
Abstract
Background
The colon has a significant site to deliver numerous active materials for colonic diseases. Highly biodegradable polymers hold significant promise among the several techniques available to deliver the drug to the colon. This research aimed to prepare chitosan, locust bean gum and xanthan gum polysaccharide composite satranidazole multiunit pellets for colonic release and assesses the bioavailability with pharmacokinetic parameters after administration of satranidazole raw drug compared to multiunit pellets. Satranidazole multiple unit pellets were prepared based on chitosan, locust bean gum and xanthan gum, which were inexpensive and harmless. The bioavailability study was done by crossover design in which satranidazole raw drug and test formulation was administered to six healthy white albino rats.
Results
The pharmacokinetic analyses were estimated using the deconvolution of the plasma profile. Compared to the satranidazole drug used as a reference, for the pellets, the maximum plasma concentration was lower (35.02 ± 3.91 ng/ml vs. 51.07 ± 1.21 ng/ml for the satranidazole drug), and the time to attain maximum concentration was 2.50 ± 0.55 h for both drugs and test formulation. Colonic drug content was significantly higher than that of free administered drug.
Conclusion
The results indicate the acquired pharmacokinetic studies and colonic analysis established the reliability of the pharmaceutical technique and the ability to release satranidazole at the colonic site.
Graphical Abstract

Similar content being viewed by others

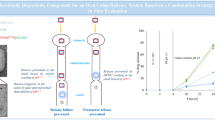

1 Background
The colon is a major site that can deliver bioactive materials to the colon for colonic diseases and transport peptides or proteins into the colonic area for systemic absorption [1]. Highly biodegradable polymers hold significant promise among the several techniques available to deliver the drug to the colon. Locust bean gum has sparked interest in the biopharmaceutical area, particularly in oral administration, which delivers a precise dose to a specific biological site. Its solubility, molecular weight and chemical structure suggest that it could be utilized for in vivo biodegradation with specialized biopharmaceutical applications [2]. Chitosan has a cationic character that delivers drugs to the colon through mucosal attachment. Drugs are controlled by a multiparticulate system containing chitosan due to their biodegradability in the colon [3]. Xanthan gum has a high molecular weight with a side chain attached to glucose residue, enhancing the fabrication of matrices and retards drug release while maintaining time-dependent properties [4]. Polysaccharides have been studied to deliver specific drugs from the colon [5]. The acceptance of these materials as pharmaceutical excipients is their most advantageous feature.
Multiunit pellet dosage forms are more important in the pharmaceutical field and have several advantages over single-unit dosage forms. Pellets that can be encapsulated or compressed into tablets are widely accepted by the patient due to reduced transport difficulties from the esophageal area, lower cost, higher production rate, higher bioavailability, reduced systemic toxicity and predictable gastric emptying [6]. The pellets, as colonic release systems are coated with natural polysaccharides, capable of controlling the release of a drug by erosion or matrix dissolution mechanism through its enzymatic degradation by the colonic microflora and allow the drug to reach quickly and be reserved in the ascending part of the colon over a longer time with its smaller particles size compared to single-unit form. Therefore, the current study focused on satranidazole powder layered colon-specific multiunit pellets [7, 8] based on the mixture of various natural polysaccharides.
Satranidazole is a 5-nitroimidazole group covered at the second position, named 1- methylsulfonyl-3-(1-methyl-5nitro-2-imidazolyl)-2-imidazolidinone and contains potential anti-disease activity protozoa and bacteria for the treatment of amoeba disease [9].
This research, based on using satranidazole drug for the colonic drug delivery system, is essential because the drug reached the colon quickly and was retained in the ascending colon for a longer time. The smaller size of the pellets made of satranidazole pellets allowed for easier drug passage through the GI tract. Satranidazole has a higher absorption rate than metronidazole [10, 11] and is more effective against protozoal infections. Satranidazole is also very patient-friendly and does not cause neurological reactions. This is helpful for patients with neurological side effects [12]. The research work aimed to develop multiunit layered satranidazole pellets based on coated polysaccharides that provided enzymatic and microbial degradation to release the drug into the colon and compare pharmacokinetic parameters after raw satranidazole drug administration to pellets. The pellets were coated with chitosan, locust bean gum and xanthan gum that were cheap and non-toxic and were organized efficiently by a multiunit system that ensured consistent medication distribution and release throughout the GIT with low variability. The requirement for in vivo investigations, particularly of pharmacokinetic characteristics with colonic analysis, revealed the pharmaceutical system's reliability and prevented satranidazole's release in the GIT, which was intended for the colonic region.
2 Methods
2.1 Materials
Alkem Laboratories Ltd., Sikkim, India, provided satranidazole. Xanthan gum, locust bean gum and chitosan gum were procured from Merck Specialities Pvt. Ltd., Mumbai, India. Anthem Biosciences, Bangalore, India, provided nonpareil sugar seeds. Loba Chemie Ltd., Mumbai, provides HPMC-K10 and PVP-K30. The solvents and chemicals that remained were of analytical grade.
2.2 Method of satranidazolemultiunit pellets
Satranidazole layered multiunit pellets were made by powder layer technology [13] using pan coating equipment that maintained all instrument parameters. Main core loading, powder application rate, type, plate speed, atomizing pressure, atomizer position, air cap type and bed temperature were taken to determine the best grades of pellets. The nonpareil sugar beads were coated with a binder polyvinyl pyrrolidone with isopropyl alcohol and purified water in a 70:30 ratio stirred at 200 rpm [14]. It was applied with spray guns and with a dispersed powder layer satranidazole drug delivered by a specially designed powder feed unit. Satranidazole layered pellets have been carried out using xanthan gum and locust bean gum blend (2:1) and locust bean gum and chitosan mixture (1:2) as film coating materials. Seal coating was used before film coating to prevent migration of the drug layer in the formulation [15] HPMC was used as the seal coating material in concentrations ranging from 5 to 15%. Eighteen batches were formed, as given in Table 1.
2.3 Drug polymer interaction studies
2.3.1 Fourier transform infrared spectroscopy study
An FTIR spectrophotometer was used to record infrared (IR) spectra between 4000 and 400 cm−1 utilizing the KBr pellet technique (PerkinElmer) [16] STZ, and a physical combination of STZ with polymers was tested to establish drug compatibility with polymers.
2.3.2 Differential scanning calorimeter study
Thermograms were achieved by a PerkinElmer differential scanning calorimeter (Pyris 6 DSC, Switzerland). Samples of 3 mg were weighed into aluminum pans and then sealed with aluminum caps [17]. Thermograms were obtained at 10 °C/min and a temperature of 50–350 °C. All of the trials were carried out twice.
2.4 In vitro drug release technique
The drug release study was selected using a USP paddle-type dissolution device with 900 ml of dissolving fluid at 37 °C ± 0.5 °C, maintaining a 50 rpm speed. The dissolution technique was performed by altering the pH of the dissolution medium at different period intervals with maintaining proper sink conditions. Here, 300 mg of pellets was weighed accurately and dissolved into 900 ml of medium. It was achieved using pH 1.2 in simulated gastric juice for 2 h, pH 4.5 in mixed simulated gastric and intestinal juice for 3 to 4 h, pH 7.4 in simulated intestinal liquid for 5–6 h and pH 7 in simulated colonic medium from 7 h onwards [18]. The process was done by withdrawing 1 ml of each sample and replacing it with the same amount of medium in the basket. The volume was increased to 10 ml and centrifuged properly. The solution was filtered using Whatman paper, and drug contents were measured at 320 nm in the UV (Shimadzu) spectrophotometer.
2.5 In vivo study
2.5.1 In vivo drug availability study
This current study evaluated the in vivo pharmacokinetic parameters [19] and compared the satranidazole from multiunit pellets with raw drugs in a randomized, two-way crossover design. The result obtained was to target the satranidazole drug in the colonic region.
2.5.1.1 Experimental animals
This study used six healthy male rats having 200 and 250 g weights. It was conducted according to animal ethics and ethical standards [20].
2.5.1.2 In vivo pharmacokinetics method design
The investigation was created with a two-way crossover design [21], a randomized study with at least a 10-day washout time between the two dosing sessions. At each dosing session, the rats acquired either the satranidazole pellet test preparation or the satranidazole raw drug reference preparation at a specified time. Each animal was further marked for identification and assigned to separate cages. Animals were orally administered both the pellet formulation and the raw drugs at a 50 mg/kg dose using an oral feeding syringe [22].
2.5.1.3 HPLC analysis of plasma samples
The HPLC method (Shimadzu Corp. HPLC) contained two VP pumps, LC 20AD, a C18 column (250 × 46 mm diameter, 5 µm particle size) and a 20 µL sample loop with a valve [23]. The mobile phase remained in phosphate buffer (pH3): Methanol (30:70), supplied at a 1 ml/min flow rate. Satranidazole and fluconazole (IS) were identified at a wavelength of 312 nm. The plasma calibration graph for satranidazole was linear at 0.5–64 mcg/ml. This study collected 0.5 ml of blood through retro-orbital puncture before dosing (0 h) and at 0.5, 1, 2, 3, 4, 6, 8, 12 and 24 h by heparinized centrifuge tubes centrifuged at 5000 rpm speed for 10 min. Blood plasma was gathered and stored at 200C [24]. 0.1 ml of blood sample mixed for 10 min by adding 0.5 ml of acetonitrile to extract the drug. 150 µl of subject plasma and 50 µl of IS were transferred to 1.5 ml polypropylene centrifuge microtubes. Deproteinization was executed by adding 300 μl of acetonitrile to the samples, shaking for 5 min and then centrifuging at 6000 rpm for 5 min. The supernatant solution was collected with a micropipette and filtered through a 0.45-micron filter, and 20 µl of the pure top layer was introduced directly into the HPLC chromatography system.
2.5.1.4 Pharmacokinetic analysis
The pharmacokinetics analysis was done using the plot between drug concentration and time to determine the pharmacokinetic parameters, such as Cmax, tmax, AUC0-t, AUC0-inf, t1/2 and Kel [25]. Various pharmacokinetics constraints calculated the relative bioavailability of satranidazole, and statistical studies were presented as the mean ± SD computed using SPSS 13 software. The significance limit was set at 0.05.
2.5.2 Colonic drug content and histopathological study of the colon
Two sets of animals (n = 3) were treated with satranidazole pellets and satranidazole drug, at 50 mg/kg rat bodyweight. These rats were killed after 12 h. The colon was then removed by cervical dislocation. The colon was ligated on both ends before being dissected longitudinally and preserved in a standard formalin solution to remove the colonic contents. The histopathological slide was prepared by using a microtome. The slide was analyzed under a microscope. At 4 °C, the colonic contents were weighed, and 20 ml buffer (pH 7.4) was added. The blend was shaken to achieve excellent homogenization, and 0.5 mL was mixed with 0.5 mL methanol under continual stirring [26]. The supernatant was maintained in a 0.05-mL container, and HPLC measured satranidazole. The colon was collected and kept in a typical formalin solution. The histopathological slides were prepared using a microtome and analyzed under a microscope.
3 Results
3.1 Method of satranidazole multiunit pellets
Multiunit pellets were prepared by the powder layer technique [27], as presented in Table 1. Here, 18 formulation batches were arranged according to formulation components.
3.2 Drug polymer interaction studies
3.2.1 Fourier transform infrared spectroscopy study
The STZ and the polymer mixtures show that there was no significant interaction between polymers and drugs, as shown in Fig. 1a.

Drug and polymers interaction studies a FTIR curve of drug &polymer mixture b DSC study of drug and polymer mixture
3.2.2 Differential scanning calorimeter study
The current study shows thermograms of pure drug, LBG, XG and CG and polymer mixtures [28] used in Fig. 1b. Major melting endotherms of pure STZ and polymer were found at 194.99 °C and 112.95 °C (LBG), 104.28 °C (CG) and 115.34 °C (XG), respectively.
3.3 In vitro drug release technique
Drug release was described using USP paddle-type dissolution device [29]. The release was 0–2% at 2 h and below 20% at 6 h, and the maximum release was at 12 h for optimized formulation. The release study of pellets [30] (Batch 1–9) is shown in Fig. 2 in a simulated gastric medium (Fig. 2a) and simulated colonic fluid (Fig. 2b) using various combinations of polysaccharides with 10–30% coating levels. In vitro release of pellets (Batch 10–18) is presented in Fig. 3 in a simulated gastric liquid (Fig. 3a) and colonic medium (Fig. 3b) [31] with maintaining a 10–30% coating level using various polysaccharides as coating material.

In vitro drug release profiles of satranidazole pellets (Batch F1-F9) at a simulated gastrointestinal fluid (pH 1.2, pH 4.5 and pH 7.4) and b simulated colonic fluid (pH 7)

In vitro drug release profiles of satranidazole pellets (Batch F10-F18) at a simulated gastrointestinal fluid (pH 1.2, pH 4.5 and pH 7.4) and b simulated colonic fluid (pH 7)
3.4 In vivo study
3.4.1 In vivo drug bioavailability study
3.4.1.1 Pharmacokinetic analysis
The plasma calibration curve of satranidazole was drawn between peak area versus concentration with its representative chromatogram of satranidazole for calibration curve [32] as shown in Fig. 4a. The representative chromatogram of the rat plasma sample administered satranidazole with the reference drug ketoconazole (IS) is revealed in Fig. 4b, and the plasma concentration–time plots of free administered satranidazole drug and the test formulation are presented in Fig. 5. The pharmacokinetic characteristics of satranidazole acquired after administration of the drug and test formulation of satranidazole and the statistical analysis results are presented [33] in Tables 2 and 3, respectively.

HPLC studies a representative chromatogram of satranidazole for calibration curve and b representative chromatogram of plasma sample of rat administered with satranidazole. *IS International standard (ketoconazole)

Mean plasma concentration–time curves of free administered satranidazole drug and test formulation
Cmax obtained for oral administration of 100 mg satranidazole and test formulation (pellets) [34] indicated considerable dissimilarities between the mean values acquired for the free administered drug and the pellet formulation as given in Table 3. tmax obtained for orally used 100 mg satranidazole and pellets showed no differences between the mean values acquired [35] for the raw drug and the pellet formulation (Table 3). The AUC0-t obtained after administration of satranidazole as a reference preparation was 381.95 ± 18.61 ng h/ml and 312.0 ± 31.66 ng h/ml for the test formulation (Table 3). The plasma elimination rate constant (Kel) of satranidazole as a drug was shown as 0.143 ± 0.006 h-1 for the reference preparation and 0.121 ± 0.004 h-1 for the test formulation (Table 3). The free administered drug and test formulation's plasma elimination half-life (t1/2) were 4.68 ± 0.19 h and 5.75 ± 0.19 h (Table 3).
3.4.2 Colonic drug content analysis and histopathological study of the colon
The colonic content of satranidazole-free administered drug and test formulation is shown by histogram plot in Fig. 6, and a statistical summary of colonic content is given in Table 4. Colonic histopathology was shown by experimental animals treated with satranidazole-free administered drug and test formulation [36] in Fig. 7a, b

Histogram diagram of mean concentration of colonic free administered drug and test formulation

Histopathological study of colon a experimental animal treated with satranidazole raw drugs b experimental animal treated with test formulation of satranidazole pellet
4 Discussion
The formulations were designed using two independent variables the ratio of coating mixture and the percentage of coating level [37, 38]. The formulations were optimized using satranidazole layered powder with various amounts of chitosan, xanthan gum and locust bean gum combination, giving relevant results compared to the conventional product.
The FTIR peak of pure drug and polymer mixtures, containing their entire characteristic peck at their position, i.e., at 1465–1470 (C–H bending), 3414–3471(O–H stretching), 1244(O–H bending by STZ and polymer blends), 815–824 (C–H rocking, C–C stretching and C–H bending) and 1076–1100 (C–C and C–O stretching) cm−1 [39]. Hence, no chemical interchange between STZ and other polymers in combination is found in the FTIR studies.
Drug mixed biopolymers showed a small broad peak at 191.16 °C and 189.93 °C for LBG + XG + STZ and LBG + CG + STZ, respectively, indicating the existence of a drug in crystalline form. The decrease in height and sensitivity of the endotherm peak is due to polymers in the mixture. An increase in the endothermic property also indicates a thermodynamically favorable interaction between polymers, indicating physical cross-linking. It is clear from the thermogram that LBG is more miscible with XG than CG. In practice, during experiments, it is found so. Previous workers have shown that LBG undergoes highly extensive H-bonding with LBG but forms a Polyelectrolyte complex with CG [40]. The charge neutralization is not so effective with LBG because LBG is a poorly ionizable biopolymer because of little no of the polarizable group compared to XG and CG, so interaction between LBG and CG is not as extensive XG and CG.
The minimum drug was discharged in the gastric fluid, the maximum drug was discharged in the colonic part, and xanthan gum with locust bean gum [41] blend was the most effective polymer mixture for targeting the drug to the colonic site.
The linearity was evaluated by linear regression with correlation coefficient, R2 = 0.9997. This method was linear in 0.5–64 ng/ml. The highest concentration was obtained from the satranidazole-free administered drug as a single dose in the provided condition at 51.07 ± 1.21 ng/ml and 35.02 ± 3.91 ng/ml for test formulation. The time to attain maximum concentration (tmax) was 2.50 ± 0.55 h for both free administered drug and test formulation. When administered as a single quantity in the fed state of free administered satranidazole drug as reference preparation, AUC0-α was 395.64 ± 18.20 ng h/ml and 312.0 ± 31.66 mcg h/ml for the pellets [42] (Table 3). There were no critical dissimilarities in the statistical analysis. Based on the pharmacokinetics parameter studied, the relative bioavailability of satranidazole pellets [43] as a test formulation was 81.69%, and log-transformed relative bioavailability was 96.60% with reference preparation of the satranidazole-free administered drug. The results show that a higher percentage of the drug was released in the colonic region with less bioavailable than the free administered drug that targets satranidazole drug in the colonic site.
The results revealed that the free administered drug colonic content was less than the formulation. The colonic drug content was significantly higher than free administered drugs [44]. The formulation was designed to release a high percentage of drugs in the colon region, and pellets could deliver high drug content into the colon. There was no necrosis and congestion below the superficial epithelium. At the submucosal level, no inflammatory cells were found. Colonic mucosa with barely visible lamina propria and rare inflammatory cells. Normal colonic mucosa with linear crypts and regular glandular arrangement. There was no space between the bottom of the colonic crypts and the muscular mucosa without any signs of epithelial damage. Colonic mucosa without any chronic inflammatory infiltrate indicates that the sediment did not induce any toxicity in the colon.
5 Conclusions
A multiunit pellets system for colonic release was developed in this study, and it was successful in achieving the goals of colon-specific satranidazole release. The pellets were designed with an HPMC-K10 as seal coating and film coating with various combinations of locust bean gum, xanthan gum and chitosan. In vitro release revealed that the pharmaceutical system was capable of preventing release in gastric medium (pH 1.2) for 2 h and also in a mixture of gastric and intestinal liquid (pH 4.5) for next 3 h, the drug release beginning after 6 h in simulated intestinal medium (pH 7.4) and a maximum drug release in 14 h approximately. The results acquired for the pharmacokinetic study, the relative bioavailability of satranidazole pellets was 81.69% which proved that a higher percentage of the drug was released in the colonic region with less bioavailable than the free administered drug to target in the colonic site. Colonic histopathology showed by treatment with test formulation that the pellet did not induce any toxicity in the colon. In conclusion, pharmacokinetic data with colonic content confirmed the reliability of the pharmaceutical technique in acquiring the colon-specific release of satranidazole.
Availability of data and materials
All necessary data generated or analyzed during this study are included in this published article. Any additional data could be available from the corresponding author upon request.
Abbreviations
- STZ:
-
Satranidazole
- HPMC:
-
Hydroxypropyl methylcellulose
- C max :
-
Maximum plasma concentration
- PVP:
-
Polyvinyl pyrrolidone
- t max :
-
Time to attain maximum concentration
- K el :
-
Plasma elimination rate constant
- FTIR:
-
Fourier transform infrared spectroscopy
- DSC:
-
Differential scanning colorimeter
- HPLC:
-
High performance liquid chromatography
- LBG:
-
Locust bean gum
- XG:
-
Xanthan gum
- CG:
-
Chitosan gum
References
Amidon S, Brown JE, Dave VS (2015) Colon-targeted oral drug delivery systems: design trends and approaches. AAPS Pharm Sci Tech 16:731–741
Marita D, Grenha A (2012) Locust bean gum: exploring its potential for biopharmaceutical applications. J Pharma Bio Allied Sci 4(3):175–185
Kulkarni N, Jain P, Shindikar A, Suryawanshi P, Thorat N (2022) Advances in the colon-targeted chitosan based multiunit drug delivery systems for the treatment of inflammatory bowel disease. Carbohydr Polym 288:119351
Ramasamy T, Kandhasami UDS, Ruttala H, Shanmugam S (2011) Formulation and evaluation of xanthan gum based aceclofenac tablets for colon targeted drug delivery. Braz J PharmaSci 7(2):300–311
Deshmukha AS, Aminabhavi TS (2015) Pharmaceutical applications of various natural gums. Polysaccharides:1933–1967
Hamman H, Hamman J, Steenekamp J (2017) Multiple-unit pellet systems (MUPS): production and applications as advanced drug delivery systems. Drug Deliv Lett 7(3):201–210
Mazumder R, Mahanti B, Majumdar S, Pal R, Chowdhury AD (2021) Response surface method for optimization of prepared satranidazole powder layered pellets. Fut J PharmaSci 7:190
Mazumder R, Mahanti B, Majumdar S, Pal R, Chowdhury AD (2020) Improved comprehensive analytical method for assessment of satranidazole in drug and product. Fut J PharmaSci 6(1):1–11
Bansal K, Rawat MK, Jain A, Rajput A, Chaturvedi TP, Singh S (2009) Development of satranidazolemucoadhesive gel for the treatment of periodontitis. AAPS Pharm Sci Tech 10:716–723
Pawar HA, Joshi PR (2014) Development and evaluation of taste masked granular formulation of satranidazole by melt granulation technique. J Pharmaceut 1:1–7
Hemphill A, Muller N, Muller J (2019) Comparative pathobiology of the intestinal protozoan parasites giardia lamblia, entamoebahistolytica, and cryptosporidium parvum. Pathogens 8(3):116
Parmar DSM, Jadav SP (2007) The concept of personal drugs in the undergraduate pharmacology practical curriculum. Ind J Pharmacol 39(3):165–167
Majumdar S, Dey S, Ganguly D, Mazumder R (2020) Enhanced topical permeability of natural flavonoid baicalein through nano liposomal gel: in-vitro and in-vivo investigation. J Drug Del SciTechnol 57:101666
Kovacevic J, Mladenovic A, Djuris J, Ibric S (2016) Evaluation of powder, solution and suspension layering for the preparation of enteric coated pellets. Eur J Pharmaceut Sci 85:84–93
Albertini B, Melegari C, Bertoni S, Dolci LS, Passerini N (2018) A novel approach for dry powder coating of pellets with ethylcellulose: Part II: evaluation of caffeine release. AAPS Pharm Sci Tech 9(3):1426–1436
Kowalczuk D, Pitucha M (2019) Application of FTIR method for the assessment of immobilization of active substances in the matrix of biomedical materials. Materials 12:2972
Mazumder R, Allamneni Y, Firdous SM, Parya H, Chowdhury AD (2013) Formulation, development and in-vitro release effects of ethyl cellulose coated pectin microspheres for colon targeting. Asian J Pharmaceut Clin Res 6(5):138–144
Ren Y, Jiang L, Yang S, Gao S, Yu H, Hu J, Hu D, Mao W, Peng H, Zhou Y (2017) Design and preparation of a novel colon-targeted tablet of hydrocortisone. Braz J Pharmaceut Sci 53(1):1–11
Hadi MA, Rao RGN, Rao SA (2015) Pharmacokinetic parameters determination and in vitro–in vivo correlation of ileocolonic-targeted pH-responsive coated mini-tablets of naproxen. Sci Pharm 83:645–658
Narayanamurthy U, Mirunalini R, Subha V, Manimekalai K, Sakthibalan K, Paul AC, Nagarajan MK, Sabarianandh VJ (2021) Acute and repeated dose toxicity study of clevira syrup-A polyherbal formulation. Biomed Pharmacol J 14(3):1459–1467
Shinada K, Ueno M, Konishi C, Takehara S, Yokoyama S, Kawaguchi Y (2008) A randomized double blind crossover placebo-controlled clinical trial to assess the effects of a mouthwash containing chlorine dioxide on oral malodor. Trials 9:71
Bordoloi R, Ahmed AB, Bhattacharya K (2021) Pharmacoscintigraphic evaluation and antidiabetic efficacy of gliclazide-loaded 99mTc-labelled mucoadhesive microspheres. Fut J Pharmaceut Sci 7:229
Seo CS, Lee MY (2020) HPLC–PDA and LC–MS/MS Analysis for the simultaneous quantification of the 14 marker components in sojadodamgangki-tang. Appl Sci 10:2804
DeWen L, Huijie Y, Yiming K, Yun Y, Yanling L, Lixin W, Yan T, Jinyu W (2018) Preparation of colon-targeted acetylharpagide tablets and its release properties in vivo and in vitro. Front Pharmacol 9:832
Nora M, Laura NM, Florencia ML, Pilar M, Pedro Z, Cecilia LF, Ignacio S, Luis EM (2016) Bioequivalence study of two long-acting formulations of oxytetracycline following intramuscular administration in bovines. Front Vet Sci 3:50
Bose A, Elyagoby A, Wong WT (2014) Oral 5-fluorouracil colon-specific delivery through in vivo pellet coating for colon cancer and aberrant crypt foci treatment. Inter J Pharmaceut 468(1–2):178–186
Ghelicha R, Jahannamab MR, Abdizadeha H, Torknikc FS, Vaezic MR (2019) Central composite design (CCD)-Response surface methodology (RSM) of effective electrospinning parameters on PVP-B-Hf hybrid nanofibrous composites for synthesis of HfB2-based composite nanofibers. Compos B 166:527–541
Gill P, Moghadam TT, Ranjbar B (2010) Differential scanning calorimetry techniques: applications in biology and nanoscience. J Bimolecul Techn 21(4):167–193
Kassem MA, Shaboury KME, Mohamed AI (2019) Application of central composite design for the development and evaluation of chitosan-based colon targeted microspheres and in-vitro characterization. Ind J Pharmaceut Sci 81(2):354–364
Pawar HA, Joshi PR (2014) Development and validation of a discriminating in vitro dissolution method for oral formulations containing satranidazole. Int J Spectrosc 3:1–7
Singh SK, Yadav AK, Prudhviraj G, Gulati M, Kaur P, Vaidya Y (2015) A novel dissolution method for evaluation of polysaccharide based colon specific delivery systems: a suitable alternative to animal sacrifice. Eur J Pharmaceut Sci 73:72–80
Jagtap O, Godse V, Deshpande S, Deodhar M (2011) HPLC determination of satranidazole in rat plasma. Asian J Chem 23(10):4317–4320
Dhat S, Pund S, Kokare C, Sharma P, Shrivastava B (2017) Risk management and statistical multivariate analysis approach for design and optimization of satranidazole nanoparticles. Eur J Pharmaceut Sci 1(96):273–283
Jackson AJ, Conner DP, Miller R (2020) First measured plasma concentration value as Cmax; impact on the Cmax confidence interval in bioequivalence studies. Biopharma Drug Dispos 21(4):139–146
Zhang Y, Huang Z, Omari-Siaw E, Lu S, Zhu Y, Jiang D, Wang M, Yu J, Xu X, Zhang W (2016) Preparation and in vitro–in vivo evaluation of sustained-release matrix pellets of capsaicin to enhance the oral bioavailability. AAPS Pharm Sci Tech 17(2):339–349
Lemmens G, Camp AV, Kourula S, Vanuytsel T, Augustijns P (2021) Drug disposition in the lower gastrointestinal tract: targeting and monitoring. Pharmaceutics 13:161
Patela S, Patelb N, Misraa M, Joshic A (2018) Controlled-release domperidone pellets compressed into fast disintegrating tablets forming a multiple-unit pellet system (MUPS). J Drug Del Sci Technol 45:220–229
Kibria G, Akhter A, Islam KMA (2010) Formulation and evaluation of domperidone pellets prepared by powder layering technology. Asian J Pharmaceut 4(1):41–47
Zhuang J, Li M, Pu M, Ragauskas AJ, Yoo CG (2020) Observation of potential contaminants in processed biomass using fourier transform infrared spectroscopy. Appl Sci 10:4345
Luna RC, Illana AM, Perez FZ, Caro RR, Veiga MD (2021) Naturally occurring polyelectrolytes and their use for the development of complex-based mucoadhesive drug delivery systems: an overview. Polymers 13:2241
Grenha A, Dionísio M (2012) Locust bean gum: Exploring its potential for biopharmaceutical applications. J Pharma Bioallied Sci 4(3):175
Priyanka N, Razdan P, Bhat MYS, Pimpale S, Chand JS, Gupta A (2017) Subgingivally delivered 3% satranidazole in the treatment of chronic periodontitis among smokers: a randomized, controlled clinical trial. J Int Acad Periodontol 19(3):189–194
Buya AB, Beloqui A, Memvanga PB, Preat V (2020) Self-nano-emulsifying drug-delivery systems: from the development to the current applications and challenges in oral drug delivery. Pharmaceutics 12:1194
Philip AK, Philip B (2010) Colon targeted drug delivery systems: a review on primary and novel approaches. Oman Med J 25(2):79–87
Acknowledgements
The authors wish to give thanks to School of Pharmacy, Techno India University, EM 4, Sector-V, Kolkata-700091, West Bengal, India, and Calcutta Institute of Pharmaceutical Technology, Howrah, West Bengal, India, given research laboratory to carry out this project work and also thanks to help and encourage by our fellow colleagues in completion of the research project work.
Funding
The authors have no funding to report.
Author information
Authors and Affiliations
Contributions
We declare that this work was done by the authors named in this article: RM, BM and RP conceived and designed the study. RM and NP carried out the laboratory work, collected and analyzed the data, and SM drafted the manuscript. BM supervised the work, and RM assisted in the data analysis. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The author’s assert that all procedure contributing to this work comply with the ethical standards of the Institutional Animal Ethics Committee “TAAB Biostudy Services,” 69 Ibrahimpur Road, Jadavpur, Kolkata–700032, and ethical approval number is 1938/PO/Rc/S/17/CPCSEA.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mazumder, R., Mahanti, B., Majumdar, S. et al. Satranidazole-loaded chitosan/locust bean gum/xanthan gum polysaccharide composite multiunit pellets for colon targeting: in vitro–in vivo investigation. Beni-Suef Univ J Basic Appl Sci 11, 151 (2022). https://doi.org/10.1186/s43088-022-00333-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43088-022-00333-w