
Abstract
Heart diseases remain a major cause of mortality and morbidity worldwide. However, terminally differentiated human adult cardiomyocytes (CMs) possess a very limited innate ability to regenerate. Directed differentiation of human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs) into CMs has enabled clinicians and researchers to pursue the novel therapeutic paradigm of cell-based cardiac regeneration. In addition to tissue engineering and transplantation studies, the need for functional CMs has also prompted researchers to explore molecular pathways and develop strategies to improve the quality, purity and quantity of hESC-derived and iPSC-derived CMs. In this review, we describe various approaches in directed CM differentiation and driven maturation, and discuss potential limitations associated with hESCs and iPSCs, with an emphasis on the role of epigenetic regulation and chromatin remodeling, in the context of the potential and challenges of using hESC-CMs and iPSC-CMs for drug discovery and toxicity screening, disease modeling, and clinical applications.
Similar content being viewed by others


Introduction
Human embryonic stem cells (hESCs), isolated from the inner cell mass of blastocysts, have the ability to propagate indefinitely in culture and can differentiate into any cell type in the body. As such, hESCs can potentially provide an unlimited supply of even highly specialized cells for restoring organ functions that have been damaged by aging, diseases, or traumas. The discovery that mature somatic cells can be reprogrammed to generate induced pluripotent stem cells (iPSCs) [1, 2] further provides investigators with a genetically diverse human model system for studying disease mechanisms, drug screening, and potential new therapeutic strategies.
In 2006, Takahashi and Yamanaka were the first to show that mouse fibroblasts can be reprogrammed to embryonic stem-like pluripotent cells by retroviral transduction with four transcription factors: OCT4 (POU5F1), SOX2, KLF4, and MYC [3]. A year later, the same four retroviral vectors were shown to be effective in reprogramming human fibroblasts [1]. Similarly, Yu and colleagues generated human induced pluripotent stem cells (hiPSCs) based on lentiviral transfer of OCT4, SOX2, LIN28, and NANOG [2]. Reprogramming has now been performed and tested with numerous somatic sources, displaying a range of kinetics and efficiencies [4], including accessible sources such as keratinocytes from skin [5], peripheral blood [6–8], mesenchymal cells in fat [9], epithelial cells in urine [10, 11], and oral mucosa [12].
Subsequent studies have further reduced the requirement to only one or two factors in the reprogramming cocktail, as small molecules or epigenetic modulating drugs can be used to replace the omitted factors [13]. For instance, the addition of valproic acid, a histone deacetylase (HDAC) inhibitor, allows reprogramming with only OCT4 and SOX2 [14]. Furthermore, to avoid permanent and random genomic integration of viral vectors that can lead to DNA aberrations, various nonintegrative or nonviral methods have been successfully employed in the generation of iPSCs. These include transient DNA transfection using episomal plasmids [15] or minicircles [16], protein delivery [17], transfection of synthetic modified mRNAs [18], or use of nonintegrating Sendai virus [19]. Although hiPSCs are comparable with hESCs in terms of morphology, surface marker expression, ability to form three germ layers, and teratoma formation capacity, mounting evidence indicates that the epigenetic landscape and gene expression profiles vary among different hESC lines and hiPSC clones, which can be indicative of incomplete reprogramming, thereby leading to differentiation potential bias and premature senescence [20–27]. Hence, the choice of reprogramming and differentiation techniques as well as stringent quality controls are critical to the prospects of pluripotent stem cell therapy regimes.
Directed cardiac differentiation
hESCs can spontaneously differentiate into cardiomyocytes (CMs) under appropriate culture conditions. When hESCs are cultured in suspension with serum for a period of 7 to 10 days, differentiation to derivatives of the three germ layers occurs and aggregates of cells called embryoid bodies (EBs) are formed. EBs can then be cultured on gelatin-coated dishes from which spontaneously contracting CMs will be observed. Within a mixed population of differentiated cells, a minority of EBs develop CMs and beating areas are visible only in 5 to 15% of EBs [28–30] with the actual yield of hESC-CMs being <1%. The efficiency has been reported to be improved by the addition of DNA demethylating agent 5-aza-cytidine [31], by incubation in hypoxic conditions [32], or by co-culture with endodermal END2 cells [33]. Yet the yields of CMs generated by these methods remain poor.
Using a series of defined growth factors to guide differentiation toward the cardiac lineage, directed differentiation protocols that significantly enhance the generation of hESC-derived and hiPSC-derived CMs have been developed [34–37]. These approaches have revealed that CM differentiation is orchestrated by sequential expression of different sets of genes in specific stages as follows: mesoderm formation (BRY, MIXL1, FOXC1, DKK1), cardiogenic mesoderm (MESP1, ISL1, KDR), cardiac-specific progenitors (NKX2.5, GATA4, TBX5, MEF2C, HAND1/2), and CM maturation (ACTN1, MYH6, TNNT2) [38]. Three families of growth factors are implicated in the control of mesoderm formation and cardiogenesis. Specifically, bone morphogenetic protein (BMP) signaling generally promotes cardiogenesis, wingless in Drosophila (Wnt) proteins are involved in cardiac specification, and fibroblast growth factors drive mesodermal cells into myocardial differentiation [39]. The timing and concentration of these growth factors are crucial for controlling signaling pathways for the induction of directed CM differentiation.
In a monolayer-based protocol for directed cardiac differentiation, H7 hESCs exposed to activin A for 1 day followed by 4 days of BMP4 in serum-free RPMI medium supplemented with B27 have been shown to yield >30% contracting CM-containing clusters at day 12 [34]. Similarly in a suspension EB protocol, the addition of BMP4, activin A, and basic fibroblast growth factor to differentiation medium for 4 days induces primitive-streak formation. Subsequent Wnt inhibition with Dickkopf homolog 1 for 4 days promotes cardiac mesoderm specification, which together with vascular endothelial growth factor promotes expansion and maturation. The differentiating cells can be maintained in medium containing basic fibroblast growth factor, Dickkopf homolog 1, and vascular endothelial growth factor to support further cardiac lineage development [35]. A specific population of kinase domain receptor (KDR)low/c-kitneg cells isolated on day 6 is found to constitute a set of cardiovascular progenitors (CPs) that are able to differentiate into all three cardiovascular lineages – namely CMs, smooth muscle cells, and endothelial cells. Cardiac genes, including those encoding cardiac troponin T (TNNT2), atrial and ventricular isoforms of myosin light chain (MYL7/MYL2), and cardiac transcription factors (NKX2.5, TBX5, TBX20), are upregulated in the KDRlow/c-kitneg cells and 50% of the population consists of contracting CMs when plated in monolayer culture. With optimal activin and BMP4 concentrations, 80% of the KDR/platelet-derived growth factor receptor-α double-positive population isolated on day 5 differentiates into CMs [36].
Recently, sequential addition of activin A and BMP4 to defined RPMI/B27 medium together with double layers of a commercially available extracellular matrix (Matrigel) on day −2 and day 0 of differentiation provided a favorable microenvironment that further promotes epithelial–mesenchymal transition for precardiac mesoderm formation. Such a matrix sandwich method results in the efficient production of CMs from multiple hESC and hiPSC lines with high yields and a purity of up to 98% cTnT+-derived cells [37].
Furthermore, as Wnt signaling activity is a key regulator of cardiogenesis, early and late Wnt signaling enhances and represses heart development, respectively. Efficient CM differentiation can be achieved via appropriate temporal modulation of regulatory elements in the Wnt signaling pathway [40]. In this robust and growth factor-free approach, the Wnt pathway is first activated by glycogen synthase kinase 3 inhibitor to induce differentiation on day 0, followed by shRNA β-catenin knockdown or the use of small molecules that block Wnt protein secretion to repress Wnt activities on day 3 of differentiation. Contracting cells are observed on day 7 and 90% of the differentiated cells are cTnT+ on day 15 in multiple hESC and hiPSC lines.
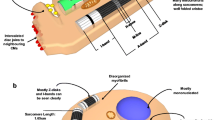
Furthermore, chemically synthesized small molecules that target other signaling pathways have also been screened for their ability to promote cardiac differentiation process. Finally, enhanced cardiogenesis of hESCs has been demonstrated through nodal pathway inhibition at day 4 to promote cardiac specification [36] and through inhibition of the p38 mitogen-activated protein kinase pathway, which favors early mesoderm formation [41]. Select methods of CM-directed differentiation are schematically summarized in Figure 1.

Methods for cardiomyocyte differentiation of human pluripotent stem cells. bFGF, basic fibroblast growth factor; BMP4, bone morphogenetic protein-4; CM, cardiomyocyte; DKK1, Dickkopf homolog 1; dox, doxycycline; IWP, inhibitor of Wnt production; IWR1, inhibitor of Wnt response 1; KSR, knockout serum replacement; MEF, mouse embryonic fibroblast; mTESR, specialized stem cell culture medium; VEGF, vascular endothelial growth factor.
All of the above CM differentiation protocols require optimization among hESC/hiPSC lines, and result in highly heterogeneous cell populations, consisting of a mixture of pacemaker, atrial and ventricular derivatives, as well as some non-CMs [42]. Functionally, the derived CMs respond to electric and chemical stimulation of the β-adrenergic signaling pathway [30, 43], and in general the isolated derived cTnT+ cells display similar gene expression profiles, ultra-structures, calcium-handling proteins, and ion channel functionality typical of immature CMs. Although a striated pattern for α-actinin and myosin light chain is observed [44], the derived CMs lack organized sacromeres and t-tubules [45, 46]. Cell surface marker signal regulatory protein-α and vascular cell adhesion molecule-1 as well as ROR2+/CD13+/KDR+/platelet-derived growth factor receptor-α+ cells derived from differentiating hESCs have been detected on CPs that form CMs [47–49], but no convenient chamber-specific surface markers have yet been identified for robust isolation of CM subtypes. Ultimately, identification of accessible chamber-specific surface markers, as opposed to the use of reporter genes, will be required for any eventual therapeutic application.
Genetic and nongenetic driven maturation of hESC cardiomyocytes
For safety and efficacy of using hESC-CMs as human heart disease models, for drug screening, or for cell-based transplantation therapies, understanding the electrophysiological functions is of paramount importance. Both genetic and nongenetic approaches have been implemented to promote hESC-CM maturation to recapitulate the properties of the adult counterparts. hESC-CMs have been structurally and functionally characterized by ourselves and several laboratories.
hESC-CMs express an array of cardiac-specific transcription factors and structural proteins [28, 30, 43, 50]. While adult ventricular CMs are normally electrically silent-yet-excitable upon stimulation, >50% of hESC-derived ventricular CMs fire spontaneously, exhibiting a high degree of automaticity [51]. The remaining quiescent cells can elicit single action potentials upon stimulation, showing an intact excitability; however, they display a prominent phase 4-like depolarization, a frequent occurrence of delayed after depolarization, and a significantly depolarized resting membrane potential. IK1 is robustly expressed in adult ventricular CMs, but is seen in neither spontaneously firing nor quiescent hESC-derived ventricular CMs. Interestingly, forced expression of Kir2.1 in immature hESC-derived ventricular CMs rendered their action potential properties adult-like, in which the percentage of quiescent ventricular CMs increased up to 100% and Kir2.1-silenced hESC-derived ventricular CMs could elicit single action potentials upon excitation, with a significantly hyperpolarized resting membrane potential indifferent from adult-like but without incomplete phase 4 and delayed after depolarization. Unfortunately, Ca2+ handling stays immature [52, 53]. Contractile apparatus and myofilaments even deteriorate, probably due to the lack of spontaneous contractions after silencing. Indeed, immature Ca2+ transient properties of hESC-CMs can be attributed to the differential developmental expression profiles of Ca2+-handling proteins [52, 53]. In a separate study, forced expression of calsequestrin improves Ca2+ transients in hESC-CMs by significantly increasing the transient amplitude, upstroke, and decay velocities as well as the sarcoplasmic reticulum content, but without altering ICa,L, suggesting the improved transient is not simply due to a higher Ca2+ influx [54]. However, calsequestrin-matured cells continue to have immature electrophysiological properties. In developing neurons, Kir2.1 expression is known to alter excitability by escalating in response to extrinsic excitation via an activity-dependent mechanism to mediate synaptic plasticity, and vice versa. Interestingly, by mimicking endogenous fetal heart pacing by field stimulation in culture, the regulated rhythmic electrical conditioning of hESC-CMs promotes in vitro electrophysiological, Ca2+ handling, as well as contractile maturation with more organized myofilaments [51].
Genetic and epigenetic manipulation and profiling of hESC/iPSC-derived cardiomyocytes
High-throughput screening allows comprehensive analysis of mRNA and miRNA expression, as well as characterization of the epigenetic landscape and detection of changes in histone modifications and DNA methylation status. More specifically, whole-genome expression profiling and RNA sequencing are commonly employed to compare and characterize transcriptomes and miRNA profiles among differentiated cell populations, as well as between iPSC and embryonic stem cell (ESC) lines (reviewed in [55]). Differences among these profiles can be informative of nonuniform epigenetic states that may exist between cell lines. DNA methylation studies and chromatin immunoprecipitation experiments (ChIP-chip or ChIP-Seq) can also reveal variations in chromatin structure and transcription factor binding. DNA methylation studies of promoter regions are informative of transcriptional activity, because active genes are generally hypomethylated, while silenced genes are hypermethylated. Similarly, genome-wide studies performed by techniques based on ChIP-chip or ChIP-Seq permit the elucidation of histone modifications that are indicative of transcriptionally active, repressed, or bivalent patterns of histone methylation. In bivalent promoters, for example, histone 3 is methylated at both lysines 4 (H3K4) and 27 (H3K27). Although H3K4 methylation is associated with gene activation and H3K27 methylation typically results in gene repression, bivalent promoters in stem cells tend to be repressed. With differentiation, this pattern switches from a bivalent state to a monovalent state, which results either in transcriptionally active genes characterized by H3K4 methylation or in nontranscribed genes with a H3K27 methylation state [56]. A number of other histone modifications are also known to affect gene activity, including the repressive H3K9me3, H4K20me3 marks, and multiple targets of histone acetylation, many of which can be assessed through genome-wide approaches. The assessment of these profiles in iPSC lines is extremely valuable when determining their suitability for therapeutic applications, as defects may lead to unintended consequences [57–59]. Principal epigenetic mechanisms of gene expression regulation are shown in Figure 2.

Major epigenetic mechanisms of gene expression regulation. RISC, RNA-induced silencing complex.
A comparative molecular, epigenetic, and biological analysis of cells differentiated from iPSCs with somatic cells from which the iPSCs originated is therefore essential to understand the translational potential of these cells. Towards this end, Xu and colleagues recently reported that reprogrammed murine ventricular myocytes form iPSCs that retain the characteristics of epigenetic memory, which is referred to as CM memory [60]. These ventricular myocyte-derived iPSCs, relative to iPSC controls derived from tail-tip fibroblasts, display a significantly greater differentiation propensity to form spontaneously beating CMs. Importantly, ventricular myocyte-derived iPSCs relative to either ESC or iPSC controls produce greater numbers of CPs at early stages of differentiation. Further analysis of both ventricular myocytes and ventricular myocyte-derived iPSCs revealed a number of genes encoding transcription factors (Nkx2.5, Irx4) and contractile proteins (Myh6, Myl2, Tnni3, Des) that appear to play a role in the specification of CPs. While potentially due to a transient state in the reprogramming of CMs to iPSCs, it is also possible that the mild DNA hypomethylation observed in these cells contributes to the process of ventriculogenesis. Somatic cells, at least during early stages, thus retain epigenetic marks on DNA or histones reminiscent of the somatic cell of origin. While these traits may be transient, this system also provides a model to identify which genes are potentially implicated in fate decisions critical to CM generation.
Genetic manipulation and alteration in epigenetic regulation through chromatin remodeling also control cell fate. Viral transduction of Gata4, Mef2c, and Tbx5 (GMT) directly transdifferentiates murine fibroblasts into CM-like cells in vitro, skipping the pluripotent stage [61]. Transduced fibroblasts are epigenetically reprogrammed with enrichment of H3K4me3 (active transcription mark) and depletion of H3K27me3 (repressed transcription mark) at the promoter regions of the sarcomeric genes, including Actn2, Ryr2, and Tnnt2. The global gene expression profile and electrophysiological properties of the transduced fibroblasts, which demonstrate spontaneous beating, resemble neonatal CMs. In vivo, delivery of GMT into fibroblasts located in the infarcted zone of murine heart also induces CM differentiation [62]. Ectopic expression of Gata4, and Tbx5 in combination with Baf60c, a cardiac-enriched subunit of the Swi/Snf-like BAF ATPase-dependent chromatin remodeling complex, also transdifferentiate cells derived from noncardiogenic mouse mesoderm into CMs [63]. With this combination, 90% of the transfected cells express cardiac α-actin (Actc1). However, GMT overexpression in murine tail tip fibroblasts and cardiac fibroblasts with myocardial lineage reporters (αMHC-Cre, Nkx2.5-Cre, cTnT-Cre) is, however, very inefficient at inducing molecular and electrophysiological phenotypes of mature CMs. While 35% of the cells infected by GMT factors expressed cTnT, the αMHC and Nkx2.5 reporters remain silenced and transduced fibroblasts transplanted into injured mouse heart fail to survive [64]. The discrepancy between these studies may be due to differences in experimental protocols, genetic background of the strain, or levels of GMT overexpression, but it is also possible that differences in the epigenetic status of these cells play an essential regulatory role.
Histone acetyltransferase and HDACs control the relaxation and condensation of chromatin structure for transcription. Treatment with HDAC inhibitor trichostatin A during differentiation of murine ESCs promotes CM differentiation [65]. The levels of acetyl-histone H3 and H4 are upregulated in EBs treated with trichostatin A when compared with the untreated controls. This is accompanied by an increase in GATA4 acetylation, which augments its DNA binding to ANF promoter. Administration of trichostatin A between days 7 and 8 of differentiation doubles the percentage of Nkx2.5-GFP+ cells and increases the expression of cardiac genes, Nkx2.5, β-MHC, and ANF. Furthermore, the introduction of transient HDAC inhibition with valproic acid in hESC-derived ventricular CMs amplifies the expression of Ca2+ handling and cardiac ion channel genes that are important for CM electrophysiological functions and induces physical maturation [50]. These pharmacological-mediated results underscore the involvement of epigenetic and post-translational modification of transcription factors in CM differentiation and heart development. Indeed, knockout models of the chromatin remodeling proteins often lead to congenital heart developmental defects or result in embryonic death (reviewed in [66, 67]). Hence, chromatin modifiers, including ATPase-dependent nucleosomal remodelers and histone-modifying enzymes, play a key role in cardiogenesis and are essential for heart development.
miRNAs are noncoding RNAs that bind to complementary sequences on target mRNA transcripts. miRNAs function as negative transcriptional regulators via translational repression, or mRNA degradation [68]. Recent reports have demonstrated in the mouse that the absence of the miRNA processing enzyme Dicer leads to differentiation and proliferation defects, highlighting the biological importance of miRNAs in stem cell research [69, 70]. Several miRNAs have been implicated in cardiovascular development of the mouse (for example, miR-1, miR-18b, miR-20b, miR-21, miR-106a, miR-126, miR-133, miR-138, and miR-208).
Specific miRNAs have also been characterized and are regulated during hESC-CM differentiation. Overexpression of miR-1 by lentiviral transduction in CPs increases the expression of mesodermal and cardiac marker genes, with accelerated occurrences of contracting areas [71, 72]. miR-1 also facilitates electrophysiological maturation of hESC-CMs, in which decreased action potential duration and hyperpolarized resting membrane potential/maximum diastolic potential due to increased Ito, Iks, and Ikr and decreased If is observed [73]. miR-133 is clustered on the same chromosome as miR-1, but they are functionally different and play opposing roles during CM differentiation. In fact, overexpression of miR-133 represses cardiac markers in hESCs and blocks CM differentiation [74]. miR-499 and miR-208 are also known to affect cardiac function. The miR-499 and miR-208 are encoded by an intron of MYH7 and MYH6, respectively, and they share many predicted targets. miR-208 plays a crucial role in stress adaptation of the adult heart [75]. miR-499 is enriched in cardiac committed CPs and hESCs, and overexpression of miR-499 reduces the proliferation of CPs and augments the formation of beating EBs, promoting differentiation of CPs into ventricular CMs [72, 73]. In contrast, downregulation of miR-499 inhibits cardiac differentiation, suggesting that miR-499 is responsible for cardiac commitment [72].
Interestingly, a recent report has demonstrated the direct conversion of mouse fibroblasts to a CM-like phenotype using single transient transfection with a combination of miRNAs (miR-1, miR-133, miR-208, and miR-499) [76]. The reprogrammed cells express genes and proteins specific for CM and electrophysiological characteristics of the CM-like phenotype can be observed. Direct administration of these miRNAs into injured myocardium likewise results in direct conversion of cardiac fibroblasts to CM-like cells in vivo.
\Interestingly, knockout of single miRNAs often does not lead to embryonic lethality, suggesting that miRNAs may be compensated by family members that differ in only a few nucleotides. In summary, different miRNAs are involved in different stages of development through repression of genes that are likely to contribute to stem cell pluripotency, stem cell renewal, differentiation, specification, lineage commitment, and maturation. Further investigation into manipulation of multiple miRNAs in combination can potentially alter physiological and pathological conditions and can reveal the complexity of miRNA–target interactions and developmental regulatory systems.
Chromatin signatures in hESC-derived cardiomyocytes
The dynamic orchestration of epigenetic factors is fundamental in regulating gene expression patterns during development. Two recent studies have examined the changes in histone modification marks during CM differentiation of mouse ESCs and hESCs, which provide a high-resolution view of the complex organization of histone modification on a genome-wide scale during cardiac development [77, 78]. As described earlier, H3K4me3 and H3K36me3 are marks associated with transcriptional initiation and elongation, respectively, whereas the H3K27me3 modification is associated with transcriptional repression. In ESCs, bivalent chromatin structures with both activating H3K4me3 and repressing H3K27me3 marks on the same promoter are found on lineage commitment genes that are poised to become either transcriptionally active or silent upon definitive cell-type differentiation [56, 79].
Using ChIP-seq technology, the H3K4me3, H3K27me3, and H3K36me3 modifications were mapped on the genome at five key developmental stages: undifferentiated hESCs (T0), mesodermal progenitors (T2), specified tripotential CPs (T5), committed cardiovascular cells (T9), and definitive cardiovascular cells (primarily CMs, T14). Interestingly, genes of different functional categories are characterized by different temporal epigenetic signatures [78]. For example, a complete reversal of active and silent histone marks is found on FGF19 and NODAL promoters. These genes are highly expressed in undifferentiated hESCs with high levels of H3K4me3 and low levels of H3K27me3, and over the course of CM differentiation they subsequently lose H3K4me3 and gain H3K27me3. The genes involved in mesodermal differentiation are highly expressed despite being heavily marked by H3K27me3. Developmental regulators, such as genes encoding NKX2.5, are highly enriched for H3K27me3 in an undifferentiated state, which gradually decreases as H3K4me3, H3K36me3, and RNA expression appear at T9 and T14. In contrast, genes encoding for CM contractile proteins, such as MYH6, do not have high levels of H3K27me3 deposition at any time [78]. These findings suggest that there are complex but distinct chromatin and gene expression patterns that are associated with lineage and cell fate decisions. The characterization of chromatin state transitions during cardiac differentiation has provided useful insights into our understanding of the transcriptional regulation in cardiac developmental programs.
Applications of hESC-derived and hiPSC-derived cardiomyocytes for disease modeling and drug development
Clinical drugs are often withdrawn from the market because of safety concerns, including many with unexpected side effects on the human heart. Harvesting human CMs is a highly invasive procedure, and the numbers of CMs that can be isolated is low. These cells are also difficult to maintain in culture, limiting their use for high-throughput drug screening. The use of animal models for cardiotoxicity screening is also not applicable as heart function differs among mammalian species. For instance, rodent hearts beat significantly faster than human hearts and use different ion channels [80].
The hESC-CMs and hiPSC-CMs provide an alternative model for drug development. Despite the fact that hESC/iPSC-CMs retain many functional and structural traits that are most analogous to embryonic or fetal heart-derived CMs, these cells do express cardiac-specific factors and structural proteins. Many essential contractile proteins, intercellular communication structures, receptors, calcium handling proteins, and ion channels for action potential repolarization are present, including ryanodine receptor, sarco/endoplasmic reticulum Ca2+-ATPase, cardiac sodium channel (SCN5A), the voltage-dependent L-type Ca2+ channel (CACNA1C), and voltage-gated K+ channels (KCN4A and KCNH2). The hESC-CMs and hiPSC-CMs exhibit depolarization patterns with action potentials typical of CMs (reviewed in [81–84]). More importantly, these cells are responsive to hormonal treatments, and positive and negative chronotropic responses can be induced by isoproterenol and carbamylcholine, respectively [85]; they therefore represent an ideal source for some toxicology and drug studies.
Patient-specific iPSC lines and differentiated CMs partially recapitulate disease phenotypes, providing new strategies for understanding disease mechanisms. We present two examples – one designed to look at morphological and structural changes, and another designed to examine electrical defects. For the first, hiPSCs were generated to model LEOPARD syndrome, an autosomal-dominant mutation in the PTPN11 gene that encodes the SHP2 phosphatase, which consequently leads to developmental disorder in multiple organ systems. The major disease phenotype of LEOPARD syndrome is hypertrophic cardiomyopathy [86]. When CMs generated from the diseased iPSCs were compared with CMs derived from hESCs or nondiseased iPSCs generated from a healthy brother, a significant enlargement in cell surface area, a higher degree of sarcomeric organization, and nuclear translocation of the NFATC4 transcription factor could be observed, all of which correlate with the hypertrophic phenotype observed in patients.
Second, CMs were derived from patients with long QT syndrome (LQTS), a cardiac disorder caused by mutations in ion channels or associated proteins and characterized by arrhythmias that can lead to sudden death [87, 88]. LQTS is a particularly apt model for cardiovascular syndromes because a risk assessment for a prolonged QT interval is part of the standard preclinical procedure for all novel drugs in development. In LQTS type 2, in which a potassium channel KCNH2 is mutated, iPSC-CMs displayed prolonged action potential and early depolarization in patch-clamp studies. Several drugs were subsequently found to prevent arrhythmias in the iPSC-derived CMs. When treated with cisapride, a drug that is banned from the market for causing lethal arrhythmias, the cells show increased susceptibility to induced arrhythmogenesis [87]. In LQTS type 1, mutations occur in the KCNQ1 gene, which encodes the repolarizing K+ channel mediating the delayed rectifier IKS current. This disease genotype is maintained in the iPSC-CMs [89]. The ventricular and atrial CMs have significantly longer QT intervals and slower repolarization velocity. The iPSC-CMs show 70 to 80% reduction in IKS current and altered channel activation and deactivation properties, with increased susceptibility to catecholamine-induced tachyarrhythmia, which can be attenuated with β-blockade [89]. The iPSC-CMs generated from patients with Timothy syndrome [90], which is caused by a mutation in an L-type Ca2+ channel CACNA1C gene, also display signatures of LQTS with irregular contraction rates. Treatment with rescovitine restores their electrical and Ca2+ signaling properties. Disease-specific iPSCs from patients are thus useful for studying disease mechanism and molecular pathways that may promote improved therapies. However, the use of iPSCs may be largely restricted to genetic diseases, as adult-onset diseases are affected by environmental and chronic conditions that are not easily modeled in two-dimensional culture systems.
Cardiac tissue bio-engineering
hESC-derived and hiPSC-derived CMs are immature, with electrophysiological properties that more closely resemble embryonic or fetal CMs. In part this may reflect their growth as individual cells or groups of cells grown on the surface of a tissue culture plate, where they are not subjected to the same mechanical forces or loads as those in a three-dimensional structure. In contrast, ventricular myocardium is a highly complex structure consisting of aligned, connected CMs, stromal cells, and a vascular network systematically embedded in a mesh of extracellular matrix [82]. In vitro differentiated and plate cells may therefore not always be a reliable model for drug testing and determining physiological endpoints [91].
Tissue engineering approaches have been suggested to better mimic the native heart tissues for better applicability and efficacy [92, 93]. Indeed, engineered heart tissue has been created by mixing neonatal rat heart cells in a fibrin matrix, attached to flexible posts [94], and engineered three-dimensional muscle strips and cardiac organoid chambers with key characteristics of cardiac physiology have been examined to calculate the rate, force, and kinetics of the contractions [95, 96]. The engineered cardiac tissue constructs are also suitable for studying the changes in CM properties upon increased exercise by mechanical stretches. When hESC-CMs were cultured on a microgrooved platform, the cells aligned and displayed typical banding patterns consistent with organized sarcomeric structure patterns [97]. The aligned hESC-CMs show characteristics of the native heart, including anisotropic conduction properties with distinct longitudinal and transverse velocities. Structural anisotropy can increase the diffusion rate in the direction of alignment and facilitate organization of ion channels. Furthermore, compared with single CMs or randomly oriented CMs, the aligned structures as shown by an increased anisotropic ratio of hESC-CMs have a lower spatial dispersion of action potential propagation through the cell syncytium, which consequently makes them more sustainable against re-entrant arrhythmia and other arrhythmogenic stimuli (Wang J and Li RA, unpublished data).
Using a triple-cell-based three-dimensional culture in scaffolds consisting of CMs, endothelial cells, and embryonic fibroblasts, highly vascularized human engineered cardiac tissue with cardiac-specific properties has been demonstrated [98]. The endothelial cells and embryonic fibroblasts did not hamper orientation and alignment of CMs, the generated tissue constructs show synchronous contraction via gap junctions, and appropriate chronotropic responses are detected following application of pharmacological agents. When coupled with the improved directed differentiation protocols described earlier, the use of three-dimensional culture systems should ultimately promote more physiological maturation events. Once achieved, it is anticipated that engineered cardiac tissues technologies will become a powerful tool for disease modeling, cardiotoxicity screening, and even cardiac regeneration and repair.
Cardiac regeneration using hESC-derived and hiPSC-derived cardiomyocytes
The ultimate goal of regenerative medicine is to repair or replace tissues that have been damaged by diseases and injuries. Unlike some organs, the human heart is unable to repair itself. The use of personalized iPSC-derived cells in regenerative medicine is therefore an attractive option for cell supplementation designed to repair the damaged heart. Indeed, ESCs and iPSCs have been reported to be almost identical at a variety of levels, through the expression of pluripotency markers, transcriptomic comparisons, and analysis of some epigenetic states; however, a number of reports have described considerable differences in epigenetic patterns, genomic imprinting, and global gene expression. Somatic mutations have also been identified between ESCs and iPSCs. Perhaps most importantly, iPSCs are believed capable of evading immune surveillance and graft rejection [99], but accumulating evidence in mice shows that iPSCs do elicit some immune response. Moreover, transplanted allogenic and xenogeneic grafts are not always immune-privileged due to expression of minor antigens that are not normally found in ESCs [100, 101] or due to generation of immunogenic neo-antigens caused by genomic instability during the reprogramming process [102]. The immunological compatibility of iPSCs is not, however, misplaced, as a recent report examining seven ESC lines and 10 iPSC lines established from bone marrow and skin tissues found negligible immunogenicity of either cell type in syngeneic situations [103]. Finally, it is noteworthy that both undifferentiated hESCs and hiPSCs have the capacity to generate teratomas, even following transplantation of fully differentiated cells [104]. More likely, however, is that these differentiated cells contain a minor population of undifferentiated ones. Hence, their use in humans remains a challenge with safety concerns.
Animal experiments have further demonstrated that the introduction of hESC-CMs into damaged areas of the heart improves cardiac function. While transplantation of undifferentiated hESCs 7 to 10 days after coronary ligation resulted in the formation of teratoma-like structures in a rat model of permanent coronary occlusion, injection of predifferentiated hESC-CMs resulted in stable engraftment in both uninjured and infarcted rat hearts [105]. The grafted CMs survived, proliferated, matured, aligned, and formed gap junctions with host cardiac tissue. Transplantation of hESC-CMs attenuated remodeling of scar tissue and improved myocardial performance. Similar results were obtained from other studies evaluating the feasibility of transplanting hESC-CMs in rodent models of myocardial infarction [106–109]. However, in a chronic model in which hESC-CMs are transplanted 1 month after myocardial infarction in the rat, no improvement in heart function or alteration in adverse remodeling was observed [110]. In other mammalian models, formation of stable engraftment of hESC-CMs in pharmacologically immunosuppressed pigs [111] and guinea pigs [43, 112] has also been described. In a guinea-pig model, the hESC-CM grafts in uninjured heart have consistent host–graft coupling, while grafts in the injured heart include both electrical-coupled and electrical-uncoupled regions. Importantly, the injured hearts are partially re-muscularized and demonstrate reduced arrhythmia susceptibility [112].
Finally, suggestions have been made that instead of using fully differentiated hESC-CMs for cardiac repair, perhaps the use of CPs would be more therapeutically appropriate [113]. CPs retain the plasticity to differentiate into other cell types needed for optimal repair, such as endothelial cells, which would contribute to vascularization of the graft, and thereby may improve the survival and integration for extensive engraftment [114]. Indeed, Isl1+ multipotent CPs from mouse and human iPSCs were shown to spontaneously differentiate into all three cardiovascular lineages after transplantation in the left ventricular wall of nude mice, without teratoma formation [115]. Engraftment of ESC-derived early population of CPs in myocardial infarcted nonhuman primate has also been demonstrated [116]. The early multipotent CP population is characterized by expression of OCT4, SSEA-1, and MESP1, and has the potential to differentiate into CMs as well as smooth muscle and endothelial cells. The grafted CPs developed into ventricular CMs and recolonized in the scar tissue. Although the adult heart possesses a population of progenitor cells capable of differentiating into functional CM, the regeneration capacity is limited and is inadequate for repairing the lost tissue in ischemic heart failure [117]. Nevertheless, by isolation and culture of adult CP cells from biopsy, cardiospheres with proliferative capacity that are capable of forming differentiated contractile CMs can be obtained [118, 119]. Injection of adult CPs also promotes cardiac regeneration and improves heart function in a mouse infarct model [119, 120]. All in all, these studies demonstrate that human myocardial grafts can potentially be used in therapies as they can repair injured heart both mechanically and electrically. Despite these encouraging results, challenges remain. The beneficial effect appears to be transient and is not sustained after 12 weeks, irrespective of the number of transplanted hESC-CMs and graft survival [107]. Long-term safety and efficacy investigation is therefore required in large animal models prior to clinical translation of hESC-based therapies [87].
Conclusion and future perspectives
Over the past few years, several major limitations in the derivation of hESC/hiPSC-CMs have been overcome. Importantly, the use of growth factors, chemically synthesized molecules, epigenetic modifiers, miRNAs, or cardiac-specific transcription factors has significantly improved the yield of cardiac differentiation to close to 100%. Furthermore, nongenetic promaturation protocols have been developed and are being fine-tuned [51]. Moreover, hESC/iPSC-CMs are beginning to be used in three-dimensional cultures that are likely to more accurately mimic the physiological state of cardiac muscle. hESC/hiPSC-CMs have therefore emerged as a powerful tool for modeling heart development and cardiac disorders. Indeed, patient-specific iPSCs that retain disease phenotypes are useful for drug cardiotoxicity screening; the diverse genetic backgrounds of the system enable such screening to be personalized.
Yet it remains unclear whether hiPSC models of diseases can be accurately interpreted because epigenetic signatures acquired during disease conditions may not be fully reset, leading to the retention of epigenetic memory. Despite advances in uncovering the molecular basis of epigenetic mechanisms, including DNA methylation, histone modifications, chromatin remodeling, and miRNA-mediated translational control, their role in cardiac differentiation, CM functions, and disease development remains poorly defined. This is largely due to the fact that regulation of CM differentiation and heart development requires complex orchestration of numerous epigenetic factors to precisely control repression of pluripotency genes, upregulation of one lineage, and suppression of other lineages. All of these processes occur simultaneously and are partially controlled by the same enzymes. Epigenetic drugs that targets DNA methylation or histone modifiers are also not gene specific. Further studies at both global and gene promoter levels are therefore necessary to fully identify the recruitment of transcription factors, histone modification enzymes, and chromatin remodelers at specific stages of cardiac differentiation or disease development for better drug discovery and disease modeling.
Regardless of these possible limitations, good quality iPSCs from the mouse are almost identical to murine ESCs. There are, however, no fully accepted criteria to assess and compare hiPSCs and hESCs. Genetic, transcriptomic, and epigenetic approaches performed at the whole-genome level together with functional assays are likely to be critical in the establishment of iPSCs useful for translational research. Transplantation studies of CMs in animal models also reveal many hurdles and challenges that must be overcome before any hESC or hiPSC products can be safely brought to the clinic, including advances in isolation and purification techniques. With better strategies to circumvent immune rejection and better understanding in long-term assessment of cell engraftment after transplantation in large animal models, the prospect of employing hESC-CMs and hiPSC-CMs as an unlimited source for cell replacement therapy to treat heart failure and other conditions will be realized.
Note
This article is part of a thematic series on Stem cell research in the Asia-Pacific edited by Oscar Lee, Songtao Shi, Yufang Shi, and Ying Jin. Other articles in the series can be found online at http://stemcellres.com/series/asiapacific.
Abbreviations
- BMP:
-
Bone morphogenetic protein
- CM:
-
Cardiomyocyte
- CP:
-
Cardiovascular progenitor
- EBs:
-
Embryoid bodies
- ESC:
-
Embryonic stem cell
- GMT:
-
Gata4, Mef2c, and Tbx5
- HDAC:
-
Histone deacetylase
- hESC:
-
Human embryonic stem cell
- hiPSC:
-
Human induced pluripotent stem cell
- iPSC:
-
Induced pluripotent stem cell
- KDR:
-
Kinase domain receptor
- LQTS:
-
Long QT syndrome
- miRNA:
-
MicroRNA
- Wnt:
-
Wingless in Drosophila.
References
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S: Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007, 131: 861-872. 10.1016/j.cell.2007.11.019.
Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA: Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007, 318: 1917-1920. 10.1126/science.1151526.
Takahashi K, Yamanaka S: Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006, 126: 663-676. 10.1016/j.cell.2006.07.024.
Yamanaka S: A fresh look at iPS cells. Cell. 2009, 137: 13-17. 10.1016/j.cell.2009.03.034.
Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F, Vassena R, Bilic J, Pekarik V, Tiscornia G, Edel M, Boué S, Izpisúa Belmonte JC: Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008, 26: 1276-1284. 10.1038/nbt.1503.
Loh YH, Hartung O, Li H, Guo C, Sahalie JM, Manos PD, Urbach A, Heffner GC, Grskovic M, Vigneault F, Lensch MW, Park IH, Agarwal S, Church GM, Collins JJ, Irion S, Daley GQ: Reprogramming of T cells from human peripheral blood. Cell Stem Cell. 2010, 7: 15-19. 10.1016/j.stem.2010.06.004.
Staerk J, Dawlaty MM, Gao Q, Maetzel D, Hanna J, Sommer CA, Mostoslavsky G, Jaenisch R: Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell Stem Cell. 2010, 7: 20-24. 10.1016/j.stem.2010.06.002.
Loh YH, Agarwal S, Park IH, Urbach A, Huo H, Heffner GC, Kim K, Miller JD, Ng K, Daley GQ: Generation of induced pluripotent stem cells from human blood. Blood. 2009, 113: 5476-5479. 10.1182/blood-2009-02-204800.
Sun N, Panetta NJ, Gupta DM, Wilson KD, Lee A, Jia F, Hu S, Cherry AM, Robbins RC, Longaker MT, Wu JC: Feeder-free derivation of induced pluripotent stem cells from adult human adipose stem cells. Proc Natl Acad Sci U S A. 2009, 106: 15720-15725. 10.1073/pnas.0908450106.
Zhou T, Benda C, Duzinger S, Huang Y, Li X, Li Y, Guo X, Cao G, Chen S, Hao L, Chan YC, Ng KM, Ho JC, Wieser M, Wu J, Redl H, Tse HF, Grillari J, Grillari-Voglauer R, Pei D, Esteban MA: Generation of induced pluripotent stem cells from urine. J Am Soc Nephrol. 2011, 22: 1221-1228. 10.1681/ASN.2011010106.
Zhou J, Wang X, Zhang S, Gu Y, Yu L, Wu J, Gao T, Chen F: Generation and characterization of human cryptorchid-specific induced pluripotent stem cells from urine. Stem Cells Dev. 2013, 22: 717-725. 10.1089/scd.2012.0260.
Miyoshi K, Tsuji D, Kudoh K, Satomura K, Muto T, Itoh K, Noma T: Generation of human induced pluripotent stem cells from oral mucosa. J Biosci Bioeng. 2010, 110: 345-350. 10.1016/j.jbiosc.2010.03.004.
Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, Okita K, Mochiduki Y, Takizawa N, Yamanaka S: Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008, 26: 101-106. 10.1038/nbt1374.
Huangfu D, Osafune K, Maehr R, Guo W, Eijkelenboom A, Chen S, Muhlestein W, Melton DA: Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol. 2008, 26: 1269-1275. 10.1038/nbt.1502.
Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II, Thomson JA: Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009, 324: 797-801. 10.1126/science.1172482.
Jia F, Wilson KD, Sun N, Gupta DM, Huang M, Li Z, Panetta NJ, Chen ZY, Robbins RC, Kay MA, Longaker MT, Wu JC: A nonviral minicircle vector for deriving human iPS cells. Nat Methods. 2010, 7: 197-199. 10.1038/nmeth.1426.
Kim D, Kim CH, Moon JI, Chung YG, Chang MY, Han BS, Ko S, Yang E, Cha KY, Lanza R, Kim KS: Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009, 4: 472-476. 10.1016/j.stem.2009.05.005.
Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F, Ebina W, Mandal PK, Smith ZD, Meissner A, Daley GQ, Brack AS, Collins JJ, Cowan C, Schlaeger TM, Rossi DJ: Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010, 7: 618-630. 10.1016/j.stem.2010.08.012.
Ban H, Nishishita N, Fusaki N, Tabata T, Saeki K, Shikamura M, Takada N, Inoue M, Hasegawa M, Kawamata S, Nishikawa S: Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc Natl Acad Sci U S A. 2011, 108: 14234-14239. 10.1073/pnas.1103509108.
Narsinh KH, Plews J, Wu JC: Comparison of human induced pluripotent and embryonic stem cells: fraternal or identical twins?. Mol Ther. 2011, 19: 635-638. 10.1038/mt.2011.41.
Narsinh KH, Sun N, Sanchez-Freire V, Lee AS, Almeida P, Hu S, Jan T, Wilson KD, Leong D, Rosenberg J, Yao M, Robbins RC, Wu JC: Single cell transcriptional profiling reveals heterogeneity of human induced pluripotent stem cells. J Clin Invest. 2011, 121: 1217-1221. 10.1172/JCI44635.
Ghosh Z, Wilson KD, Wu Y, Hu S, Quertermous T, Wu JC: Persistent donor cell gene expression among human induced pluripotent stem cells contributes to differences with human embryonic stem cells. PLoS One. 2010, 5: e8975-10.1371/journal.pone.0008975.
Bar-Nur O, Russ HA, Efrat S, Benvenisty N: Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell. 2011, 9: 17-23. 10.1016/j.stem.2011.06.007.
Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LI, Yabuuchi A, Takeuchi A, Cunniff KC, Hongguang H, McKinney-Freeman S, Naveiras O, Yoon TJ, Irizarry RA, Jung N, Seita J, Hanna J, Murakami P, Jaenisch R, Weissleder R, Orkin SH, Weissman IL, Feinberg AP, Daley GQ: Epigenetic memory in induced pluripotent stem cells. Nature. 2010, 467: 285-290. 10.1038/nature09342.
Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD, Ziller M, Croft GF, Amoroso MW, Oakley DH, Gnirke A, Eggan K, Meissner A: Reference maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell. 2011, 144: 439-452. 10.1016/j.cell.2010.12.032.
Feng Q, Lu SJ, Klimanskaya I, Gomes I, Kim D, Chung Y, Honig GR, Kim KS, Lanza R: Hemangioblastic derivatives from human induced pluripotent stem cells exhibit limited expansion and early senescence. Stem Cells. 2010, 28: 704-712. 10.1002/stem.321.
Huang C, Wu JC: Epigenetic modulations of induced pluripotent stem cells: novel therapies and disease models. Drug Discov Today Dis Models. 2012, 9: e153-e160. 10.1016/j.ddmod.2012.02.004.
Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, Gepstein A, Livne E, Binah O, Itskovitz-Eldor J, Gepstein L: Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest. 2001, 108: 407-414.
Xu C, Police S, Rao N, Carpenter MK: Characterization and enrichment of cardiomyocytes derived from human embryonic stem cells. Circ Res. 2002, 91: 501-508. 10.1161/01.RES.0000035254.80718.91.
He JQ, Ma Y, Lee Y, Thomson JA, Kamp TJ: Human embryonic stem cells develop into multiple types of cardiac myocytes: action potential characterization. Circ Res. 2003, 93: 32-39. 10.1161/01.RES.0000080317.92718.99.
Yoon BS, Yoo SJ, Lee JE, You S, Lee HT, Yoon HS: Enhanced differentiation of human embryonic stem cells into cardiomyocytes by combining hanging drop culture and 5-azacytidine treatment. Differentiation. 2006, 74: 149-159. 10.1111/j.1432-0436.2006.00063.x.
Niebruegge S, Bauwens CL, Peerani R, Thavandiran N, Masse S, Sevaptisidis E, Nanthakumar K, Woodhouse K, Husain M, Kumacheva E, Zandstra PW: Generation of human embryonic stem cell-derived mesoderm and cardiac cells using size-specified aggregates in an oxygen-controlled bioreactor. Biotechnol Bioeng. 2009, 102: 493-507. 10.1002/bit.22065.
Mummery C, Ward-van Oostwaard D, Doevendans P, Spijker R, van den Brink S, Hassink R, van der Heyden M, Opthof T, Pera M, de la Riviere AB, Passier R, Tertoolen L: Differentiation of human embryonic stem cells to cardiomyocytes: role of coculture with visceral endoderm-like cells. Circulation. 2003, 107: 2733-2740. 10.1161/01.CIR.0000068356.38592.68.
Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, Reinecke H, Xu C, Hassanipour M, Police S, O'Sullivan C, Collins L, Chen Y, Minami E, Gill EA, Ueno S, Yuan C, Gold J, Murry CE: Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007, 25: 1015-1024. 10.1038/nbt1327.
Yang L, Soonpaa MH, Adler ED, Roepke TK, Kattman SJ, Kennedy M, Henckaerts E, Bonham K, Abbott GW, Linden RM, Field LJ, Keller GM: Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature. 2008, 453: 524-528. 10.1038/nature06894.
Kattman SJ, Witty AD, Gagliardi M, Dubois NC, Niapour M, Hotta A, Ellis J, Keller G: Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell. 2011, 8: 228-240. 10.1016/j.stem.2010.12.008.
Zhang J, Klos M, Wilson GF, Herman AM, Lian X, Raval KK, Barron MR, Hou L, Soerens AG, Yu J, Palecek SP, Lyons GE, Thomson JA, Herron TJ, Jalife J, Kamp TJ: Extracellular matrix promotes highly efficient cardiac differentiation of human pluripotent stem cells: the matrix sandwich method. Circ Res. 2012, 111: 1125-1136. 10.1161/CIRCRESAHA.112.273144.
Thorrez L, Sampaolesi M: The future of induced pluripotent stem cells for cardiac therapy and drug development. Curr Pharm Des. 2011, 17: 3258-3270. 10.2174/138161211797904154.
Mummery CL, Zhang J, Ng ES, Elliott DA, Elefanty AG, Kamp TJ: Differentiation of human embryonic stem cells and induced pluripotent stem cells to cardiomyocytes: a methods overview. Circ Res. 2012, 111: 344-358. 10.1161/CIRCRESAHA.110.227512.
Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, Raval KK, Zhang J, Kamp TJ, Palecek SP: Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A. 2012, 109: E1848-E1857. 10.1073/pnas.1200250109.
Graichen R, Xu X, Braam SR, Balakrishnan T, Norfiza S, Sieh S, Soo SY, Tham SC, Mummery C, Colman A, Zweigerdt R, Davidson BP: Enhanced cardiomyogenesis of human embryonic stem cells by a small molecular inhibitor of p38 MAPK. Differentiation. 2008, 76: 357-370. 10.1111/j.1432-0436.2007.00236.x.
Moore JC, Fu J, Chan YC, Lin D, Tran H, Tse HF, Li RA: Distinct cardiogenic preferences of two human embryonic stem cell (hESC) lines are imprinted in their proteomes in the pluripotent state. Biochem Biophys Res Commun. 2008, 372: 553-558. 10.1016/j.bbrc.2008.05.076.
Xue T, Cho HC, Akar FG, Tsang SY, Jones SP, Marban E, Tomaselli GF, Li RA: Functional integration of electrically active cardiac derivatives from genetically engineered human embryonic stem cells with quiescent recipient ventricular cardiomyocytes: insights into the development of cell-based pacemakers. Circulation. 2005, 111: 11-20. 10.1161/01.CIR.0000151313.18547.A2.
Dick E, Rajamohan D, Ronksley J, Denning C: Evaluating the utility of cardiomyocytes from human pluripotent stem cells for drug screening. Biochem Soc Trans. 2010, 38: 1037-1045. 10.1042/BST0381037.
Novak A, Barad L, Zeevi-Levin N, Shick R, Shtrichman R, Lorber A, Itskovitz-Eldor J, Binah O: Cardiomyocytes generated from CPVTD307H patients are arrhythmogenic in response to beta-adrenergic stimulation. J Cell Mol Med. 2012, 16: 468-482. 10.1111/j.1582-4934.2011.01476.x.
Lieu DK, Liu J, Siu CW, McNerney GP, Tse HF, Abu-Khalil A, Huser T, Li RA: Absence of transverse tubules contributes to non-uniform Ca2+ wavefronts in mouse and human embryonic stem cell-derived cardiomyocytes. Stem Cells Dev. 2009, 18: 1493-1500. 10.1089/scd.2009.0052.
Elliott DA, Braam SR, Koutsis K, Ng ES, Jenny R, Lagerqvist EL, Biben C, Hatzistavrou T, Hirst CE, Yu QC, Skelton RJ, Ward-van Oostwaard D, Lim SM, Khammy O, Li X, Hawes SM, Davis RP, Goulburn AL, Passier R, Prall OW, Haynes JM, Pouton CW, Kaye DM, Mummery CL, Elefanty AG, Stanley EG: NKX2-5(eGFP/w) hESCs for isolation of human cardiac progenitors and cardiomyocytes. Nat Methods. 2011, 8: 1037-1040. 10.1038/nmeth.1740.
Dubois NC, Craft AM, Sharma P, Elliott DA, Stanley EG, Elefanty AG, Gramolini A, Keller G: SIRPA is a specific cell-surface marker for isolating cardiomyocytes derived from human pluripotent stem cells. Nat Biotechnol. 2011, 29: 1011-1018. 10.1038/nbt.2005.
Ardehali R, Ali SR, Inlay MA, Abilez OJ, Chen MQ, Blauwkamp TA, Yazawa M, Gong Y, Nusse R, Drukker M, Weissman IL: Prospective isolation of human embryonic stem cell-derived cardiovascular progenitors that integrate into human fetal heart tissue. Proc Natl Acad Sci U S A. 2013, 110: 3405-3410. 10.1073/pnas.1220832110.
Chow MZ, Geng L, Kong CW, Keung W, Fung JC, Boheler K, Li RA: Epigenetic regulation of the electrophysiological phenotype of human embryonic stem cell-derived ventricular cardiomyocytes: insights for driven maturation and hypertrophic growth. Stem Cells Dev. 2013, Epub ahead of print
Lieu DK, Fu JD, Chiamvimonvat N, Tung KC, McNerney GP, Huser T, Keller G, Kong CW, Li RA: Mechanism-based facilitated maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Arrhythm Electrophysiol. 2013, 6: 191-201. 10.1161/CIRCEP.111.973420.
Liao SY, Tse HF, Chan YC, Mei-Chu Yip P, Zhang Y, Liu Y, Li RA: Overexpression of Kir2.1 channel in embryonic stem cell-derived cardiomyocytes attenuates posttransplantation proarrhythmic risk in myocardial infarction. Hear Rhythm. 2013, 10: 273-282. 10.1016/j.hrthm.2012.10.008.
Fu JD, Jung Y, Chan CW, Li RA: An inducible transgene expression system for regulated phenotypic modification of human embryonic stem cells. Stem Cells Dev. 2008, 17: 315-324. 10.1089/scd.2007.0114.
Liu J, Lieu DK, Siu CW, Fu JD, Tse HF, Li RA: Facilitated maturation of Ca2+ handling properties of human embryonic stem cell-derived cardiomyocytes by calsequestrin expression. Am J Physiol Cell Physiol. 2009, 297: C152-C159. 10.1152/ajpcell.00060.2009.
Müller GA, Tarasov KV, Gundry RL, Boheler KR: Human ESC/iPSC-based ‘omics’ and bioinformatics for translational research. Drug Discov Today Dis Models. 2012, 9: e161-e170.
Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES: A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006, 125: 315-326. 10.1016/j.cell.2006.02.041.
Ringrose L, Ehret H, Paro R: Distinct contributions of histone H3 lysine 9 and 27 methylation to locus-specific stability of polycomb complexes. Mol Cell. 2004, 16: 641-653. 10.1016/j.molcel.2004.10.015.
Marion RM, Strati K, Li H, Tejera A, Schoeftner S, Ortega S, Serrano M, Blasco MA: Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell. 2009, 4: 141-154. 10.1016/j.stem.2008.12.010.
Mali P, Chou BK, Yen J, Ye Z, Zou J, Dowey S, Brodsky RA, Ohm JE, Yu W, Baylin SB, Yusa K, Bradley A, Meyers DJ, Mukherjee C, Cole PA, Cheng L: Butyrate greatly enhances derivation of human induced pluripotent stem cells by promoting epigenetic remodeling and the expression of pluripotency-associated genes. Stem Cells. 2010, 28: 713-720. 10.1002/stem.402.
Xu H, Yi BA, Wu H, Bock C, Gu H, Lui KO, Park JH, Shao Y, Riley AK, Domian IJ, Hu E, Willette R, Lepore J, Meissner A, Wang Z, Chien KR: Highly efficient derivation of ventricular cardiomyocytes from induced pluripotent stem cells with a distinct epigenetic signature. Cell Res. 2012, 22: 142-154. 10.1038/cr.2011.171.
Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, Srivastava D: Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010, 142: 375-386. 10.1016/j.cell.2010.07.002.
Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu JD, Srivastava D: In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature. 2012, 485: 593-598. 10.1038/nature11044.
Takeuchi JK, Bruneau BG: Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature. 2009, 459: 708-711. 10.1038/nature08039.
Chen JX, Krane M, Deutsch MA, Wang L, Rav-Acha M, Gregoire S, Engels MC, Rajarajan K, Karra R, Abel ED, Wu JC, Milan D, Wu SM: Inefficient reprogramming of fibroblasts into cardiomyocytes using Gata4, Mef2c, and Tbx5. Circ Res. 2012, 111: 50-55. 10.1161/CIRCRESAHA.112.270264.
Kawamura T, Ono K, Morimoto T, Wada H, Hirai M, Hidaka K, Morisaki T, Heike T, Nakahata T, Kita T, Hasegawa K: Acetylation of GATA-4 is involved in the differentiation of embryonic stem cells into cardiac myocytes. J Biol Chem. 2005, 280: 19682-19688. 10.1074/jbc.M412428200.
Vallaster M, Vallaster CD, Wu SM: Epigenetic mechanisms in cardiac development and disease. Acta Biochim Biophys Sin. 2012, 44: 92-102. 10.1093/abbs/gmr090.
van Weerd JH, Koshiba-Takeuchi K, Kwon C, Takeuchi JK: Epigenetic factors and cardiac development. Cardiovasc Res. 2011, 91: 203-211. 10.1093/cvr/cvr138.
Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM: Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005, 433: 769-773. 10.1038/nature03315.
Wang Y, Medvid R, Melton C, Jaenisch R, Blelloch R: DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat Genet. 2007, 39: 380-385. 10.1038/ng1969.
Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, Mills AA, Elledge SJ, Anderson KV, Hannon GJ: Dicer is essential for mouse development. Nat Genet. 2003, 35: 215-217. 10.1038/ng1253.
Wilson KD, Hu S, Venkatasubrahmanyam S, Fu JD, Sun N, Abilez OJ, Baugh JJ, Jia F, Ghosh Z, Li RA, Butte AJ, Wu JC: Dynamic microRNA expression programs during cardiac differentiation of human embryonic stem cells: role for miR-499. Circ Cardiovasc Genet. 2010, 3: 426-435. 10.1161/CIRCGENETICS.109.934281.
Sluijter JP, van Mil A, van Vliet P, Metz CH, Liu J, Doevendans PA, Goumans MJ: MicroRNA-1 and −499 regulate differentiation and proliferation in human-derived cardiomyocyte progenitor cells. Arterioscler Thromb Vasc Biol. 2010, 30: 859-868. 10.1161/ATVBAHA.109.197434.
Fu JD, Rushing SN, Lieu DK, Chan CW, Kong CW, Geng L, Wilson KD, Chiamvimonvat N, Boheler KR, Wu JC, Keller G, Hajjar RJ, Li RA: Distinct roles of microRNA-1 and −499 in ventricular specification and functional maturation of human embryonic stem cell-derived cardiomyocytes. PLoS One. 2011, 6: e27417-10.1371/journal.pone.0027417.
Ivey KN, Srivastava D: MicroRNAs as regulators of differentiation and cell fate decisions. Cell Stem Cell. 2010, 7: 36-41. 10.1016/j.stem.2010.06.012.
van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN: Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007, 316: 575-579. 10.1126/science.1139089.
Jayawardena TM, Egemnazarov B, Finch EA, Zhang L, Payne JA, Pandya K, Zhang Z, Rosenberg P, Mirotsou M, Dzau VJ: MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ Res. 2012, 110: 1465-1473. 10.1161/CIRCRESAHA.112.269035.
Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, Ding H, Wylie JN, Pico AR, Capra JA, Erwin G, Kattman SJ, Keller GM, Srivastava D, Levine SS, Pollard KS, Holloway AK, Boyer LA, Bruneau BG: Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012, 151: 206-220. 10.1016/j.cell.2012.07.035.
Paige SL, Thomas S, Stoick-Cooper CL, Wang H, Maves L, Sandstrom R, Pabon L, Reinecke H, Pratt G, Keller G, Moon RT, Stamatoyannopoulos J, Murry CE: A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell. 2012, 151: 221-232. 10.1016/j.cell.2012.08.027.
Rada-Iglesias A, Wysocka J: Epigenomics of human embryonic stem cells and induced pluripotent stem cells: insights into pluripotency and implications for disease. Genome Med. 2011, 3: 36-10.1186/gm252.
Callaway E: Cells snag top modelling job. Nature. 2011, 469: 279-10.1038/469279a.
Vidarsson H, Hyllner J, Sartipy P: Differentiation of human embryonic stem cells to cardiomyocytes for in vitro and in vivo applications. Stem Cell Rev. 2010, 6: 108-120. 10.1007/s12015-010-9113-x.
Poon E, Kong CW, Li RA: Human pluripotent stem cell-based approaches for myocardial repair: from the electrophysiological perspective. Mol Pharm. 2011, 8: 1495-1504. 10.1021/mp2002363.
Kong CW, Akar FG, Li RA: Translational potential of human embryonic and induced pluripotent stem cells for myocardial repair: insights from experimental models. Thromb Haemost. 2010, 104: 30-38. 10.1160/TH10-03-0189.
Lui KO, Stachel MW, Lieu DK, Li RA, Bu L: Induced pluripotent stem cells as a disease model for studying inherited arrhythmias: promises and hurdles. Drug Discov Today Dis Models. 2012, 9: e199-e207. 10.1016/j.ddmod.2012.09.001.
Zwi L, Caspi O, Arbel G, Huber I, Gepstein A, Park IH, Gepstein L: Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation. 2009, 120: 1513-1523. 10.1161/CIRCULATIONAHA.109.868885.
Carvajal-Vergara X, Sevilla A, D'Souza SL, Ang YS, Schaniel C, Lee DF, Yang L, Kaplan AD, Adler ED, Rozov R, Ge Y, Cohen N, Edelmann LJ, Chang B, Waghray A, Su J, Pardo S, Lichtenbelt KD, Tartaglia M, Gelb BD, Lemischka IR: Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature. 2010, 465: 808-812. 10.1038/nature09005.
Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G, Hammerman H, Boulos M, Gepstein L: Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011, 471: 225-229. 10.1038/nature09747.
Kamp TJ: An electrifying iPSC disease model: long QT syndrome type 2 and heart cells in a dish. Cell Stem Cell. 2011, 8: 130-131. 10.1016/j.stem.2011.01.010.
Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flugel L, Dorn T, Goedel A, Hohnke C, Hofmann F, Seyfarth M, Sinnecker D, Schomig A, Laugwitz KL: Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010, 363: 1397-1409. 10.1056/NEJMoa0908679.
Hoekstra M, Mummery CL, Wilde AA, Bezzina CR, Verkerk AO: Induced pluripotent stem cell derived cardiomyocytes as models for cardiac arrhythmias. Front Physiol. 2012, 3: 346-
Kim C, Majdi M, Xia P, Wei KA, Talantova M, Spiering S, Nelson B, Mercola M, Chen HS: Non-cardiomyocytes influence the electrophysiological maturation of human embryonic stem cell-derived cardiomyocytes during differentiation. Stem Cells Dev. 2010, 19: 783-795. 10.1089/scd.2009.0349.
Lieu DK, Turnbull IC, Costa KD, Li RA: Engineered human pluripotent stem cell-derived cardiac cells and tissues for electrophysiological studies. Drug Discov Today Dis Models. 2012, 9: e209-e217. 10.1016/j.ddmod.2012.06.002.
Turnbull IC, Lieu DK, Li RA, Costa KD: Cardiac tissue engineering using human stem cell-derived cardiomyocytes for disease modeling and drug discovery. Drug Discov Today Dis Models. 2012, 9: e219-e227.
Hansen A, Eder A, Bonstrup M, Flato M, Mewe M, Schaaf S, Aksehirlioglu B, Schwoerer AP, Uebeler J, Eschenhagen T: Development of a drug screening platform based on engineered heart tissue. Circ Res. 2010, 107: 35-44. 10.1161/CIRCRESAHA.109.211458.
Kim do E, Lee EJ, Martens TP, Kara R, Chaudhry HW, Itescu S, Costa KD: Engineered cardiac tissues for in vitro assessment of contractile function and repair mechanisms. Conf Proc IEEE Eng Med Biol Soc. 2006, 1: 849-852.
Lee EJ, Kim Do E, Azeloglu EU, Costa KD: Engineered cardiac organoid chambers: toward a functional biological model ventricle. Tissue Eng Part A. 2008, 14: 215-225. 10.1089/tea.2007.0351.
Luna JI, Ciriza J, Garcia-Ojeda ME, Kong M, Herren A, Lieu DK, Li RA, Fowlkes CC, Khine M, McCloskey KE: Multiscale biomimetic topography for the alignment of neonatal and embryonic stem cell-derived heart cells. Tissue Eng Part C Methods. 2011, 17: 579-588. 10.1089/ten.tec.2010.0410.
Caspi O, Lesman A, Basevitch Y, Gepstein A, Arbel G, Habib IH, Gepstein L, Levenberg S: Tissue engineering of vascularized cardiac muscle from human embryonic stem cells. Circ Res. 2007, 100: 263-272. 10.1161/01.RES.0000257776.05673.ff.
Koch CA, Geraldes P, Platt JL: Immunosuppression by embryonic stem cells. Stem Cells. 2008, 26: 89-98. 10.1634/stemcells.2007-0151.
Swijnenburg RJ, Schrepfer S, Cao F, Pearl JI, Xie X, Connolly AJ, Robbins RC, Wu JC: In vivo imaging of embryonic stem cells reveals patterns of survival and immune rejection following transplantation. Stem Cells Dev. 2008, 17: 1023-1029. 10.1089/scd.2008.0091.
Zhao T, Zhang ZN, Rong Z, Xu Y: Immunogenicity of induced pluripotent stem cells. Nature. 2011, 474: 212-215. 10.1038/nature10135.
Mullally A, Ritz J: Beyond HLA: the significance of genomic variation for allogeneic hematopoietic stem cell transplantation. Blood. 2007, 109: 1355-1362. 10.1182/blood-2006-06-030858.
Araki R, Uda M, Hoki Y, Sunayama M, Nakamura M, Ando S, Sugiura M, Ideno H, Shimada A, Nifuji A, Abe M: Negligible immunogenicity of terminally differentiated cells derived from induced pluripotent or embryonic stem cells. Nature. 2013, 494: 100-104. 10.1038/nature11807.
Yoshida Y, Yamanaka S: Recent stem cell advances: induced pluripotent stem cells for disease modeling and stem cell-based regeneration. Circulation. 2010, 122: 80-87. 10.1161/CIRCULATIONAHA.109.881433.
Caspi O, Huber I, Kehat I, Habib M, Arbel G, Gepstein A, Yankelson L, Aronson D, Beyar R, Gepstein L: Transplantation of human embryonic stem cell-derived cardiomyocytes improves myocardial performance in infarcted rat hearts. J Am Coll Cardiol. 2007, 50: 1884-1893. 10.1016/j.jacc.2007.07.054.
van Laake LW, Passier R, Monshouwer-Kloots J, Verkleij AJ, Lips DJ, Freund C, den Ouden K, Ward-van Oostwaard D, Korving J, Tertoolen LG, van Echteld CJ, Doevendans PA, Mummery CL: Human embryonic stem cell-derived cardiomyocytes survive and mature in the mouse heart and transiently improve function after myocardial infarction. Stem Cell Res. 2007, 1: 9-24. 10.1016/j.scr.2007.06.001.
van Laake LW, Passier R, Doevendans PA, Mummery CL: Human embryonic stem cell-derived cardiomyocytes and cardiac repair in rodents. Circ Res. 2008, 102: 1008-1010. 10.1161/CIRCRESAHA.108.175505.
Leor J, Gerecht S, Cohen S, Miller L, Holbova R, Ziskind A, Shachar M, Feinberg MS, Guetta E, Itskovitz-Eldor J: Human embryonic stem cell transplantation to repair the infarcted myocardium. Heart. 2007, 93: 1278-1284. 10.1136/hrt.2006.093161.
Kofidis T, Lebl DR, Swijnenburg RJ, Greeve JM, Klima U, Robbins RC: Allopurinol/uricase and ibuprofen enhance engraftment of cardiomyocyte-enriched human embryonic stem cells and improve cardiac function following myocardial injury. Eur J Cardiothorac Surg. 2006, 29: 50-55. 10.1016/j.ejcts.2005.10.015.
Fernandes S, Naumova AV, Zhu WZ, Laflamme MA, Gold J, Murry CE: Human embryonic stem cell-derived cardiomyocytes engraft but do not alter cardiac remodeling after chronic infarction in rats. J Mol Cell Cardiol. 2010, 49: 941-949. 10.1016/j.yjmcc.2010.09.008.
Kehat I, Khimovich L, Caspi O, Gepstein A, Shofti R, Arbel G, Huber I, Satin J, Itskovitz-Eldor J, Gepstein L: Electromechanical integration of cardiomyocytes derived from human embryonic stem cells. Nat Biotechnol. 2004, 22: 1282-1289. 10.1038/nbt1014.
Shiba Y, Fernandes S, Zhu WZ, Filice D, Muskheli V, Kim J, Palpant NJ, Gantz J, Moyes KW, Reinecke H, Van Biber B, Dardas T, Mignone JL, Izawa A, Hanna R, Viswanathan M, Gold JD, Kotlikoff MI, Sarvazyan N, Kay MW, Murry CE, Laflamme MA: Human ES-cell-derived cardiomyocytes electrically couple and suppress arrhythmias in injured hearts. Nature. 2012, 489: 322-325. 10.1038/nature11317.
Lui KO, Stachel MW, Li RA, Bu L: Human pluripotent stem cell-derived cardiovascular progenitors for heart regeneration. Drug Discov Today Dis Models. 2012, 9: e189-e197. 10.1016/j.ddmod.2012.08.003.
Wong SS, Bernstein HS: Cardiac regeneration using human embryonic stem cells: producing cells for future therapy. Regen Med. 2010, 5: 763-775. 10.2217/rme.10.52.
Moretti A, Bellin M, Jung CB, Thies TM, Takashima Y, Bernshausen A, Schiemann M, Fischer S, Moosmang S, Smith AG, Lam JT, Laugwitz KL: Mouse and human induced pluripotent stem cells as a source for multipotent Isl1+ cardiovascular progenitors. FASEB J. 2010, 24: 700-711. 10.1096/fj.09-139477.
Blin G, Nury D, Stefanovic S, Neri T, Guillevic O, Brinon B, Bellamy V, Rucker-Martin C, Barbry P, Bel A, Bruneval P, Cowan C, Pouly J, Mitalipov S, Gouadon E, Binder P, Hagege A, Desnos M, Renaud JF, Menasche P, Puceat M: A purified population of multipotent cardiovascular progenitors derived from primate pluripotent stem cells engrafts in postmyocardial infarcted nonhuman primates. J Clin Invest. 2010, 120: 1125-1139. 10.1172/JCI40120.
Urbanek K, Torella D, Sheikh F, De Angelis A, Nurzynska D, Silvestri F, Beltrami CA, Bussani R, Beltrami AP, Quaini F, Bolli R, Leri A, Kajstura J, Anversa P: Myocardial regeneration by activation of multipotent cardiac stem cells in ischemic heart failure. Proc Natl Acad Sci U S A. 2005, 102: 8692-8697. 10.1073/pnas.0500169102.
Messina E, De Angelis L, Frati G, Morrone S, Chimenti S, Fiordaliso F, Salio M, Battaglia M, Latronico MV, Coletta M, Vivarelli E, Frati L, Cossu G, Giacomello A: Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ Res. 2004, 95: 911-921. 10.1161/01.RES.0000147315.71699.51.
Smith RR, Barile L, Cho HC, Leppo MK, Hare JM, Messina E, Giacomello A, Abraham MR, Marban E: Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation. 2007, 115: 896-908. 10.1161/CIRCULATIONAHA.106.655209.
Bauer M, Kang L, Qiu Y, Wu J, Peng M, Chen HH, Camci-Unal G, Bayomy AF, Sosnovik DE, Khademhosseini A, Liao R: Adult cardiac progenitor cell aggregates exhibit survival benefit both in vitro and in vivo. PLoS One. 2012, 7: e50491-10.1371/journal.pone.0050491.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Chow, M.Z., Boheler, K.R. & Li, R.A. Human pluripotent stem cell-derived cardiomyocytes for heart regeneration, drug discovery and disease modeling: from the genetic, epigenetic, and tissue modeling perspectives. Stem Cell Res Ther 4, 97 (2013). https://doi.org/10.1186/scrt308
Published:
DOI: https://doi.org/10.1186/scrt308