
Abstract
Background
The field of laser-based photochemical reactions receives a great promising for additional applications especially for targeting cells, pathogens, or molecules. Limited studies have investigated the impact of light-emitting diode on stem cell behavior. Thus, the aim of the present study was to assess the effect of diode laser irradiations on the proliferation of stem cells isolated from the human dental pulp. Isolation procedures were according to previously developed protocols for dental pulp stem cells (DPSCs). Low-level laser therapy irradiation (LLLT) was applied in two doses (0.5 J/cm2 and 1 J/cm2 for 20 s) into 96-well plates by the diode laser device (970 nm) through the fiber optic (SiroLaser fibers 320) at a distance from the opening of the wells to be accurate for equal distribution of the laser irradiation. To assess the proliferation capacity of the isolated stem cells, MTT assay was performed 24 h, 48 h, and 72 h.
Results
There was no significant difference among the different groups (gp 1 control, gp II dose 0.5 J/cm2, gp III dose 1 J/cm2) on day 1. While on day 2, the optical density of DPSCs subjected to dose 1 was found to be significantly higher than that of those subjected to dose 2. This was also demonstrated on day 3. It was also demonstrated that the proliferation of DPSCs subjected to dose 1 increased compared to that of DPSCs subjected to dose 2 from day 1 to day 2. There was a significant decrease in the cell number in both groups by 72 h.
Conclusion
In conclusion, the use of LLLT as a stimulatory factor for enhancing and proliferation of the stem cells is very promising.
Similar content being viewed by others

Background
Surface treatments such as radiation grafting, plasma surface modification, and the implementation of various coatings in biological applications are becoming more widely used within science and industry. These surface treatments increase the ability to enhance surface properties and enhance the bio-functionality of the material (Waugh et al. 2016). Laser technology has evolved at a high rate of advance for both the hard tissue and soft tissue application in the last decade of life. The field of laser-based photochemical reactions receives a great promising for additional applications especially for targeting cells, pathogens, or molecules (Sethi et al. 2012). Recently, diode lasers have undergone tremendous developments that showed novel ways for the researchers, and have some interesting attributes like the coast effectiveness, size, high reliability, and simplicity for use which make diode lasers good candidates for utilization in different applications (Nasim and Jamil 2014). In recent years, it was used to improve the life of patients with conditions that range from Alzheimer’s disease to cardiac ischemia and regenerative medicine, like bone or tooth loss. Stem cell researches have grown exponentially. It is also now recognized that stem cell-based therapies have the potential to increase the life of diseased patients. Based on their ability to rescue and/or repair injured tissue and partially restore organ function, multiple types of stem/progenitor cells have been speculated (Balic et al. 2010). Growing evidence demonstrates that stem cells are primarily found in niches and that certain tissues contain more stem cells than others. Mesenchymal stem cells (MSCs), which are found in adult tissues, are an attractive cell therapy for the regeneration of the damaged tissues due to their ability to self-renew and differentiate into various cells and tissues (Mvula et al. 2010). Among these tissues, the dental tissues are considered a rich source of mesenchymal stem cells that are suitable for tissue engineering applications. Dental pulp stem cells (DPSCs), like the mesenchymal stem cells, and stem cells from human exfoliated deciduous teeth (SHED) can undergo self-renewal and have multipotent differentiation ability. Unlike embryonic stem cells, which involve the destruction of human embryo, DPSC and SHED have limited ethical concerns as they are readily and easily accessible, non-invasive, and disposed of naturally. Not only can DPSC and SHED be used for cell-based therapies and bio-artificial tissue constructs but can also be induced to differentiate into other cell types (Kim et al. 2012). Both DPSCs from permanent teeth and stem cells from SHED have attracted tremendous interest recently by playing a major role in tissue engineering and regenerative medicine. However, since stem cell technology is still in its infancy, interdisciplinary cooperation between medicine, basic biological research, nanotechnology, and materials science is needed to achieve successful clinical applications (Miura et al. 2003). Promoting the proliferation of different cells has shown to be effective with low-intensity laser irradiation. Low-intensity laser mechanism induces bio-stimulation of the cell activity and formation of a transmembrane electromechanical proton in mitochondria (Farea et al. 2014). Laser was applied to hard tissue for caries prevention, bleaching, restoration removal, curing, cavity preparation, etc., whereas the soft tissue laser application was used in accelerating wound healing, surgical removal of soft tissue, photodynamic therapy of malignancies, photostimulation of herpetic lesions, and increase proliferation and differentiation of stem cells (Friedmann et al. 1991). Thus, the aim of the present study was to assess the effect of diode laser irradiations on the proliferation of stem cells isolated from the human dental pulp.
Methods
Sample collection and stem cell isolation
Teeth were collected from the outpatient clinic of the Excellence Medical unit of the National Research Centre. All teeth were premolars indicated for extraction as a part of the Orthodontic treatment plan of the children involved. All cases were approved by the National Research Centre’s ethical committee. All donors were apparently healthy aged from 16 to 20 years. The collected human dental pulp tissues were obtained from the extracted premolars teeth following the protocols described by Eduardo et al. (2008) and Gronthos et al. (2000). Prior to extraction, patients received chlorhexidine mouth rinse to reduce oral microbial flora. Extraction was as atraumatic as possible in complete aseptic condition. The freshly extracted premolar was then grooved using a diamond stone at nearly the level of the cemento-enamel junction under sterile conditions using sterile saline coolant to facilitate later splitting. The groove surrounded the whole crown till half the depth of dentin without exposing the pulp. The tooth was immediately placed in a sterile tube containing phosphate buffer solution (PBS) and transferred to the laboratory in less than 2 h using a sterile cement spatula under the laminar; the crown was split; and the pulp was gently removed in sterilized conditions using sterile tweezers, a small excavator, and K-files and then immediately placed in PBS.
Sample preparation
-
The extirpated pulp tissues were washed in PBS three times, and the specimens were placed in a small plate and minced into small pieces 1 mm3 using sterile scissors and surgical blades.
-
The minced pieces were collected in sterile, labeled 1.5-ml Eppendorf tubes.
-
Enzyme digestion was carried out according to Gronthos et al. (2000). The digested solution used was 3 mg/ml collagenase type I (Sigma-Aldrich, USA) for 60 min at 37 °C.
-
The digestive reaction was stopped by the addition of culture media. The culture media consisted of alpha modified Eagle’s medium (αMEM) with l-glutamine (Gibco, Invitrogen Life Technologies, USA) supplemented with 10% fetal bovine serum (Gibco, Invitrogen Life Technologies, USA), antibiotics, penicillin/streptomycin (penicillin G100 unit/ml and streptomycin 100 μg/ml), and finally antimycotic agent (Fungizone, 0.25 μg/ml).
-
Single-cell suspensions were obtained by passing the digested tissues through a 70-μm cell strainer (Becton/Dickinson, USA). The strainer also ensured the removal of debris.
-
The tubes were centrifuged for 20 min at room temperature to obtain a cell pellet of pulp-derived cells.
-
The supernatant was discarded, and then the cells in the pellet were resuspended in complete culture media by successive pipetting.
At this point, 1 ml of the tube was taken and 0.2% Trypan Blue stain (Sigma–Aldrich, UK) was mixed with the cell suspension and incubated for 5 min at room temperature, then placed on a Nubarau hemocytometer. The plates were labeled by cell type and date and incubated at 37 °C in a humidified atmosphere of 5% CO2. The medium was changed once every 4 days.
Sub-culturing “passaging”
Passaging was performed when the primary cell culture of adherent cells reached 70% confluence and was named passage zero (P0), and later passages were named accordingly.
-
Primary cell cultures of adherent cells were detached by treatment with a sterile solution of trypsin/EDTA for 5–10 min at 37 °C in the incubator and shaken intermittently.
-
The used trypsin was prepared with the dissolution of 2500 mg trypsin powder in 100 ml PBS solution and filtered onto 2-mm filters. Fetal bovine (100 μl) serum was added to inactivate the trypsin.
-
The cells were then collected by centrifugation at 200 rpm for 10 min, and the cell pellet obtained was resuspended in 1-ml complete medium and divided into two plates (passaging) both followed by immersion in complete culture medium to increase cell numbers.
-
Thus, the primary cell culture was propagated and expanded in repeated cell cultures.
-
Cells were sub-cultured every other week, and the culture medium was replaced every 3 days over a 10–14-day period. All the previous procedures were conducted under aseptic conditions in an air-filtered laminar flow safety cabinet using sterile instruments.
Experimental groups
The culture was divided into 3 groups according to the LLLT energy density of irradiation:
-
Group I: no irradiation (control)
-
Group II: irradiation at dose 0.5 J/cm2for 20 s
-
Group III: irradiation at dose 1 J/cm2for 20 s
Low-level laser therapy irradiation
- Stem cells were seeded in a 96-well plate at a density of 1 × 104 cells/well and allowed to attach overnight.
- Low-level laser therapy irradiation (LLLT) irradiation was applied in two doses into the 96 wells by the diode laser device (970 nm) through the fiber optic (SiroLaser fibers 320) at a distance from the opening of the wells to be accurate for equal distribution of the laser irradiation.
The energy density (fluence) calculated was equal to:
-All doses were executed in continuous wave and noncontact mode
- Prior to irradiation, the media of all wells were replaced by fresh media supplemented with 15% FBS, to ensure the removal of any dead cells.
-
Each group was then evaluated after 24, 48, and 72 h after irradiation using MTT assay.
Assessing cell morphology and proliferation capability
Daily follow-up of stem cells was done to ensure cell viability. Estimation of the cell morphology and proliferation of pulp-derived stem cells were monitored in primary cultures and sub-cultures. The proliferation capacity was judged by close follow-up of confluence rate, i.e., culture plates reaching 70% confluence according to culture days. Cultures from DPSC were monitored using an inverted light microscope (Olympus, USA) with a digital camera for capturing images (Nikon, Japan). Furthermore, the cells were tested for the ability to form colonies where MTT assay was performed to compare the proliferation of the cells. A suspension of 100 cell/ml was cultured in a 3.5-cm dish in complete culture media, and the cultures were observed under an inverted light microscope. Aggregates with more than 50 cells were scored as colonies.
MTT assay
MTT assay according to the standard procedure after the cells were irradiated was performed. An indirect method measuring the metabolic activity of mitochondrial enzymes was used to determine viable cell number in the cultures. The assay is based on the cellular conversion of a tetrazolium salt [MTT: 3(-4, 5-dimethylthiazol-2-yl) 2, 5-diphenyltetrazolium bromide] to formazan. Control and treated cells were incubated with MTT (0.2 mg/ml) diluted in the appropriate media, for 60 min at 37 °C in 96-well plates. The culture medium was removed and formazan was solubilized. The extent of reduction of MTT to formazan within cells was quantified by using a spectrophotometer at a wavelength of 572 nm. Absorbance is directly proportional to the number of living cells in culture. Cell proliferation was measured by MTT assay which was performed after 24 h, 48 h, and 72 h of irradiation according to the cell proliferation kit protocol (Vybrant, Invitrogen). The experiment was repeated in triplicates. This protocol is optimized for dental pulp stem cells.
Results
Morphological appearance of irradiated stem cells
The morphological appearance of the irradiated cells was monitored and demonstrated characteristic features of stem cells similar in appearance to those cells in the control group. Irradiated DPSCS demonstrated the characteristic properties of these cells (spindle, stellate-shaped cells started to appear) (Fig. 1). eThos cells started to form colonies of adherent cells (Fig. 2). Also, these cells increased in number till reaching the confluence by 10–14 days (Fig. 3).

Photomicrograph of DPSC illustrating an increase in the number of stem cells and spindle-shaped cells (magnification × 100)

Photomicrograph of colony formation of the DPSCs illustrating an increase in the number of stem cells and colony formation (magnification × 100)

Photomicrograph of confluence of the DPSCs after reaching confluence, cells illustrate compressed appearance due to increase in number (magnification × 100)
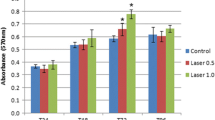
Assessment of cell proliferation via MTT assay
Following laser irradiation, MTT assay was performed after 24 h, 48 h, and 72 h. Stem cell proliferation was increased gradually in all tested groups. However, the highest proliferation capacity was at group III which was significantly higher than the other groups. Statistical analysis (mean, standard deviation, and standard error) for each group was calculated, after 24 h, 48 h, and 72 h. There was no significant difference among the different groups on day 1. While on day 2, the optical density of DPSCs subjected to dose 1 was found to be significantly higher than that of those subjected to dose 2. This was also demonstrated on day 3. Moreover, both DPSCs from both groups proliferated fast in the first 24 h, and then after 48 h (2 days). It was also demonstrated that the proliferation of DPSCs subjected to dose 1 increased as compared to that of DPSCs subjected to dose 2 from day 1 to day 2. There was a significant decrease in cell number in both groups by 72 h. Figure 4 illustrates an increase in the number of cells and proliferation between group I and the other groups after laser irradiation groups II and III. An increase in the number of stem cells at different times and different groups is also demonstrated, where a marked increase in the number and proliferation of the stem cells after 48 h and a marked decrease in the number of the proliferative stem cells after 72 h (Fig. 4) were observed. The effect of different doses of laser irradiation on DPSCs over the tested duration is illustrated in Fig. 5. It was noted that the proliferation of both doses indicated in the number of stem cells was at its maximum at 48 h (2nd day) and that group II was significantly higher than group III (P < 0.005).

Histogram illustrating the number of cells in different times and doses

Histogram illustrating comparison between different doses
Discussion
Due to ethical issues associated with the use of embryonic stem cells, recent attention has focused on stem cells derived from adult tissues. It is thought that adult stem cells have a more restricted differentiation potential compared to embryonic stem cells. Adult stem cells still fulfill the basic characteristics of stem cells’ abilities to self-renew, generate large numbers of progeny, and differentiate into multiple mature cell types. The ability of these cells to regenerate tissues of all germ layers leads to the thought that they may find great use in stem cell regenerative therapy of tissues damaged by disease, aging, or trauma (Bongso et al., 2010). Dental pulp represents a source of stem cells that offers numerous research potentials, if these cells are applied in tissue engineering. It is obvious now that stem cell biology became an important field for tissue regeneration understanding and regenerative medicine implementation (Ginani et al. 2017). Researchers have reported in the bio-stimulating effect of LLLT and how to develop the best laser therapy protocol that will promote positive effects on stem cells (Souzy et al. 2013). Al Ghamdi et al. (2016) (Fernandes et al. 2016) confirmed that the laser wavelengths may be red or near infrared (600–1200 nm) for biological effects, and the energy fluences may range from 0.05 to 10 J/cm2 for bio-stimulatory effects to induce cell proliferation. But if the energies are more than 10 J/cm2, they may produce anti-proliferative effects. In the present study, a model of isolated stem cells from permanent pulp of the premolar teeth was used, and DPSC populations were assessed regarding their proliferation under the influence of laser irradiation. It was found that the proliferation increased at both 24 and 48 h in irradiated cells (gp II and gp III) as compared to non-irradiated cells (gp I), and the proliferation was greater after 48 h than 24 h. Our results were in agreement with Mvula et al. (2008) who found that the number of cells increased at both 24 and 48 h in irradiated cells as compared to non-irradiated cells, and the proliferation was greater after 48 h than 24 h. Their results were observed at 5 J/cm2 and power density of 5.5 mW/cm2, and a wavelength of 635 nm can affect positively in ADSCs in vitro by increase cell viability and cell proliferation. Also, the effect of LLLT on the dental pulp stem cells which are isolated from either normal and inflamed dental pulps using red low-level laser (660 nm) and four different energy fluences (0.05, 0.30, 7, and 42 J/cm2) was reported. It was hypothesized that the first three energy densities would increase the cell proliferation and differentiation where the highest dose (42 J/cm2) would cause an anti-proliferative effect. Also, the same steps in methodology were used in harvesting the normal and inflamed pulp from the extracted teeth, transferring into a sterile regular medium, and incubating at 37 °C in 5% Co2 and 80% humidity environment (Peplow et al., 2010). The laser irradiation was applied at the top of the 96 well at a constant distance. It was found that the phosphate buffer solution (PBS) concentration in the medium plays an important role in the mesenchymal stem cell differentiation (Zaccara et al. 2015). Ferreira et al. (2009) reported that, under nutritional deficiency, no proliferation was observed for stem cells irradiated with low-energy GsAlAs 660-nm and with In GaAlP 780-nm lasers of different powers and wavelengths. P.R. Arany et al.’s (2014) investigations demonstrated that LLLT irradiation (diode laser 810 nm) can be used as minimal invasive tools to activate growth factor complex in the dental pulp. Recently, the best results were obtained in using the visible light spectrum (600–700 nm); in contrast, using the infrared light spectrum (810–830 nm) may lead to inhibition of the proliferation (Mvula et al. 2010). Eduardo et al. (2008) have investigated the effect of LLLT on DPSCs and noted that the energy density (3.0 J/cm2) with a wavelength of 600 nm positively stimulated cell proliferation. It was observed that the bio-stimulatory effect of energy density (1.0 J/cm2) on periodontal ligament stem cells causes stimulation and proliferation of cells. So, the energy density is a very important factor influencing the cell proliferation. Any increase in the energy density may cause damage in the photoreceptors that lead to inhibition of the biomodulatory effect of LLLT on stem cells, but energy density ranged of 0.5–1.0 J/cm2 promoted the bio-stimulation of DPSCs (Soares et al. 2013). In recent studies, it was found that when the cells were irradiated with dose 1.0 J/cm2, there was a more pronounced response compared to the cells irradiated with 0.5 J/cm2 due to insufficient energy density that is applied, and so, no cellular response due to the decrease in the threshold reached to cells (more applying of energy, more threshold achieved), causing positive bio-stimulation (Zaccara et al. 2015). Ginani et al. (2018) found a higher proliferation after 72 h and 96 h of irradiation with a dose of 1.0 J/cm2 in comparison with the non-irradiated control group. On the other hand, for the stem cells derived from the periodontal ligaments, the rate of proliferation was significantly higher when irradiated with a dose of 1.0 J/cm2 after 48 h and 72 h from irradiation. Because of the recent studies, the laser therapy will permit a significant increase in the number of stem cells before differentiation that will lead to an increase in the number of differentiated cells for tissue engineering and healing processes. In our study work, we have two doses of the LLLI by using diode 970 nm, at different power density (0.1 and 0.2 J/cm2) for the same time (20 s). We found that both doses are effective for the proliferation of the stem cells after 24 h and 48 h which were in agreement with previous studies by the researchers (Al Ghamdi et al. 2016). But we also noticed that there was a decrease in the cell proliferation on the third day (after 72 h), in both doses, which was different from what was observed by Park et al. (2004). This could be due to the increase and proliferation in the number of stem cells in relation to 96-well surface area and diameter, which causes triggered apoptosis of the cells.
Conclusion
In conclusion, the use of LLLT as a stimulatory factor for enhancing and proliferation of the stem cells is very promising. It will represent a safe and convenient method for enhancing and accelerating the stem cell proliferation rendering it a promising and beneficial regenerative therapeutic modality.
Availability of data and materials
This article is under the terms of the creative commons Attribution Noncommercial-No Derives License, which permits the use and distribution in any medium, provided the original work is properly cited, the use is non-commercial, and no modifications or adaptations are made. Materials were available in the market, and data was collected in a smooth manner.
Abbreviations
- LLLT:
-
Low-level laser therapy irradiation
- DPSCs:
-
Dental pulp stem cells
- MSCs:
-
Mesenchymal stem cells
- SHED:
-
Stem cells from human exfoliated deciduous teeth
- PBS:
-
Phosphate buffer solution
- MTT:
-
3(-4, 5-Dimethylthiazol-2-yl) 2, 5-diphenyltetrazolium bromide
References
Al Ghamdi AS, Shibly O, Ciancio SG (2016) Osseous grafting part I autografts and allografts for periodontal regeneration – a literature review. J Int Acad Periodontal 12:34–38
Arany P, Mooney D (2014) Photoactivation of endogenous latent transforming growth factor- B1 direct dental stem cells proliferation for regeneration. Sci Transl Med:238–269
Balic R, Mummery C, Wilmut IS (2010) Stem cells: scientific facts and fiction. Academic Press
Bongso A, Fong C-y, Subramanian A, Biswas A (2010) Deviation efficiency, cell proliferation, Freeze-thaw survival, stem cells properties and differetiation of human whartons jelly stem cells. Reprod Biomed Online. 21:391–401
Eduardo F d P, Bueno DF, de Freitas PM, Passo-Bueno MR, Eduardo C d P, Mayanazatz (2008) Stem cells proliferation under low intensity laser irradiation: a preliminary study. Lasers Surg Med 40:433–438
Farea M, Hussein A, Sukarihalim A, Abdullah NA, Keonglim C, Berahim Z, Mokhtar K (2014) Synergistic effects of chitosan scaffold and TGFB on the proliferation and osteogenic differentiation of DPSCs derived from human exfoliated deciduous teeth. Arch Oral Biol 59:1400–1411
Fernandes R, Lisiane B, Simone B, Tiago P, Jose AB, Anna CM (2016) The isolation of stem cells from human deciduous teeth pulp is related to the physiological process of resorption. J Endodontics 37:973–979
Ferreira LS, Netto MC, Maranduba CM, Mello-Moura AC, Marqyes MM (2009) Low Intensity laser phototherapy enhances the proliferation of dental pulp stem cells under nutritional defciency. Braz Oral Res 30
Friedmann H, Lubart R, Rochkind S (1991) A possible explanation of laser induced stimulation and damage of cell culture. J Photochem Photobiol:87–91
Ginani F, Soares DM, Barboza AG (2018) Low level laser irradiation promotes proliferation of cryopreservedadipose derived stem cells. Einestien, Sao Paulo, pp 334–338
Ginani F, Soares DM, Barboza CAG (2017) Low level laser irradiation promotes proliferation of cryopreserved adipose derived stem cells. Einestien, Sao Paulo, pp 334–338
Gronthos S, Mankani M, Brahim J, Robey PG (2000) Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Nat Acad U S 97:13625030
Kim H, Choi K, Kweon O-k, Heekim W (2012) Enhanced wound healing effect of canine adipose-derived mesenchymal stem cells with low level laser therapy in athymic mice. J Dermatol Sci 68:149–156
Miura M, Gronthos S, Zhao M, Lu B, Fisher LW, Robey PG, Shi S (2003) SHED: Stem cells from human exfoliated deciduous teeth. PNAS 100:5807–5812
Mvula B, Moore TJ, Abrahamse H (2008) The effect of low level laser irradiation on adult human adipose stem cells. Lasers Med Sci 25:33–39
Mvula B, Moore TJ, Abrahamse H (2010) The effect of low level laser irradiation on adult human adipose derived stem cells. Lasers Med Sci 25:33–39
Nasim H, Jamil Y (2014) Diode laser: from laboratory to industry. Opt Laser Technol 56:211–222
Park SH, Hsiao GY, Huang GT (2004) Role of substance P and calcitonin gene-related peptide in the regulation of interlukin-8 and monocytes chemotactic protein-1 expression in human dental pulp. Int Endod J 37:185–192
Peplow PV, Baxter GD (2010) Gene expression and release of growth factors duringdelayed wound healing: a review of studies in diabetic animals and possiblecombined laser phototherapy and growth factor treatment to enhance healing. Photomed Laser Surg
Sethi S, Vrana JA, Theis JD, Leung N, Sethi A, Nasr SH, Fervenza FC, Cornell LD, Fidler ME, Dodan A (2012) Laser microdessection and mass spectrometry based proteomics aids the diagnosis and typing of renal amyloidosis. Kidney Int:226–234
Soares DM, Ginani F, Barboza AG (2013) Low level laser irradiation promotes proliferation of cryopreserved adipose derived stem cells. Einestin, Sao Paulo, pp 334–338
Souzy FS, Nadia LS, Effat AA, Reham MA, Reham MA (2013) Isolation and osteogenic differentiation of human adult dental pulp stem cells. J Appl Sci Res
Waugh DG, Hussain I, Lawrence J, Smith GC, Cosgrove D, Toccaceli C (2016) In vitro mesenchymal stem cell response to a CO2 laser modified polymeric material. Mater Sci Eng C 67:727–736 https://doi.org/10.1016/j.msec.2016.05.055
Zaccara IM, Fernanda G, Haroldo GF, Aguida CH, Carlos AG (2015) Effect of low level laser irradiation on proliferation and viability of human dental pulp stem cells. Lasers Med Sci 30:2259–2264
Acknowledgements
- National Research Centre (NRC), Cairo, Egypt
- National Institute of Laser Enhanced Sciences, Cairo University
Funding
Not applicable
Author information
Authors and Affiliations
Contributions
All authors certify that they have participated sufficiently in contributing to the intellectual content, concept, and design of this work. MD carried out the laser application, dosage adjustments, and isolation of stem cells in addition to writing the manuscript. MG helped in the laser application and dose adjustment. RA carried out culturing and maintenance of dental pulp stem cell. NS performed the analysis and interpretation of the data, as well as the revision of the manuscript. All authors have agreed to have their names in the listed order as a contributor of this research. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experiments conducted in this study have been reviewed and were granted approval by the Ethical Committee of the National Research Centre, Cairo, Egypt (reference no: 15023). All patients were informed about the use of their extracted teeth, and they all submitted a written informed consent before enrolment, which was carried out in accordance with the Declaration of Helsinki.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abo El-Dahab, M.M., Gheith, M., Soliman, N.L. et al. Effect of diode laser potentiality on proliferation of dental pulp stem cells (in vitro study). Bull Natl Res Cent 44, 170 (2020). https://doi.org/10.1186/s42269-020-00414-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42269-020-00414-9