol. 2000, 18: 959-963. 10.1038/79449.
Breitbart EA, Meade S, Azad V, Yeh S, Al-Zube L, Lee YS, Benevenia J, Arinzeh TL, Lin SS: Mesenchymal stem cells accelerate bone allograft incorporation in the presence of diabetes mellitus. J Orthop Res. 2010, 28: 942-949.
Bulman SE, Barron V, Coleman CM, Barry F: Enhancing the mesenchymal stem cell therapeutic response: cell localization and support for cartilage repair. Tissue Eng Part B Rev. 2013, 19: 58-68. 10.1089/ten.teb.2012.0101.
Berner A, Reichert JC, Woodruff MA, Saifzadeh S, Morris AJ, Epari DR, Nerlich M, Schuetz MA, Hutmacher DW: Autologous vs. allogenic mesenchymal progenitor cells for the reconstruction of critical sized segmental tibial bone defects in aged sheep. Acta Biomater. 2013, 9: 7874-7884. 10.1016/j.actbio.2013.04.035.
Bensaïd W, Oudina K, Viateau V, Potier E, Bousson V, Blanchat C, Sedel L, Guillemin G, Petite H: De novo reconstruction of functional bone by tissue engineering in the metatarsal sheep model. Tissue Eng. 2005, 11: 814-824. 10.1089/ten.2005.11.814.
Cipitria A, Reichert JC, Epari DR, Saifzadeh S, Berner A, Schell H, Mehta M, Schuetz MA, Duda GN, Hutmacher DW: Polycaprolactone scaffold and reduced rhBMP-7 dose for the regeneration of critical-sized defects in sheep tibiae. Biomaterials. 2013, 34: 9960-9968. 10.1016/j.biomaterials.2013.09.011.
Friedenstein AJ, Chailakhyan RK, Latsinik NV, Panasyuk AF, Keiliss-Borok IV: Stromal cells responsible for transferring the microenvironment of the hemopoietic tissues, Cloning in vitro and retransplantation in vivo. Transplantation. 1974, 17: 331-340. 10.1097/00007890-197404000-00001.
Horwitz EM, Dominici M: How do mesenchymal stromal cells exert their therapeutic benefit?. Cytotherapy. 2008, 10: 771-774. 10.1080/14653240802618085.
Sacchetti B, Funari A, Michienzi S, Di Cesare S, Piersanti S, Saggio I, Tagliafico E, Ferrari S, Robey PG, Riminucci M, Bianco P: Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007, 131: 324-336. 10.1016/j.cell.2007.08.025.
Morikawa S, Mabuchi Y, Kubota Y, Nagai Y, Niibe K, Hiratsu E, Suzuki S, Miyauchi-Hara C, Nagoshi N, Sunabori T, Shimmura S, Miyawaki A, Nakagawa T, Suda T, Okano H, Matsuzaki Y: Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J Exp Med. 2009, 206: 2483-2496. 10.1084/jem.20091046.
Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, Nagasawa T: The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity. 2010, 33: 387-399. 10.1016/j.immuni.2010.08.017.
Méndez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma’ayan A, Enikolopov GN, Frenette PS: Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010, 466: 829-834. 10.1038/nature09262.
Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, Nagasawa T, Link DC: CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013, 495: 227-230. 10.1038/nature11926.
Park D, Spencer JA, Koh BI, Kobayashi T, Fujisaki J, Clemens TL, Lin CP, Kronenberg HM, Scadden DT: Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell. 2012, 10: 259-272. 10.1016/j.stem.2012.02.003.
Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK, Espeli M, Kraman M, McKenna B, Wells RJ, Zhao Q, Caballero OL, Larder R, Coll AP, O’Rahilly S, Brindle KM, Teichmann SA, Tuveson DA, Fearon DT: Depletion of stromal cells expressing fibroblast activation protein-alpha from skeletal muscle and bone marrow results in cachexia and anemia. J Exp Med. 2013, 210: 1137-1151. 10.1084/jem.20122344.
Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CC, Restifo NP, Rosenberg SA: Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med. 2013, 210: 1125-1135. 10.1084/jem.20130110.
Jones E, McGonagle D: Human bone marrow mesenchymal stem cells in vivo. Rheumatology (Oxford). 2008, 47: 126-131.
Tormin A, Li O, Brune JC, Walsh S, Schütz B, Ehinger M, Ditzel N, Kassem M, Scheding S: CD146 expression on primary nonhematopoietic bone marrow stem cells is correlated with in situ localization. Blood. 2011, 117: 5067-5077. 10.1182/blood-2010-08-304287.
Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, Andriolo G, Sun B, Zheng B, Zhang L, Norotte C, Teng PN, Traas J, Schugar R, Deasy BM, Badylak S, Buhring HJ, Giacobino JP, Lazzari L, Huard J, Péault B: A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008, 3: 301-313. 10.1016/j.stem.2008.07.003.
Nakamura Y, Arai F, Iwasaki H, Hosokawa K, Kobayashi I, Gomei Y, Matsumoto Y, Yoshihara H, Suda T: Isolation and characterization of endosteal niche cell populations that regulate hematopoietic stem cells. Blood. 2010, 116: 1422-1432. 10.1182/blood-2009-08-239194.
Corselli M, Chin CJ, Parekh C, Sahaghian A, Wang W, Ge S, Evseenko D, Wang X, Montelatici E, Lazzari L, Crooks GM, Péault B: Perivascular support of human hematopoietic stem/progenitor cells. Blood. 2013, 121: 2891-2901. 10.1182/blood-2012-08-451864.
Wu Y, Zhao RC: The role of chemokines in mesenchymal stem cell homing to myocardium. Stem Cell Rev. 2012, 8: 243-250. 10.1007/s12015-011-9293-z.
Nervi B, Link DC, DiPersio JF: Cytokines and hematopoietic stem cell mobilization. J Cell Biochem. 2006, 99: 690-705. 10.1002/jcb.21043.
Liu H, Liu S, Li Y, Wang X, Xue W, Ge G, Luo X: The role of SDF-1-CXCR4/CXCR7 axis in the therapeutic effects of hypoxia-preconditioned mesenchymal stem cells for renal ischemia/reperfusion injury. PLoS One. 2012, 7: e34608-10.1371/journal.pone.0034608.
Kitaori T, Ito H, Schwarz EM, Tsutsumi R, Yoshitomi H, Oishi S, Nakano M, Fujii N, Nagasawa T, Nakamura T: Stromal cell-derived factor 1/CXCR4 signaling is critical for the recruitment of mesenchymal stem cells to the fracture site during skeletal repair in a mouse model. Arthritis Rheum. 2009, 60: 813-823. 10.1002/art.24330.
Ito H: Chemokines in mesenchymal stem cell therapy for bone repair: a novel concept of recruiting mesenchymal stem cells and the possible cell sources. Mod Rheumatol. 2011, 21: 113-121. 10.3109/s10165-010-0357-8.
Taguchi K, Ogawa R, Migita M, Hanawa H, Ito H, Orimo H: The role of bone marrow-derived cells in bone fracture repair in a green fluorescent protein chimeric mouse model. Biochem Biophys Res Commun. 2005, 331: 31-36. 10.1016/j.bbrc.2005.03.119.
Pereira RF, Halford KW, O’Hara MD, Leeper DB, Sokolov BP, Pollard MD, Bagasra O, Prockop DJ: Cultured adherent cells from marrow can serve as long-lasting precursor cells for bone, cartilage, and lung in irradiated mice. Proc Natl Acad Sci U S A. 1995, 92: 4857-4861. 10.1073/pnas.92.11.4857.
Nilsson SK, Dooner MS, Weier HU, Frenkel B, Lian JB, Stein GS, Quesenberry PJ: Cells capable of bone production engraft from whole bone marrow transplants in nonablated mice. J Exp Med. 1999, 189: 729-734. 10.1084/jem.189.4.729.
Granero-Moltó F, Weis JA, Miga MI, Landis B, Myers TJ, O’Rear L, Longobardi L, Jansen ED, Mortlock DP, Spagnoli A: Regenerative effects of transplanted mesenchymal stem cells in fracture healing. Stem Cells. 2009, 27: 1887-1898. 10.1002/stem.103.
Horwitz EM, Prockop DJ, Fitzpatrick LA, Koo WW, Gordon PL, Neel M, Sussman M, Orchard P, Marx JC, Pyeritz RE, Brenner MK: Transplantability and therapeutic effects of bone marrow-derived mesenchymal cells in children with osteogenesis imperfecta. Nat Med. 1999, 5: 309-313. 10.1038/6529.
Myers TJ, Yan Y, Granero-Molto F, Weis JA, Longobardi L, Li T, Li Y, Contaldo C, Ozkan H, Spagnoli A: Systemically delivered insulin-like growth factor-I enhances mesenchymal stem cell-dependent fracture healing. Growth Factors. 2012, 30: 230-241. 10.3109/08977194.2012.683188.
Burastero G, Scarfì S, Ferraris C, Fresia C, Sessarego N, Fruscione F, Monetti F, Scarfò F, Schupbach P, Podestà M, Grappiolo G, Zocchi E: The association of human mesenchymal stem cells with BMP-7 improves bone regeneration of critical-size segmental bone defects in athymic rats. Bone. 2010, 47: 117-126. 10.1016/j.bone.2010.03.023.
Tortelli F, Tasso R, Loiacono F, Cancedda R: The development of tissue-engineered bone of different origin through endochondral and intramembranous ossification following the implantation of mesenchymal stem cells and osteoblasts in a murine model. Biomaterials. 2010, 31: 242-249. 10.1016/j.biomaterials.2009.09.038.
Scotti C, Tonnarelli B, Papadimitropoulos A, Scherberich A, Schaeren S, Schauerte A, Lopez-Rios J, Zeller R, Barbero A, Martin I: Recapitulation of endochondral bone formation using human adult mesenchymal stem cells as a paradigm for developmental engineering. Proc Natl Acad Sci U S A. 2010, 107: 7251-7256. 10.1073/pnas.1000302107.
Scotti C, Piccinini E, Takizawa H, Todorov A, Bourgine P, Papadimitropoulos A, Barbero A, Manz MG, Martin I: Engineering of a functional bone organ through endochondral ossification. Proc Natl Acad Sci U S A. 2013, 110: 3997-4002. 10.1073/pnas.1220108110.
von Bahr L, Batsis I, Moll G, Hägg M, Szakos A, Sundberg B, Uzunel M, Ringden O, Le Blanc K: Analysis of tissues following mesenchymal stromal cell therapy in humans indicates limited long-term engraftment and no ectopic tissue formation. Stem Cells. 2012, 30: 1575-1578. 10.1002/stem.1118.
Rundle CH, Wang H, Yu H, Chadwick RB, Davis EI, Wergedal JE, Lau KH, Mohan S, Ryaby JT, Baylink DJ: Microarray analysis of gene expression during the inflammation and endochondral bone formation stages of rat femur fracture repair. Bone. 2006, 38: 521-529. 10.1016/j.bone.2005.09.015.
Gerstenfeld LC, Cho TJ, Kon T, Aizawa T, Tsay A, Fitch J, Barnes GL, Graves DT, Einhorn TA: Impaired fracture healing in the absence of TNF-alpha signaling: the role of TNF-alpha in endochondral cartilage resorption. J Bone Miner Res. 2003, 18: 1584-1592. 10.1359/jbmr.2003.18.9.1584.
Alblowi J, Kayal RA, Siqueira M, McKenzie E, Krothapalli N, McLean J, Conn J, Nikolajczyk B, Einhorn TA, Gerstenfeld L, Graves DT: High levels of tumor necrosis factor-alpha contribute to accelerated loss of cartilage in diabetic fracture healing. Am J Pathol. 2009, 175: 1574-1585. 10.2353/ajpath.2009.090148.
Le Blanc K, Frassoni F, Ball L, Locatelli F, Roelofs H, Lewis I, Lanino E, Sundberg B, Bernardo ME, Remberger M, Dini G, Egeler RM, Bacigalupo A, Fibbe W, Ringdén O: Developmental Committee of the European Group for Blood and Marrow Transplantation: Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet. 2008, 371: 1579-1586. 10.1016/S0140-6736(08)60690-X.
Goshima J, Goldberg VM, Caplan AI: The origin of bone formed in composite grafts of porous calcium phosphate ceramic loaded with marrow cells. Clin Orthop Relat Res. 1991, 269: 274-283.
Tasso R, Fais F, Reverberi D, Tortelli F, Cancedda R: The recruitment of two consecutive and different waves of host stem/progenitor cells during the development of tissue-engineered bone in a murine model. Biomaterials. 2010, 31: 2121-2129. 10.1016/j.biomaterials.2009.11.064.
Lazarus HM, Haynesworth SE, Gerson SL, Rosenthal NS, Caplan AI: Ex vivo expansion and subsequent infusion of human bone marrow-derived stromal progenitor cells (mesenchymal progenitor cells): implications for therapeutic use. Bone Marrow Transplant. 1995, 16: 557-564.
Quarto R, Mastrogiacomo M, Cancedda R, Kutepov SM, Mukhachev V, Lavroukov A, Kon E, Marcacci M: Repair of large bone defects with the use of autologous bone marrow stromal cells. N Engl J Med. 2001, 344: 385-386. 10.1056/NEJM200102013440516.
Marcacci M, Kon E, Moukhachev V, Lavroukov A, Kutepov S, Quarto R, Mastrogiacomo M, Cancedda R: Stem cells associated with macroporous bioceramics for long bone repair: 6- to 7-year outcome of a pilot clinical study. Tissue Eng. 2007, 13: 947-955. 10.1089/ten.2006.0271.
Morishita T, Honoki K, Ohgushi H, Kotobuki N, Matsushima A, Takakura Y: Tissue engineering approach to the treatment of bone tumors: three cases of cultured bone grafts derived from patients’ mesenchymal stem cells. Artif Organs. 2006, 30: 115-118. 10.1111/j.1525-1594.2006.00190.x.
Liebergall M, Schroeder J, Mosheiff R, Gazit Z, Yoram Z, Rasooly L, Daskal A, Khoury A, Weil Y, Beyth S: Stem cell-based therapy for prevention of delayed fracture union: a randomized and prospective preliminary study. Mol Ther. 2013, 21: 1631-1638. 10.1038/mt.2013.109.
Giannotti S, Trombi L, Bottai V, Ghilardi M, D’Alessandro D, Danti S, Dell’Osso G, Guido G, Petrini M: Use of autologous human mesenchymal stromal cell/fibrin clot constructs in upper limb non-unions: long-term assessment. PLoS One. 2013, 8: e73893-10.1371/journal.pone.0073893.
Tasso R, Ulivi V, Reverberi D, Lo Sicco C, Descalzi F, Cancedda R: In vivo implanted bone marrow-derived mesenchymal stem cells trigger a cascade of cellular events leading to the formation of an ectopic bone regenerative niche. Stem Cells Dev. 2013, 22: 3178-3191. 10.1089/scd.2013.0313.
Hernigou P, Poignard A, Beaujean F, Rouard H: Percutaneous autologous bone-marrow grafting for nonunions. Influence of the number and concentration of progenitor cells. J Bone Joint Surg Am. 2005, 87: 1430-1437. 10.2106/JBJS.D.02215.
Acknowledgments
Funding was provided by REDDSTAR: European Union FP7 HEALTH-F2-2012-305736.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article

Cite this article
Watson, L., Elliman, S.J. & Coleman, C.M. From isolation to implantation: a concise review of mesenchymal stem cell therapy in bone fracture repair. Stem Cell Res Ther 5, 51 (2014). https://doi.org/10.1186/scrt439
Published:
DOI: https://doi.org/10.1186/scrt439