
Abstract
Background
Indonesia has started the big project of COVID-19 vaccination program since 13 January 2021 by employing the first shot of vaccine to the President of Indonesia as the outbreak and rapid transmission of COVID-19 have endangered not only Indonesian but the global health and economy. This study aimed to investigate the full-length genome mutation analysis of 166 Indonesian SARS-CoV-2 isolates as of 12 January 2021.
Results
All data of the isolates were extracted from the Global Initiative on Sharing All Influenza Data (GISAID) EpiCoV database. CoVsurver platform was employed to investigate the full-length genome mutation analysis of all isolates. This study also focused on the phylogeny analysis in unlocking the mutation of S protein in Indonesian SARS-CoV-2 isolates. WIV04 isolate that was originated from Wuhan, China was used as the virus reference according to the CoVsurver default. The result showed that a full-length genome mutation analysis of 166 Indonesian SARS-CoV-2 isolates was successfully generated. Every single mutation in S protein was described and then visualized by utilizing BioRender platform. Furthermore, it also found that D614G mutation appeared in 103 Indonesian SARS-CoV-2 isolates.
Conclusions
To sum up, this study helped to observe the spread of COVID-19 transmission. However, it also proposed that the epidemiological surveillance and genomics studies might be improved on COVID-19 pandemic in Indonesia.
Similar content being viewed by others


Background
SARS-CoV-2 firstly occurred in China and then transmitted sporadically worldwide. In March 2020, WHO announced that its infection was a pandemic. COVID-19 outbreak and rapid transmission have endangered global health and economy (Khan et al. 2020a, b; Wu et al. 2020) including Indonesia (Gunadi et al. 2020, 2021). This crisis has called for an extensive scientific mobilization of researches on SARS-CoV-2 focusing on its clinical aspects, its characteristics, and its mechanism of transmission, with the ultimate aim of counteracting the devastating outcomes (Huang et al. 2020; Li et al. 2020). Previously, there were six coronaviruses that infected humans, these are HCoV-229E (1966), HCoV-OC43 (1967), SARS-CoV (2002), HCoV-NL63 (2004), HCoV-HKU1 (2005), and MERS-CoV (2012) (Artika et al. 2020). Recently, around 196 million people globally have been infected by the seventh coronavirus called SARS-CoV-2 (COVID-19), with more than 4 million deaths as a result of this pandemic. In Indonesia, there are more than 3.3 million cases and around 92,000 people died. These data were derived from CSSE at Johns Hopkins University online website as 30 July 2021 which tracks COVID-19 cases in real-time (Dong et al. 2020).
As for the coronaviruses themselves, the family Coronaviridae is categorised into four different genera: Gammacoronavirus, Deltacoronavirus, Betacoronavirus, and Alphacoronavirus. Both animals and humans can be infected by coronaviruses (Ou et al. 2020). SARS-CoV-2 genome is a single-stranded positive-sense RNA of roughly 30,000 nucleotides with four structural proteins encoded by the genome and spike (S) protein is the most important one (Shereen et al. 2020). It is because S protein is the primary target antigen in the SARS-CoV-2 vaccine (Phan 2020). Previously, our study revealed the candidate for a peptide-based vaccine against the virus was identified based on the four structural proteins (Ansori et al. 2020; Normalina et al. 2020). Thus, it is very important to investigate the S protein from Indonesian SARS-CoV-2 isolates.
Many significant variants of SARS-CoV-2 appeared in the end of 2020 (Ansori et al. 2021). D614G mutation ever became the spotlight in the early time of COVID-19 pandemic and had a high correlation with the widespread infection and virulence besides the changes of antigenicity (Korber et al. 2020; Nidom et al. 2020a, b). Mutations on SARS-CoV-2 need to be highly supervised and mapped with the sole goal in overcoming COVID-19 pandemic (Tegally et al. 2021). These days WHO had mapped four variants which needed to be attention, there were the variants of Alpha, Beta, Gamma, and Delta (Duong 2021). Delta variant has caused an increasing crisis in Indonesia after hitting India for about two months (May 2021). The number of cases in Indonesia has sharply arisen over the last month (July 2021) and kept rising (Dyer 2021; Kupferschmidt and Wadman 2021).
Recently, many attempts have been made by scientists to generate vaccines to fight against SARS-CoV-2 worldwide with protein-based vaccines as the most advanced types and the private sector is at the forefront of these studies (Belete 2020; Callaway 2020; Nidom et al. 2020b; van Riel and de Wit 2020). However, the mutation rate of Coronaviridae is reminding high as various studies reported the mutation might implicate the efficacy of vaccines or other therapeutic strategies. Mutations on variants or lineages have appeared in several countries around the world which need to be mapped (Chen et al. 2021; Greaney et al. 2021; Weisblum et al. 2020) including in Indonesia. Thus, investigating the phylogenetic and full-length genome mutation analysis of 166 Indonesian SARS-CoV-2 isolates became the goal of this study.
Methods
SARS-CoV-2 isolates
The data extraction of all Indonesian SARS-CoV-2 isolates (166 isolates) from Global Initiative on Sharing All Influenza Data (GISAID) EpiCoV database (https://www.gisaid.org/) was completed on 12 January 2021. This study only used the complete genome and high coverage criteria according to the GISAID EpiCoV standard. All of 166 Indonesian SARS-CoV-2 isolates were derived from various provinces in Indonesia, for example: Special Region of Aceh, North Sumatra, Lampung, Banten, Special Capital Region of Jakarta, West Java, Central Java, Special Region of Yogyakarta, East Java, and so on (Additional file 1). This study identified the total isolates in every province, GISAID clades, and lineages. All data were visualized using GraphPad Prism software v.9.2 (GraphPad Software, Inc., California, USA). Additionally, WIV04 isolate (GISAID clade: L; lineage: B) was collected from a female retailer at Huanan Seafood Wholesale Market, submitted by the Wuhan Institute of Virology, Chinese Academy of Sciences in China and applied as a virus reference based on CoVsurver default in this study (Sengupta et al. 2021).
Full-length genome mutation analysis
Every single gene of 166 Indonesian SARS-CoV-2 isolates was investigated, such as NSP1-16, S, NS3, E, M, NS6, NS7a, NS7b, NS8, and N. In addition, the application of CoVsurver platform (https://www.gisaid.org/epiflu-applications/covsurver-mutations-app) was applied to investigate the full-length genome mutation analysis of all isolates (Sengupta et al. 2021). Besides, the mapping of D614G mutation from the isolates derived from the database was also performed in this study based of the study from Korber et al. (2020). All data were visualized using GraphPad Prism software v.9.2 (GraphPad Software, Inc., California, USA) and BioRender platform (https://www.biorender.com).
3D structure visualization
In this study, 3D structure visualization of SARS-CoV-2 S protein was rendered by utilizing SWISS-MODEL web server (https://swissmodel.expasy.org) and PyMOL v2.4 (Schrödinger, Inc, New York, USA) with professional license for academic (Gurung 2020; Raj 2020). Then, the schematic diagram was edited with BioRender platform (https://www.biorender.com) (Ansori et al. 2020). This method was employed to easily identify the location of various mutations in SARS-CoV-2 S protein.
Molecular phylogenetic analysis
The molecular phylogenetic modelling and tree visualization was rendered by applying MEGA X software (Pennsylvania State University, USA) to the maximum likelihood method using the Indonesian and other coronavirus isolates from other places such as Nepal, Sri Lanka, Russia, Nigeria, Serbia, Bangladesh, Greece, Poland, Morocco, Czech Republic, the Netherland, Germany, Saudi Arabia, Egypt, South Africa, Malaysia, Guam, France, Australia, Puerto Rico, Tunisia, Chile, Vietnam, United States, Uruguay, Thailand, Timor-Leste, Spain, Kenya, Japan, Pakistan, Kazakhstan, Colombia, Jamaica, Hong Kong, Israel, Italy, Brazil, China, Turkey, South Korea, India, Sweden, and Iran. Furthermore, coronaviruses derived from bat, pangolin, mink, and other coronaviruses isolated which had infected human previously were also utilized as the comparison. The molecular phylogenetic was tested by 1000 bootstrapped input datasets and cross-referencing it with the Tamura-Nei substitution model (Ansori et al. 2021; Normalina et al. 2020; Sohpal 2020; Stefanelli et al. 2020).
Results
Full-length genome mutation analysis and 3D structure visualization
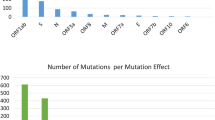
This study took 166 SARS-CoV-2 isolates from various regional areas in Indonesia, such as Special Region of Aceh (n = 2), North Sumatra (n = 3), Riau Islands (n = 1), Bengkulu (n = 1), Lampung (n = 2), Banten (n = 8), Special Capital Region of Jakarta (n = 42), West Java (n = 25), Central Java (n = 7), Special Region of Yogyakarta (n = 15), East Java (n = 31), South Kalimantan (n = 2), North Kalimantan (n = 1), Central Kalimantan (n = 2), East Kalimantan (n = 3), North Sulawesi (n = 8), Bali (n = 2), West Nusa Tenggara (n = 1), East Nusa Tenggara (n = 3), North Papua (n = 1), West Papua (n = 1), and Papua (n = 5) (Fig. 1A and Additional file 1). Various lineages of SARS-CoV-2 are also mapped in this study. As a result, this study identified 14 lineages from 166 isolates and the Lineage B (n = 54) was dominant in Indonesia (Fig. 1B). Significantly, the isolates were grouped in the G (n = 3), GH (n = 73), GR (n = 20), L (n = 55), and O (n = 15) clades. This result also revealed those lineages of SARS-CoV-2 isolates in Indonesia according to the accumulated data from GISAID EpiCoV database and found that Clade GH was dominant in Indonesia (Fig. 1C).

All Indonesian SARS-CoV-2 isolates data. A The origin of isolates; B the lineages distribution of the isolates; C the GISAID EpiCoV clades; and D the full-length genome mutation analysis of all isolates used in this study. All data were visualized using GraphPad Prism software
This study also analyzed the full-length genome mutation analysis of all isolates using a heat map data (Fig. 1D and Additional file 1). Based on the data, non-structural protein 3 (NSP3) became the most frequent mutation compared to all genes in 166 Indonesian SARS-CoV-2 isolates. In line with this, various genes, such as NSP12, NS3, and S protein, also were found to have high mutation numbers.
In this study, D614G mutation was detected in 103 Indonesian SARS-CoV-2 isolates. All the isolates were mostly from the West Java (n = 22), East Java (n = 20), Special Region of Yogyakarta (n = 12), Special Capital Region of Jakarta (n = 11), and Central Java (n = 7), respectively (Fig. 2A). The results also demonstrated the mapping of amino acid mutation sites in S protein of all SARS-CoV-2 isolates (Fig. 2B, C). However, this study could not find any various important novel mutations or its variants like 484K, 501Y, and 681H. The 3D visualization of structure from SARS-CoV-2 S protein was developed using WIV04 isolate. It also was marked with the red dots in every amino acid mutation of S protein occurred as mentioned previously in this study (Fig. 2B and Additional file 1).

Mutation analysis originated from S protein of Indonesian SARS-CoV-2 isolates. A Distribution of D614G mutation from various provinces in Indonesia; B 3D structure visualization of SARS-CoV-2 S protein. Red dots are the location of various mutations in S protein; C The mapping of amino acid mutation sites in S protein of 166 Indonesian SARS-CoV-2 isolates
Molecular phylogenetic analysis
This study developed a molecular phylogenetic tree and presented the relationship between Indonesian SARS-CoV-2 isolates, many isolates from various nations around the world, and the coronaviruses originating from humans, mink, bats, and pangolins (Fig. 3). Here, this research reported an advanced studied to construct the Indonesian virus isolates’ molecular phylogenetic.

Molecular phylogenetic of SARS-CoV-2 isolates and other coronaviruses. Indonesian SARS-CoV-2 isolates and other SARS-CoV-2 isolates from various countries are grouping into a clade and closely related to CoV RATG13 from bats, CoV from pangolin, and SARS-CoV from human, respectively
Discussion
Until the end of 2019, there were six identified coronaviruses to be causative agents of infection in humans. The seventh, SARS-CoV-2, emerged in China (Khan et al. 2020a, b; Wu et al. 2020). To date, according to the CSSE at Johns Hopkins University online website, there are more than 195 million people infected with the virus globally (Dong et al. 2020). Moreover, the reports said that human-to-human transmission has occurred and WHO has acknowledged the chance of aerosol infection (Tellier et al. 2019). Based on our study, the identification of the virus was generated from several collection methods through swabbing activities in saliva, throat, sputum, bronchoalveolar-lavage, oropharyngeal and nasopharyngeal area.
The data of 166 Indonesian SARS-CoV-2 isolates were retrieved prior to the beginning of COVID-19 vaccination program on 13 January 2021 from the database used to pool the virus samples collected from sputum, oropharyngeal and nasopharyngeal swabs by many collaborations among research centres and universities in Indonesia.
Recent updates show that GISAID EpiCoV database has acknowledged seven subtypes of SARS-CoV-2, specifically V, S, O, L, GR, GH, GV, and G clades. Significantly, the isolates from Indonesia in this study were grouped into the G, GH, GR, L, and O clades. This study also reveals those above lineages of SARS-CoV-2 isolates in Indonesia according to the accumulated data from GISAID EpiCoV database (Fig. 1). In this study, 14 lineages from 166 isolates were displayed in this study (Fig. 1). However, based on this data, there were no any novel lineages identified related to new variants, while many reports stated that novel variants of SARS-CoV-2 occurred in various countries, such as UK, Brazil, and South Africa (Ali et al. 2021; Chaillon and Smith 2021; Tegally et al. 2021; Volz et al., 2021; Nonaka et al. 2021). These novel variants might be more transmissible and suspected to be accountable for the rise of COVID-19 patient numbers in those countries (Tegally et al. 2021; Volz et al., 2021).
Meanwhile, S protein mediates the entry and membrane fusion of the new virus and is the main target for many studies of antiviral drugs and vaccines (Jean et al. 2020; Syahniar et al. 2020). S1 and S2 are the two domains of the virus S protein. S1 is conscientious for binding to host cellular receptors. Besides the efficacy of several therapies which include disrupting protease inhibitors, small RNAs, neutralizing antibodies, fusion blockers, S protein inhibitors, ACE2 blockers, however, the in vitro studies on S protein inhibitors have been unsatisfactory (Yin 2020). Many methods have been employed to produce vaccines using S protein as an antigen (Normalina et al. 2020; Watanabe et al. 2020).
Scientists have demonstrated that mutations occur in the virus genome globally (Ansori et al. 2020; Benvenuto et al. 2020; Joob and Wiwanitkit 2020; Phan 2020). Previously, Phan et al. performed a genetic analysis in 86 virus genomes and reported many mutations. One of the most important mechanisms proposed for the evolution of viruses in nature is nucleotide substitution (Phan 2020). Yadav et al. (2020) also reported a study to analyze the first two virus isolates from India, while Garcés-Ayala et al. (2020) who conducted a study with the reference sequence for fully describing the novel SARS-CoV-2 complete genome in Mexico. Khailany et al. (2020) successfully retrieved 94 SARS-CoV-2 genomes and checked the molecular variation between them. Furthermore, Kim et al. (2020) revealed that the quick transmission and infectivity of the virus correlated with specific mutations in the genome. This study reported various S protein mutations such as A222, S477, D614, Q677, and so on (Fig. 2). Further research was highly considered that S protein mutations to affect vaccination program worldwide (Le Page 2021; Xie et al. 2021; Zhang et al. 2020).
Besides, recent publications show that one of the most notable amino acid mutations is D614G (Korber et al. 2020; Nidom et al. 2020a, b). Based on these recent studies, the virus virulence and the increase of viral loads in COVID-19 patients characterize the occurrence of D614G mutation (Korber et al. 2020; Zhang et al. 2020), while, based on the current available information, the infectivity as well as the receptor binding, fusion activation, or ADE enhancement can be influenced by D614G mutation in several ways (Ulrich et al. 2020; Wang and Zand. 2020; Nidom et al. 2020a). An antibody escape is considered as another mutation mechanism like the upcoming form of D614G which can be accelerated by an antigenic drift. If the sensitivity of neutralizing antibody can be affected by D614G mutation in SARS-CoV-2 or vice versa, then the ADE activity also can be monitored in the SARS-CoV study; thus, D614G can be considered as an intermediate antibody escape which puts people to be more vulnerable for second infections (Cloutier et al. 2020; Zhang et al. 2020; Nidom et al. 2020a).
A study by Zhang et al. on D614G mutation which discovered that S1 residue 614 is in a close proximity to S2 domain. An altered release or shedding of S1 domain after cleavage at S1/S2 junction might be displayed by the ratio between S1 and S2 domains in the virion. Glycine amino acid found at residue 614 of S protein G614 secures the interaction between S1 and S2 domains and limits S1 shedding. D614G mutation has been previously speculated in raising an open configuration of S protein that is more advantageous to ACE2 association (Zhang et al. 2020). Therefore, SARS-CoV-2 S protein D614G mutation is highly believed in promoting the virion spike density and infectivity and it is also highly speculated that this mutation might be influence further mutations.
Previous studies on the molecular phylogenetic tree revealed that the relationship of SARS-CoV-2 and other Coronaviridae is based on the four structural protein genes. In accordance with this, SARS-CoV-2 is considered to be the closest to Rhinolophus affinis coronavirus RaTG13 and followed by pangolin coronavirus (Andersen et al. 2020). Thus, Malayan pangolin is assumed as the intermediate host before infecting to humans (Tu et al. 2020). In addition, the previous study also reported that the type of mutation emerged in the virus isolates were originated from canine, environment, Felis catus, mice, Mustela lutreola, and Panthera tigris jacksoni (Nidom et al. 2020a) (Fig. 3). It further advised in promoting the surveillance researches to be implemented in many mammals in their native habitats including bats and pangolins, especially in East Asia; thus, the risk of the forthcoming zoonotic diseases can be well-predicted.
Compared to most other microorganisms, the rates of RNA viruses’ mutations are much higher (Chen and Chen. 2020). An elevated mutation rate can lead to an increase in virulence and a high potential for adaptive evolution (Chen and Chen 2020; Khan et al. 2020a, b; Wang et al. 2020). This capability boosts the chance of zoonotic viral pathogens to establish human-to-human transmission and permits them to enhance their virulence (Wang et al. 2020).
This study provides the fundamental data for accomplished studies into the medication and prevention of COVID-19. Indonesian SARS-CoV-2 genomic data extraction would be valuable in vaccine construction and options in medication. In fact, mining the data from the Indonesian SARS-CoV-2 variants and molecular epidemiology could enable the mapping of its origin and the tracking of its transmission (Setiawaty et al. 2020). In line with this, the sequence investigation performs an important role in viral surveillance, public health policy problems, and host identification (Álvarez-Díaz et al. 2020; Setiawaty et al. 2020). Thus, high-speed detection of mutations from the Indonesian SARS-CoV-2 is mandatory in the unlocking to the COVID-19 pandemic in Indonesia.
As the availability of COVID-19 vaccine is limited, not many people can access it. In countries that have not implemented large-scale active case testing and isolation, controlling the spread of the virus can be very challenging. In this case, transmission suppression relies primarily on the community adherence to non-pharmacological strategies such as social distancing, the mandatory mask using, and hand washing (Bedford et al. 2020; Güner et al. 2020; Lewnard and Lo 2020; Qian and Jiang 2020). Inevitably, as the result of SARS-CoV-2 outbreaks, many countries declared medical emergency which led to the economic emergency since those countries enforcing limited or strict mobility both regionally and nationally (Ahmad et al. 2020; Hiscott et al. 2020; Palacios Cruz et al. 2021). Therefore, regulating and containing further transmission of COVID-19 is a fundamental move to discover the characteristics of SARS-CoV-2 genome and constitute the systems for observing SARS-CoV-2 during this pandemic. The recognition of genotypes related to temporal infectious clusters and specific geographic areas suggests that the employment of genomic data is highly recommended in observing and tracking the further spreading of SARS-CoV-2. Researchers might be able to introduce the origin of a specific variant and observe the virus transmission by acknowledging the specific SARS-CoV-2 variants and connecting them using a molecular epidemiology approach. Ergo, it can be argued that this study might become an important tool in regulating the COVID-19 pandemic in Indonesia.
Conclusion
In conclusion, this study successfully identified the full-length genome mutation analysis of 166 Indonesian SARS-CoV-2 isolates. This study helps in observing the spread of the COVID-19 transmission. However, we proposed that the epidemiological surveillance and genomics studies might be improved on COVID-19 pandemic in Indonesia.
Availability of data and materials
All data on which abstracted of the study have been drawn are presented in the main manuscript.
Abbreviations
- ACE2:
-
Angiotensin-converting enzyme 2
- ADE:
-
Antibody-dependent enhancement
- COVID-19:
-
Coronavirus disease 2019
- CSSE:
-
Center for Systems Science and Engineering
- GISAID:
-
Global Initiative on Sharing All Influenza Data
- RNA:
-
Ribonucleic acid
- S:
-
Spike
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- WHO:
-
World Health Organization
References
Ahmad T, Haroon Baig M, Hui J (2020) Coronavirus disease 2019 (COVID-19) pandemic and economic impact. Pak J Med Sci. 36(COVID19-S4):S73–S78
Ali F, Kasry A, Amin M (2021) The new SARS-CoV-2 strain shows a stronger binding affinity to ACE2 due to N501Y mutant. Med Drug Discov. 10:100086
Álvarez-Díaz DA, Franco-Muñoz C, Laiton-Donato K, Usme-Ciro JA, Franco-Sierra ND, Flórez-Sánchez AC et al (2020) Molecular analysis of several in-house rRT-PCR protocols for SARS-CoV-2 detection in the context of genetic variability of the virus in Colombia. Infect Genet Evol. 84:104390
Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF (2020) The proximal origin of SARS-CoV-2. Nat Med 26:450–452
Ansori ANM, Kusala MKJ, Normalina I, Indrasari S, Alamudi MY, Nidom RV et al (2020) Immunoinformatic investigation of three structural protein genes in Indonesian SARS-CoV-2 isolates. Syst Rev Pharm 11(7):422–434
Ansori ANM, Nidom RV, Kusala MKJ, Indrasari S, Normalina I, Nidom AN et al (2021) Viroinformatics investigation of B-cell epitope conserved region in SARS-CoV-2 lineage B.1.1.7 isolates originated from Indonesia to develop vaccine candidate against COVID-19. J Pharm Pharmacogn Res 9(6):766–779
Artika IM, Dewantari AK, Wiyatno A (2020) Molecular biology of coronaviruses: current knowledge. Heliyon. 6(8):e04743
Bedford J, Enria D, Giesecke J, Heymann DL, Ihekweazu C, Kobinger G et al (2020) COVID-19: towards controlling of a pandemic. Lancet 395(10229):1015–1018
Belete TM (2020) A review on promising vaccine development progress for COVID-19 disease. Vacunas 21(2):121–128
Benvenuto D, Angeletti S, Giovanetti M, Bianchi M, Pascarella S, Cauda R et al (2020) Evolutionary analysis of SARS-CoV-2: How mutation of non-structural protein 6 (NSP6) could affect viral autophagy. J Infect S0163–4453(20):30186–30189
Callaway E (2020) The race for coronavirus vaccines: a graphical guide. Nature 580:576–577
Chaillon A, Smith DM (2021) Phylogenetic analyses of SARS-CoV-2 B.1.1.7 lineage suggest a single origin followed by multiple exportation events versus convergent evolution. Clin Infect Dis ciab265
Chen JW, Chen JM (2020) Potential of live pathogen vaccines for defeating the COVID-19 pandemic: history and mechanism. J Med Virol 92(9):1469–1474
Chen RE, Zhang X, Case JB, Winkler ES, Liu Y, VanBlargan LA et al (2021) Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat Med 27(4):717–726
Cloutier M, Nandi M, Ihsan AU, Chamard HA, Ilangumaran S, Ramanathan S (2020) ADE and hyperinflammation in SARS-CoV2 infection- comparison with dengue hemorrhagic fever and feline infectious peritonitis. Cytokine 136:155256
Dong E, Du H, Gardner L (2020) An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis S1473–3099(20):30120–30121
Duong D (2021) Alpha, Beta, Delta, Gamma: What’s important to know about SARS-CoV-2 variants of concern? CMAJ 193(27):E1059–E1060
Dyer O (2021) Covid-19: Indonesia becomes Asia’s new pandemic epicentre as delta variant spreads. BMJ. 374:n1815
Garcés-Ayala F, Araiza-Rodríguez A, Mendieta-Condado E, Rodríguez-Maldonado AP, Wong-Arámbula C, Landa-Flores M et al (2020) Full genome sequence of the first SARS-CoV-2 detected in Mexico. Arch Virol 165:2095–2098
Greaney AJ, Loes AN, Crawford KHD, Starr TN, Malone KD, Chu HY et al (2021) Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 29(3):463-476.e6
Gunadi Wibawa H, Marcellus Hakim MS, Daniwijaya EW, Rizki LP, Supriyati E et al (2020) Full-length genome characterization and phylogenetic analysis of SARS-CoV-2 virus strains from Yogyakarta and Central Java, Indonesia. PeerJ. 8:e10575
Gunadi WH, Hakim MS, Marcellus TI, Khair RE et al (2021) Molecular epidemiology of SARS-CoV-2 isolated from COVID-19 family clusters. BMC Med Genomics 14(1):144
Güner R, Hasanoğlu I, Aktaş F (2020) COVID-19: prevention and control measures in community. Turk J Med Sci. 50(SI-1):571–577
Gurung AB (2020) In silico structure modelling of SARS-CoV-2 Nsp13 helicase and Nsp14 and repurposing of FDA approved antiviral drugs as dual inhibitors. Gene Rep. 21:100860
Hiscott J, Alexandridi M, Muscolini M, Tassone E, Palermo E, Soultsioti M et al (2020) The global impact of the coronavirus pandemic. Cytokine Growth Factor Rev 53:1–9
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y et al (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395(10223):497–506
Jean SS, Lee PI, Hsueh PR (2020) Treatment options for COVID-19: the reality and challenges. J Microbiol Immunol Infect 53(3):436–443
Joob B, Wiwanitkit V (2020) Genetic variant severe acute respiratory syndrome coronavirus 2 isolates in Thailand. J Pure Appl Microbiol 14:6314
Khailany RA, Safdar M, Ozaslan M (2020) Genomic characterization of a novel SARS-CoV-2. Gene Rep. 19:100682
Khan MI, Khan ZA, Baig MH, Ahmad I, Farouk AE, Song YG et al (2020) Comparative genome analysis of novel coronavirus (SARS-CoV-2) from different geographical locations and the effect of mutations on major target proteins: An in silico insight. PLoS ONE 15(9):e0238344
Khan M, Adil SF, Alkhathlan HZ, Tahir MN, Saif S, Khan M et al (2020a) COVID-19: A global challenge with old history, epidemiology and progress so far. Molecules 26(1):39
Kim JS, Jang JH, Kim JM, Chung YS, Yoo CK, Han MG (2020) Genome-wide identification and characterization of point mutations in the SARS-CoV-2 genome. Osong Public Health Res Perspect 11(3):101–111
Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W et al (2020) Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 182(4):812-827.e19
Kupferschmidt K, Wadman M (2021) Delta variant triggers new phase in the pandemic. Science 372(6549):1375–1376
Le Page M (2021) Threats from new variants. New Sci 249(3316):8–9
Lewnard JA, Lo NC (2020) Scientific and ethical basis for social-distancing interventions against COVID-19. Lancet Infect Dis 20(6):631–633
Li X, Giorgi EE, Marichannegowda MH, Foley B, Xiao C, Kong XP et al (2020) Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci Adv. 6(27):eabb9153
Nidom RV, Ansori ANM, Indrasari S, Normalina I, Kusala MKJ, Saefuddin A et al (2020a) Recent updates on COVID-19 vaccine platforms and its immunological aspects: a review. Syst Rev Pharm 11(10):807–818
Nidom RV, Indrasari S, Normalina I, Kusala MKJ, Ansori ANM, Nidom CA (2020b) Investigation of the D614G mutation and antibody-dependent enhancement sequences in Indonesian SARS-CoV-2 isolates and comparison to Southeast Asian isolates. Syst Rev Pharm 11(8):203–213
Nonaka CKV, Franco MM, Gräf T, de Lorenzo Barcia CA, de Ávila Mendonça RN, de Sousa KAF et al (2021) Genomic evidence of SARS-CoV-2 reinfection involving E484K spike mutation, Brazil. Emerg Infect Dis 27(5):1522–1524
Normalina I, Indrasari S, Nidom RV, Kusala MKJ, Alamudi MY, Santoso KP et al (2020) Characterization of the spike glycoprotein and construction of an epitope-based vaccine candidate against Indonesian SARS-CoV-2: In silico study. Syst Rev Pharm 11(7):404–413
Ou X, Liu Y, Lei X, Li P, Mi D, Ren L et al (2020) Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun 11:1620
Palacios Cruz M, Santos E, Velázquez Cervantes MA, León Juárez M (2021) COVID-19, a worldwide public health emergency. Rev Clin Esp 221(1):55–61
Phan T (2020) Genetic diversity and evolution of SARS-CoV-2. Infect Genet Evol. 81:104260
Qian M, Jiang J (2020) COVID-19 and social distancing. Z Gesundh Wiss
Raj R (2020) Analysis of non-structural proteins, NSPs of SARS-CoV-2 as targets for computational drug designing. Biochem Biophys Rep. 25:100847
Sengupta A, Hassan SS, Choudhury PP (2021) Clade GR and clade GH isolates of SARS-CoV-2 in Asia show highest amount of SNPs. Infect Genet Evol. 89:104724
Setiawaty V, Kosasih H, Mardian Y, Ajis E, Prasetyowati EB, Siswanto et al (2020) The identification of first COVID-19 cluster in Indonesia. Am J Trop Med Hyg. 103(6):2339–2342
Shereen MA, Khan S, Kazmi A, Bashir N, Siddique R (2020) COVID-19 infection: origin, transmission, and characteristics of human coronaviruses. J Adv Res 24:91–98
Sohpal VK (2020) Computational analysis of SARS-CoV-2, SARS-CoV, and MERS-CoV genome using MEGA. Genomics Inform. 18(3):e30
Stefanelli P, Faggioni G, Lo Presti A, Fiore S, Marchi A, Benedetti E et al (2020) Whole genome and phylogenetic analysis of two SARS-CoV-2 strains isolated in Italy in January and February 2020: additional clues on multiple introductions and further circulation in Europe. Euro Surveill 25(13):2000305
Syahniar R, Purba MB, Bekti HS, Mardhia M (2020) Vaccines against coronavirus disease: target proteins, immune responses, and status of ongoing clinical trials. J Pure Appl Microbiol 14(4):2253–2263
Tegally H, Wilkinson E, Lessells RJ, Giandhari J, Pillay S, Msomi N et al (2021) Sixteen novel lineages of SARS-CoV-2 in South Africa. Nat Med 27(3):440–446
Tellier R, Li Y, Cowling BJ, Tang WJ (2019) Recognition of aerosol transmission of infectious agents: a commentary. BMC Infect Dis 19(1):101
Tu YF, Chien CS, Yarmishyn AA, Lin YY, Luo YH, Lin YT et al (2020) A review of SARS-CoV-2 and the ongoing clinical trials. Int J Mol Sci 21(7):2657
Ulrich H, Pillat MM, Tárnok A (2020) Dengue fever, COVID-19 (SARS-CoV-2), and antibody-dependent enhancement (ADE): a perspective. Cytometry A 97(7):662–667
van Riel D, de Wit E (2020) Next-generation vaccine platforms for COVID-19. Nat Mater 19(8):810–812
Volz E, Mishra S, Chand M, Barrett JC, Johnson R, Geidelberg L et al (2021) Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature 593(7858):266–269
Wang J, Zand MS (2020) The potential for antibody-dependent enhancement of SARS-CoV-2 infection: translational implications for vaccine development. J Clin Transl Sci 1–4.
Wang C, Liu Z, Chen Z, Huang X, Xu M, He T et al (2020) The establishment of reference sequence for SARS-CoV-2 and variation analysis. J Med Virol. 92(6):667–674
Watanabe Y, Allen JD, Wrapp D, McLellan JS, Crispin M (2020) Site-specific glycan analysis of the SARS-CoV-2 spike. Science 369(6501):330–333
Weisblum Y, Schmidt F, Zhang F, DaSilva J, Poston D, Lorenzi JC et al (2020) Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife. 9:e61312
Wu JT, Leung K, Leung GM (2020) Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: A modelling study. Lancet 395(10225):689–697
Xie X, Liu Y, Liu J, Zhang X, Zou J, Fontes-Garfias CR et al (2021) Neutralization of SARS-CoV-2 spike 69/70 deletion, E484K and N501Y variants by BNT162b2 vaccine-elicited sera. Nat Med 27:620–621
Yadav PD, Potdar VA, Choudhary ML, Nyayanit DA, Agrawal M, Jadhav SM et al (2020) Full-genome sequences of the first two SARS-CoV-2 viruses from India. Indian J Med Res 151(2 & 3):200–209
Yin C (2020) Genotyping coronavirus SARS-CoV-2: methods and implications. Genomics S0888–7543(20):30318–30319
Zhang L, Jackson CB, Mou H, Ojha A, Peng H, Quinlan BD et al (2020) SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat Commun 11:6013
Acknowledgements
We would like to take this opportunity to express our deepest condolences to the victims of COVID-19. We would also like to pay tribute to our frontline heroes who have fought continuously to this day. Thankfully, we also recognize the authors from the originating and submitting laboratories of GISAID database on which the samples were derived from. We would like to thank to National Institute of Health Research and Development, Ministry of Health, Indonesia (Balitbangkes Kemenkes, Republic of Indonesia) for the supports. Additionally, we also thank Dewi Sartika, M.Ed. for her help in editing the manuscript.
Funding
This research is financially supported by the Professor Nidom Foundation, Surabaya, Indonesia (Grant Number: 005/PNF-RF/07/2020).
Author information
Authors and Affiliations
Contributions
RVN, SI, IN, ANMA, and MKJK completed the sample collection, the data analysis, and the writing of the draft of the manuscript. RVN, ANN, BA, LD, AKP, ANMA, and CAN were designed the research work and involved in the result interpretations and reviewed the manuscript. MYA and CAN were involved in reviewing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Table of 166 Indonesian SARS-CoV-2 Isolates and Its Mutations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nidom, R.V., Indrasari, S., Normalina, I. et al. Phylogenetic and full-length genome mutation analysis of SARS-CoV-2 in Indonesia prior to COVID-19 vaccination program in 2021. Bull Natl Res Cent 45, 200 (2021). https://doi.org/10.1186/s42269-021-00657-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42269-021-00657-0