
Abstract
Maladaptation in sensory neural plasticity of nociceptive pathways is associated with various types of chronic pain through central sensitization and remodeling of brain connectivity. Within this context, extensive research has been conducted to evaluate the mechanisms and efficacy of certain non-pharmacological pain treatment modalities. These include neurostimulation, virtual reality, cognitive therapy and rehabilitation. Here, we summarize the involved mechanisms and review novel findings in relation to nociceptive desensitization and modulation of plasticity for the management of intractable chronic pain and prevention of acute-to-chronic pain transition.
Similar content being viewed by others


Introduction
Sensory plasticity encodes environmental experiences through functional and structural reorganizational processes that shape memory, perceptual sensitivity and behavior [1]. However, pathological alterations in sensory system’s nociceptive pathways can cause corresponding plasticity to become chronically maladaptive, in response to particularly intense or repetitive noxious triggers, and mediate pain chronification as a form of memory [2]. This is especially evident for the somatosensory system in specific types of neuropathic pain through potentiation of nociception, central sensitization and altered brain connectivity [3, 4]. Since plasticity represents an intrinsic activity-dependent neuronal ability, this suggests that it is essentially reversible or at least modifiable and that sensory experience, learning mechanisms and psychology are critical treatment factors affecting pain perception [2, 5]. Accordingly, a promising therapeutic approach would be to interfere with these alterations or induce the reversal process as “physiologically” as possible; that is, accelerating recovery by resetting the system to the original baseline state via activity or use-dependent mechanisms, reflecting sensory experience, such as neurostimulation [6, 7]. Certain non-pharmacological interventions including neurostimulation and other modalities show significant potentials in the management of maladaptive plasticity of chronic pain and the prevention of acute-to-chronic pain transition. In this review we discuss novel research findings on the efficacy of these treatment modalities in nociceptive desensitization and associated neural plasticity mechanisms.
Nociceptive pathways
Noxious mechanical, thermal and chemical stimuli of cutaneous and visceral tissues are detected by afferent sensory neurons known as nociceptors, which are generally classified into fast Aδ and slow C-type fibers. Acute tissue injury triggers the local release of various inflammatory mediators including adenosine triphosphate (ATP), bradykinins, histamine, prostaglandins, neurotrophic factors and cytokines [8]. These mediators activate nociceptors to generate action potential impulses to transmit detected inputs to central neurons. In addition, activation of C-fibers initiates neurogenic inflammation characterized by the upregulated production and release of neuropeptides such as neurokinin A, substance P and calcitonin gene-related peptide (CGRP) in a retrograde manner [9]. Together, these alterations lead to reversible sensitization of peripheral nociceptors with reduction in activation threshold and increased stimulus-induced and spontaneous discharge [10]. Within the laminae of the spinal dorsal horn the primary afferents, except sensory cranial nerves, synapse with second-order afferents, which include three groups: proprioceptive, nociceptive and wide dynamic range (WDR) neurons. The nociceptive signals are transmitted from primary to secondary afferents via the release of different neurotransmitters, particularly glutamate and substance P. However, dorsal horn nociceptive transmission is also regulated by inhibitory mechanisms involving endogenous opioids, descending inhibitory pathways and inhibitory interneurons releasing γ-amino butyric acid (GABA) and glycine. The secondary afferents form ascending pathways that project to the brain stem and medulla and terminate in the thalamus and cerebral cortex. Lastly, an extensive cortical network commonly referred to as the pain matrix processes nociceptive and other salient sensory inputs [11].
Nociceptive sensitization
Sensitization of nociception is a temporary adaptive process that occurs following inflammatory and noxious tissue insults and involves lowered pain threshold and amplified responses due to nociceptive neuronal hyperexcitability [12]. However, maladaptation in this protective response can arise from central or peripheral pathologies; for instance neuropathy, and lead to chronic pain characterized by allodynia and hyperalgesia [13]. The enhanced responsiveness of central nociceptive neurons is known as central sensitization, which is hypothesized to result in chronic amplification of pain associated with ongoing tissue inflammation, following neuronal injury or even in the absence of peripheral pathology such as in migraine, fibromyalgia and irritable bowel syndrome [14]. Central sensitization is mediated by neural plasticity mechanisms that involve increased neuronal activity, potentiated synaptic efficacy, enlarged receptive fields and reduced inhibition [15]. Therefore, pain “perception” would no longer be coupled to the presence, intensity or duration of noxious inputs, rendering localized peripheral treatments less effective; thus, central sensitization is thought to account, at least partly, for unexplained chronic pain [16]. Peripheral sensitization processes on the level of primary afferents and free nerve endings have also been identified and involve plasticity changes of nociceptors leading to primary hyperalgesia [17]. The maladaptive plasticity processes outlasting tissue healing are, in certain subgroups of patients, associated with various forms of intractable chronic pain; accordingly, central sensitization and associated psychocognitive factors should be taken into account for developing individualized treatments [18].
Neural plasticity of pain
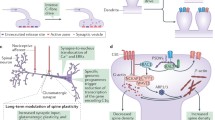
Plasticity changes occur along pain pathways throughout the neuroaxis and mediate peripheral nociceptor sensitization, central (spinal) sensitization and brain remodeling (Fig. 1). Peripherally, inflammatory mediators and retrograde neuropeptides “sensitize” nociceptors leading to upregulation of substance P, transient receptor potential vanilloid (TRPV) and purinergic receptors; in addition to altered membrane ion channels, protein kinase activity and growth factor expression resulting in hypersensitivity and primary hyperalgesia [17]. The main excitatory transmitter of nociceptive neurons is glutamate, which acts upon 3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), N-methyl-d-aspartate (NMDA), kainic acid (KA) and metabotropic glutamate (mGlu) receptors. Under baseline conditions (resting membrane potential), NMDAR channel pore is blocked by Mg2+ ions [19]. During intense, repeated or sustained nociceptor activation, as with neuroinflammation and nerve injury, the continued release of neuropeptides such as substance P and CGRP from primary afferents on dorsal horn neurons provides enough postsynaptic depolarization to expel Mg2+ ions and relieve NMDAR channel blockade. Subsequently, the binding of glutamate to NMDA receptors generates a strong inflow of Ca2+ ions and mediates long-term potentiation (LTP) of dorsal horn excitatory transmission [20]. Other sources for intracellular Ca2+ in dorsal horn neurons include the Ca2+-permeable GluA1 subunit-lacking AMPARs and the mobilization of Ca2+ from intracellular stores, the latter of which is mediated by the activation of group-I mGluRs. The generated Ca2+ signals and the activity of various peptides including substance P, CGRP and brain-derived neurotrophic factor (BDNF) activate protein kinases such as calcium/calmodulin-dependent protein kinase II (CAMKII), protein kinase A (PKA) and protein kinase C (PKC) [15]. These kinases are found to mediate the induction and early-phase of LTP through AMPA receptor phosphorylation and synaptic insertion following C-fiber tetanization, while the maintenance or late-phase of LTP requires de novo protein synthesis [21, 22]. Other mechanisms of functional plasticity in dorsal horn neurons include disinhibition, glial activation and nitric oxide (NO)-dependent retrograde signaling, which leads to increased neurotransmitter release probability from C-fiber terminals [15]. In addition, delayed structural plasticity changes are observed on the level of dendritic spine size and density and involve alterations in gene expression and connectivity [23]. Within the brain, similar functional and structural plasticity alterations associated with multiple chronic pain syndromes have been documented in various regions including the brain stem, thalamus, insular cortex, cingulate cortex and primary somatosensory cortex [24]. While it remains unclear if brain remodeling is a cause or consequence of chronic pain, it was proposed that pain-associated plasticity changes within brain circuits resemble associative learning and memory trace formation; thus, rendering pain perception more affective than somatic in nature [25]. Accordingly, structural spinal and supraspinal remodeling are hypothesized to mediate pain chronification with representational shifting towards emotional than nociceptive circuits [26]. It should be noted; however, that not all forms of chronic pain, and not all patients with a specific type of pain, show these reorganizations. As previously shown, patients with orofacial neuropathic pain exhibit cortical somatosensory remodeling; however, patients with chronic non-neuropathic orofacial pain do not [27]. Therefore, individualized targeting of central sensitization and brain plasticity may provide the means for preventing acute-to-chronic pain transitioning and the reversal of pathologic plasticity in specific forms of chronic pain. Various modalities have been investigated for potential effectiveness in nociceptive desensitization as modulators of neural activity and plasticity such as neurostimulation, virtual reality, cognitive therapy and rehabilitation.

Nociceptive pathways and the main loci of maladaptive plasticity. Activation of peripheral nociceptors generates action potentials that are transmitted to secondary afferents in the dorsal horn, which then project to the brain for processing. Particularly intense tissue insults result in maladaptive plasticity including peripheral sensitization in primary afferents, central sensitization in dorsal horn secondary afferents and brain remodeling. AP action potential, AMPAR 3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, DRG dorsal root ganglion, Glu glutamate, mGluR metabotropic glutamate receptor, NK1 neurokinin receptor, NMDAR N-methy-d-aspartate receptor, PKA/PKC protein kinase A/C, SP substance-P, VGCC voltage-gated calcium channel
Modulating maladaptive plasticity
Neurostimulation
Neurostimulation is a neuromodulatory method based on the delivery of electrical impulses to stimulate specific neurological sites within the body intended for various diagnostic and treatment purposes [28]. Multiple invasive and non-invasive neurostimulation techniques have been developed for the management of pain, especially chronic forms, which can be used to stimulate peripheral nerves, the spinal cord or specific brain regions. Peripherally, the techniques include transcutaneous electrical nerve stimulation (TENS), peripheral nerve stimulation (PNS) and peripheral nerve field stimulation (PNFS) [29]. Central neurostimulation techniques include spinal cord stimulation (SCS), non-invasive brain stimulation (NIBS) and invasive brain stimulation techniques such as deep brain stimulation (DBS). NIBS techniques include transcranial magnetic stimulation (TMS) and transcranial direct current stimulation (tDCS) [30]. In this review, we will focus on SCS and NIBS techniques in relation to maladaptive plasticity of central sensitization and brain remodeling in chronic pain.
Spinal cord stimulation
The basic principle behind SCS development is the stimulation of ascending non-nociceptive Aβ fibers to block or close the gate for C-fiber nociceptive transmission, through activating inhibitory interneurons, based on the Gate Control Theory of pain [31]. However, the analgesic mechanism of SCS is complex and affects various aspects of pain through spinal and supraspinal mechanisms, which together with the analgesic efficacy can vary across different stimulation protocols [32]. These include tonic (conventional), high-frequency (paresthesia-free or high-dose stimulation), burst and closed-loop SCS waveforms [33]. In relation to clinical efficacy, high-frequency (10 kHz) and burst SCS are shown to be effective and superior to tonic or conventional SCS in chronic back and leg pain, failed back surgery syndrome (FBSS) and intractable and diabetic neuropathy [34,35,36,37,38]. In addition, various clinical studies demonstrate effectiveness of high-frequency SCS in relieving other forms of pain including chronic neck and upper limb pain [39], thoracic back pain [40], chronic pelvic pain [41], chronic post-surgical pain [42] and chronic widespread pain / fibromyalgia [43]. In relation to central maladaptive plasticity, high-frequency SCS is clinically found, at 3 months of application, to enhance the functional connectivity between the insula, frontoparietal and central executive networks in patients with FBSS, suggesting potential influence on affective saliency and thus emotional awareness of pain [44]. Furthermore, magnetic resonance imaging (MRI) in FBSS patients who received high-frequency SCS for 3 months revealed significant volumetric alterations of white and grey matter in various brain regions, which correlated with pain relief [45]. These studies confirm the supraspinal modulatory effects of SCS and further support the reversibility of chronic pain-induced alterations of brain connectivity. Regarding central sensitization of secondary spinal afferents, multiple in vitro studies investigated the effects of SCS using rat models. Early findings using rodent models showed that tetanization, nerve injury and acute noxious stimuli (chemical, mechanical and thermal) induce C-fiber synaptic LTP on dorsal horn neurons [46] including WDR neurons [47] with subsequent hyperexcitability of WDR neurons [48]. Appropriately, the application of SCS is found to block these effects by inhibiting dorsal horn C-fiber LTP on WDR neurons with no effect on A-fiber responses [49], decreasing spinal excitatory amino acid release via a GABAergic mechanism in neuropathic rats experiencing allodynia [50] and attenuating the increased WDR neuronal excitability without affecting induced or spontaneous discharge in control non-allodynic rats [51]. To unravel the underlying mechanisms, extensive research has been recently conducted providing novel insights into the molecular basis corresponding to SCS excitability normalization and reversal of central sensitization. A study by Tilley and colleagues (2021) showed, through proteomic analysis, that conventional SCS influenced the expression of over 150 proteins, many of which are involved in stress, nociception and neuroglial interactions [52]. Accordingly, the results not only show the reversal of pain-associated proteomic profiles but further indicate that the mechanism of SCS is not solely dependent on the interruption of electrical transmission. Another study by Liao and colleagues (2020) revealed that spared nerve injury (SNI) model of neuropathic pain results in mechanical hyperalgesia and increased expression and phosphorylation of extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinases (JNKs), and p38 mitogen-activated protein kinase (p38-MAPK), which are important regulators of neuronal activity and plasticity. Importantly, early application of high-frequency SCS was able to prevent these alterations in dorsal root ganglia (DRG) and spinal dorsal horn, which associated with the attenuation of hyperalgesia [53]. Furthermore, Shinoda and colleagues (2020) investigated the effects of SCS on SNI, applied at 60 Hz for 6 h on the third day of SNI, as well. The findings show that SCS suppressed mechanical hypersensitivity, microglial activation and dorsal horn nociceptive hyperexcitability; additionally, SCS reduced somatosensory neuronal activity [54]. Interestingly, it is also reported that conventional SCS can activate microglia and thus compromise its own analgesic efficacy as preventing microglial activation prolonged the pain inhibition induced by SCS [55]. As microglia are important modulators of neurotransmission, neuroglial interactions and pain signaling with hypothesized roles in the pathogenesis of neuropathic pain [56], these results highlight the regulation of microglial activity by SCS as a potentially essential analgesic mechanism in neuropathic pain. Lastly, the application of high-frequency SCS in rats with SNI-induced neuropathic pain was observed to restore, at least partially, spinal glutamate uptake activity, spinal glutamate levels and miniature excitatory postsynaptic current (mEPSC) frequency [57]. Therefore, SCS induces various spinal and supraspinal modifications that reverse pain-associated maladaptive plasticity in relation to brain connectivity and central sensitization.
Non-invasive brain stimulation
NIBS techniques have significant potentials in the study and treatment of various psychiatric and neurological disorders including pain [58]. However, it must be stressed that the underlying mechanisms are complex and the clinical effects depend on various factors leading to high degree of variability [59]. Therefore, the best clinical practice for the use NIBS techniques in pain management should be derived from standardized guidelines [60, 61]. The two main studied NIBS techniques are TMS and tDCS. The principle of TMS is the non-invasive application of magnetic field pulses, which carry a current through the skull and excite superficial layers of the cortex. The two main cortical region to which TMS is applied are the primary motor (M1) cortex and the dorsolateral prefrontal cortex (DLPFC), both of which are associated with chronic pain and affect various aspects of pain processing [62, 63]. The repetitive application of TMS (rTMS) produces long-lasting, stimulation frequency-dependent excitability effects [64, 65] and leads to widespread activity changes in connected cortical and subcortical regions, yet the alterations of functional connectivity remain network-specific [66, 67]. The rTMS-induced excitability changes are believed to be mediated through neural plasticity mechanisms [68]. Indeed, evidence shows that rTMS-induced changes are NMDAR-dependent, lead to enhanced BDNF function [69] and able to induce potentiation [70] and depression [71] of excitability. Furthermore, rTMS application in rodents is observed to enhance cognition, facilitate hippocampal plasticity and increase the levels of various plasticity markers [72]. Accordingly, accumulating evidence supports the hypothesis that rTMS accelerates recovery of sensory and motor functions after stroke, incomplete spinal cord injury and nerve injury by promoting synaptic plasticity and thereby reversing maladaptive plasticity [73,74,75]. In addition, the LTP-like plasticity induced by rTMS treatment correlates with cognitive function improvement in Alzheimer’s disease patients [76]. Similarly, early treatment with rTMS is proposed to block pain-associated maladaptive plasticity induced by surgery, spinal injury and brain trauma; thus, preventing acute-to-chronic pain transitioning [77]. In a recent meta-analysis, Che and colleagues (2021) found that rTMS exerts a short-term analgesic effect that is specific to neuropathic pain, a long-term (average of 3 month) analgesic effect across multiple chronic pain conditions and significant analgesia of provoked pain, which could model either acute pain or acute-to-chronic pain transition [78]. These findings support the general consensus that rTMS exerts a multitude of mechanisms that could differentially modulate specific types of pain and indicate an acute analgesic effect that could be independent from the maladaptive plasticity associated with chronic pain. Indeed, rTMS application is shown to activate opioid-mediated analgesia of acute pain in healthy individuals [79], induce dose-dependent immediate analgesia following stimulation in patients with intractable neuropathic pain [80] and elevate electrical pain thresholds up to 40 min following application over the somatosensory cortex of healthy subjects without altering the excitability of the M1 cortex [81]. Therefore, rTMS carries significant potentials in both: prevention of acute-to-chronic pain transition, through acute analgesia and prevention of maladaptive plasticity, and treatment of chronic pain through reversal of maladaptive plasticity. On the other hand, the principle of tDCS is the passage of current between two electrodes; thus, anodal tDCS leads to depolarization while cathodal stimulation causes hyperpolarization. In contrast to TMS, which can stimulate cortical neuronal axons to fire action potentials, the effects of tDCS are more electrically subtle. This is due to weaker current pulses affecting membrane excitability (subthreshold potential alterations); however, depending on the duration and frequency of application it can also induce long-lasting effects mediated by intracortical inhibition and facilitation [82]. In relation to neural plasticity, the application of tDCS is found in rodent models to promote BDNF-dependent synaptic plasticity [83], enhance synaptic plasticity and memory [84] and improve plasticity deficits and cognitive dysfunction associated with diabetes [85]. Clinically, tDCS facilitates the formation of long-term motor memory, reflecting experience-dependent plasticity [86], improves motor performance in the elderly via enhanced facilitation and reduced inhibition [87] and, at short stimulation intervals, leads to LTP-like excitability enhancements in healthy participants [88]. These findings indicate significant facilitatory effects of tDCS on neuronal plasticity, which could thereby accelerate the recovery from, or prevent the development of, maladaptive plasticity similar to rTMS. Various studies support the potential analgesic efficacy of tDCS in multiple chronic neuropathic pain conditions [89] as well as migraine, osteoarthritis and capsaicin-induced mechanical sensitivity [90,91,92]. Furthermore, tDCS has no impact on pain thresholds and mechanical detection in healthy individuals [93]. However, the analgesic response to tDCS depends on many factors; hence, it is not effective in all patients with neuropathic pain [94]. The mechanisms underlying direct tDCS-induced analgesia are not completely understood; however, the effects may not only be related to increased or decreased neuronal firing rates as reports suggest the engagement of endorphins [95, 96] and, in addition to modulation of glutamatergic and GABAergic balance [97], the alteration of certain neuromodulators such as dopamine [98]. In rodent models, other neuromodulators are also found to significantly affect tDCS responses including serotine [99] and norepinephrine [100]. These neuromodulators are known to regulate neuronal activity, synaptic plasticity, input processing and associated neurological functions [101]. Lastly, many other tDCS effects were proposed to mediate analgesia in relation to altered pain processing and modulation of its emotional aspects [102].
Virtual reality
Virtual reality (VR) is a technology that provides an immersive experience in a simulated and interactive environment via multimodal sensory stimuli including visual, auditory and tactile inputs using computer hardware. The potential VR applications in the medical field were recognized over two decades ago, such as education, surgery and rehabilitation [103]. Since then, extensive research has been conducted to evaluate the therapeutic application of VR in various conditions. These include recovery from stroke [104], improving motor function in cerebral palsy [105], managing post-traumatic stress disorder [106], alleviating perioperative pain and anxiety [107], treatment of phobias [108] and management of acute and chronic pain [109] especially phantom-limb pain [110]. The effectiveness of VR in pain management, commonly termed VR analgesia, can be generally attributed to distraction, or the shifting of attention away from pain, with potential affective aspects. Early evidence demonstrated that increased pain vigilance and awareness in patients with chronic pain is associated with higher feelings of distress and disability [111] while distraction through cognitively demanding tasks reduces perceived pain intensity and neuronal activity in brain structures associated with pain processing [112] and produces even greater analgesia in high catastrophizing patients [113]. Indeed, the actual process of “pain perception” is not solely a somatic reflection but rather dependent on emotion, cognition and attention as well [114,115,116,117,118]. Accordingly, the use of VR demonstrates significant analgesic efficacy, during or immediately following the “VR experience”, in different types of acute and chronic pain [119,120,121,122]. However, targeting central sensitization and brain remodeling of chronic pain through VR would essentially require evidence of long-lasting improvement of perceived pain intensity. Indeed, VR produced lasting analgesia in patients with fibromyalgia at 6 months follow up [123] and chronic headache pediatrics at 3 months post-treatment [124]. Furthermore, Mehesz and colleagues (2021) showed that an immersive VR experience in healthy participants is able to produce efficient conditioned pain modulation and, in a surrogate central sensitization model, alleviate mechanical pain sensitivity [125]. Additionally, a recent case report by Orakpo and colleagues (2021) showed that VR, fused with neurofeedback therapy, achieved adequate analgesia that was sustained for 1 year in a patient with chronic spondylolisthesis pain, indicating further neuromodulation promise of VR in centralized pain syndromes [126]. Moreover, immersive VR is shown to not only reduce perception of capsaicin-induced ongoing pain, but also to elevate pain thresholds of corresponding secondary hyperalgesia [127]. These observations provide direct evidence supporting the effectiveness of VR in the management of central sensitization and modulation of pain processing. It should be noted; however, that effective patient distraction would entail being comfortable with and willing to use VR, which might vary across different demographics, available VR hardware and simulated VR environments. Accordingly, the production of a “VR pharmacy” to provide individualized or patient-tailored experiences was previously proposed [128]. On the other hand, the use of VR in phantom limb pain and pain associated with certain musculoskeletal disorders relies on additional mechanisms other than distraction. Phantom limb pain is a form of neuropathic pain that is highly prevalent among amputees, which results from representational mismatching and subsequent central pain mechanisms [129]. Indeed, phantom limb pain is associated with reduced thermal pain thresholds in various body parts, indicating central alterations [130], correlated with mechanical wind-up pain and thermal allodynia [131], and the altered pain processing and wind-up of phantom limb pain are positively correlated with catastrophizing indicating roles for cognitive and emotional sensitization [132]. Furthermore, phantom limb pain involves reorganizations or regional, amputated limb, boundary re-mapping; however, maladaptive plasticity of preserved representation and activity despite the lack of sensory input results in multiple painful and non-painful, illusory, amputated limb perceptions [133]. In order to correct, or account for, the representational mismatching in phantom limp pain; various techniques, mainly based on enhanced visual input, have been developed including mirror therapy, motor imagery, and virtual visual feedback, all of which are able to reduce phantom limb pain [134]. Through immersive VR systems, embodiment of a virtual limb or body part allows the modulation of perceptual disturbances and control of phantom limb pain and other types of chronic pain [135]. Accordingly, somatic VR experiences represent a novel form of rehabilitation. Indeed, the use of immersive VR in phantom limb pain patients is shown in various studies and case reports to elevate pain thresholds [136], decrease pain and improve anxiety [137] and provide sustained pain reductions [138,139,140]. These findings support a significant promise for VR in the modulation of central processing in chronic pain and management of phantom limb pain; however, larger studiers are still required.
Cognitive therapy
As discussed previously, cognition and emotion are important factors influencing the process of pain perception. In addition, catastrophizing and maladjusted pain cognitions are associated with higher pain scores, anxiety, central sensitization and maladaptive processing of pain [141,142,143,144]. This is also observed in neuropathic pain conditions; for instance, catastrophizing is commonly observed in patients with orofacial neuropathic pain, for which only select pharmacological options are available, and is associated with higher pain intensity [145,146,147]. Accordingly, various studies investigated the potentials of cognitive-based therapies in the management of chronic pain conditions. These mainly include cognitive behavioral therapy (CBT), mindfulness-based therapies (MBT) and acceptance and commitment therapy (ACT). Current evidence indicates that these thee approaches lead to incremental but statistically significant reductions in chronic pain scores [148,149,150,151]. Despite these improvements, the aim of cognitive therapy should be to affect pain processing and modulate central mechanisms of sensitization to improve responses to pharmacological therapy. Indeed, CBT is found to decrease induced-pain unpleasantness but not intensity; however, it significantly reduced secondary hyperalgesia; thus, central sensitization [152]. Accordingly, extensive research has been recently conducted to evaluate the neural mechanisms of cognitive therapies. It was shown that catastrophizing is associated with higher functional connectivity between the insula and primary somatosensory (S1) cortex in fibromyalgia patients. However, CBT intervention led to significant and long-term improvements in pain intensity and catastrophizing, which were associated with restorations of lower resting-state functional connectivity levels between the insula and S1 cortex [153]. In addition, chronic pain is associated with reduced grey matter volume of the prefrontal cortex [154] while CBT intervention causes increased grey matter volume in various cortical regions, and the volume increase in the prefrontal and somatosensory cortices is associated with reduced catastrophizing [155]. These findings are functionally reflected as well in fibromyalgia patients undergoing CBT therapy in whom CBT led to significant elevation in pain-evoked neuronal activity in the prefrontal cortex with suggested alterations in pain processing loops relating to pain reappraisal [156]. Further neuroimaging evidence shows that 11 weeks of CBT in chronic pain patients caused significant elevations in connectivity between the somatosensory cortex and basal ganglia while causing reductions in connectivity of default mode network with limbic regions such as the amygdala, which were accompanied with clinical improvements and improved pain-coping [157]. The connectivity alterations of CBT in chronic pain patients also involve resting-state brain networks, especially the orbitofrontal cortex, which has important roles in the cognitive processing of pain [158]. On the other hand, MBCT is another form of psychotherapy that relates to CBT but focuses on mindfulness through certain interventions such as meditation and other practices. It was found that cognitive therapies including CBT and MBCT, in patients with various chronic pain conditions, alter neuronal function throughout brain networks and reduce affective aspects of the pain experience [159]. In addition, mindfulness meditation in chronic pain, when compared to sham controls and placebo analgesia, is found to cause significantly higher reductions in pain intensity and unpleasantness and cause different brain activity alterations. These include enhanced activity of cognition-dependent pain-modulating cortical regions including the anterior insular, orbitofrontal and subgenual anterior cingulate cortices [160]. Therefore, MBCT-induced modulation of pain is different from and relies on different mechanisms compared to placebo analgesia. Positive findings are also observed with ACT interventions in relation to pain, behavior and connectivity alterations across emotion, cognition and pain processing networks [161, 162]. These novel findings provide key insights into the neural plasticity mechanisms by which cognitive therapies modulate central pain processing. Lastly, some reports indicate that perioperative CBT can decrease postsurgical pain and catastrophizing [163, 164], which in principle, and based on preliminary findings [165], aid in the prevention of post-surgical acute-to-chronic pain transition; however, further investigations are needed.
Exercise rehabilitation
Rehabilitation encompasses a multitude of interventions; however, in relation to pain management it mainly includes physical or exercise therapy, dietary control, stress management and other lifestyle modifications. Within the scope of this review, the focus on rehabilitation will be directed towards physical or exercise therapy in chronic pain. It is well established that exercise, within appropriate limits, has beneficial impact on pain and associated symptoms [166]. In addition, exercise-induced analgesia is a known phenomenon; however, the underlying mechanisms are complex and multiple hypotheses have been proposed [167]. On the other hand, various reports suggest that patients with chronic pain may not benefit from post-exercise analgesia as healthy individuals [168]. The pattern and not necessarily type of exercise; however, is a major outcome determinant such that sudden bouts of heavy exercise result in pain exacerbation, while regular moderate physical activity improves pain, decreases central neuronal excitability and promotes central inhibition [169]. In relation to central sensitization, various studies investigated the effects of exercise on pain sensitivity in patients with chronic pain and accumulating evidence demonstrates beneficial effects for exercise-induced hypoalgesia. In osteoarthritis, education and exercise lead to pain reduction and lower analgesic use post-exercise, while additional strength exercise reduces hyperalgesia but attenuates pain reductions [170, 171]. In chronic back pain, aerobic exercise results in significant reductions of chronic pain intensity, induced-pain sensitivity and interference, potentially due to activation of endogenous opioid analgesia [172]. Other specific types of exercise are also effective for chronic low-back pain; for instance, McKenzie exercise program was found more effective than conventional physiotherapy and led to significant reduction of central sensitization markers, pain intensity and disability; however, trunk muscular endurance did not improve [173]. However, effective exercise-induced recruitment of endogenous analgesia is not observed in all chronic pain conditions; for instance, exercise is effective in rheumatoid arthritis but not in chronic fatigue syndrome and fibromyalgia [174]. Therefore, a moderate physical activity, unless contraindicated, can be generally recommended; however, specific rehabilitation and exercise programs should be selected in an individualized manner.
Discussion and clinical considerations
Accumulating evidence indicates a significant role for maladaptive plasticity in the pathophysiology of various forms of chronic pain through functional and structural connectivity alterations. In this regard, non-pharmacological interventions including the discussed neuromodulation techniques, cognitive therapies and rehabilitation carry significant potentials to counteract maladaptive plasticity to help alleviate chronic pain or prevent acute-to-chronic pain transition. However, the functional and structural plasticity alterations associated with chronic pain show significant discrepancies across a wide array of chronic pain conditions. In addition, the molecular mechanisms by which different neuromodulation techniques impact neuronal plasticity vary widely as well; thus, each intervention would have differential efficacy across different pain conditions. Furthermore, inter-individual variability as well as associated psychocognitive factors must be taken into account as not all patients develop central sensitization, exhibit connectivity alterations or equally respond, or develop tolerance, to the various therapeutic interventions. Therefore, the importance of individualized treatment and patient-tailored selection of appropriate treatment options must be stressed. Clinical tools such as the central sensitization inventory [175] have been developed, which can help identify patients with central components of sensitization and corresponding severity [176], and shown to be valid even in the outpatient setting [177]. The choice of treatment intervention should be based on guideline recommendations derived from clinical evidence supporting the application of each treatment modality. The use of brain stimulation techniques such as rTMS and tDCS largely remains investigational with weak or inconclusive recommendations in neuropathic pain, fibromyalgia and spinal cord injury pain [60, 61]. This is due to inconsistent clinical evidence mainly attributable to randomized controlled trials (RCTs) with low study sample sizes [178]. On the other hand, an expert consensus panel in 2020 recommended the use of rTMS, applied to the M1 cortex, for neuropathic pain, post-traumatic brain injury-related headache, postoperative pain and prevention of migraine [179]. Other neurostimulation techniques have been more widely applied in the clinical setting such as high-frequency SCS, which is approved by the U.S. Food and Drug Administration as aid for the management of chronic back and limb pain as well as diabetic neuropathy [180]. Multiple clinical trials on the use of high-frequency SCS have been done with robust evidence to support its use for persistent back and radicular pain especially following failed back surgery [181], as also recommended by the National Institute for Health and Care Excellence for chronic neuropathic pain [182]. In relation to psychological therapy, particularly CBT, and exercise therapy, alone or as part of multi-disciplinary rehabilitation programs, clinical evidence supports slight improvements of function and pain scores over short (< 6 months), intermediate and long-term (> 12 months) follow-up in various chronic pain conditions including fibromyalgia [183]. Despite that rehabilitation and cognitive therapies provide modest improvements of pain scores, their psychological impact on pain cognition and brain connectivity could prove to be essential for patients with centralized pain syndromes. Therefore, CBT and exercise should be considered for all adult patients with primary chronic pain as recommended by the National Institute for Health and Care Excellence [184]. Lastly, the use of VR is yet to be approved for pain management as more robust clinical evidence is required.
Conclusions
Over the last two decades, the impact of maladaptive plasticity of central sensitization and brain remodeling has been highlighted and identified as a major component of various chronic pain conditions. Accordingly, neuromodulation research targeting maladaptive plasticity has been gaining momentum and shown tremendous usefulness in managing various forms of chronic pain that would otherwise be considered intractable and unresponsive. While pharmacological agents are still considered the cornerstone in the treatment of acute and chronic pain, novel neuromodulation techniques and protocols are continuously advancing with significant future potentials. Further large clinical trials are required to establish the long-term clinical safety and efficacy of these techniques, the results of which could reshape the scope of pain management in various chronic pain conditions.
Availability of data and materials
Not applicable.
Abbreviations
- ACT:
-
Acceptance and commitment therapy
- AMPA:
-
3-Hydroxy-5-methyl-4-isoxazolepropionic acid
- ATP:
-
Adenosine triphosphate
- BDNF:
-
Brain-derived neurotrophic factor
- CAMKII:
-
Calcium/calmodulin-dependent protein kinase II
- CBT:
-
Cognitive behavioral therapy
- CGRP:
-
Calcitonin gene-related peptide
- DBS:
-
Deep brain stimulation
- DLPFC:
-
Dorsolateral prefrontal cortex
- DRG:
-
Dorsal root ganglia
- ERKs:
-
Extracellular signal-regulated kinases
- FBSS:
-
Failed back surgery syndrome
- GABA:
-
γ-Amino butyric acid
- JNKs:
-
C-Jun N-terminal kinases
- KA:
-
Kainic acid
- LTP:
-
Long-term potentiation
- M1:
-
Primary motor cortex
- MBT:
-
Mindfulness-based therapy
- mEPSC:
-
Miniature excitatory postsynaptic current
- mGluR:
-
Metabotropic glutamate receptor
- MRI:
-
Magnetic resonance imaging
- NIBS:
-
Non-invasive brain stimulation
- NMDA:
-
N-methyl-d-aspartate
- NO:
-
Nitric oxide
- p38-MAPK:
-
P38 mitogen-activated protein kinase
- PKA:
-
Protein kinase A
- PKC:
-
Protein kinase C
- PNFS:
-
Peripheral nerve field stimulation
- PNS:
-
Peripheral nerve stimulation
- RCT:
-
Randomized controlled trial
- rTMS:
-
Repetitive transcranial magnetic stimulation
- S1:
-
Primary somatosensory cortex
- SCS:
-
Spinal cord stimulation
- SNI:
-
Spared nerve injury
- tDCS:
-
Transcranial direct current stimulation
- TENS:
-
Transcutaneous electrical nerve stimulation
- TMS:
-
Transcranial magnetic stimulation
- TRPV:
-
Transient receptor potential vanilloid
- VR:
-
Virtual reality
- WDR:
-
Wide dynamic range
References
McGann JP. Associative learning and sensory neuroplasticity: how does it happen and what is it good for? Learn Mem. 2015;22(11):567–76.
McCarberg B, Peppin J. Pain pathways and nervous system plasticity: learning and memory in pain. Pain Med. 2019;20(12):2421–37.
Grau JW, Huang YJ, Turtle JD, Strain MM, Miranda RC, Garraway SM, et al. When pain hurts: nociceptive stimulation induces a state of maladaptive plasticity and impairs recovery after spinal cord injury. J Neurotrauma. 2017;34(10):1873–90.
Chao CC, Tseng MT, Lin YH, Hsieh PC, Lin CH, Huang SL, et al. Brain imaging signature of neuropathic pain phenotypes in small-fiber neuropathy: altered thalamic connectome and its associations with skin nerve degeneration. Pain. 2021;162(5):1387–99.
Nees F, Becker S. Psychological processes in chronic pain: influences of reward and fear learning as key mechanisms–behavioral evidence, neural circuits, and maladaptive changes. Neuroscience. 2018;387:72–84.
Naro A, Milardi D, Russo M, Terranova C, Rizzo V, Cacciola A, et al. Non-invasive brain stimulation, a tool to revert maladaptive plasticity in neuropathic pain. Front Hum Neurosci. 2016;10:376.
Meyers EC, Kasliwal N, Solorzano BR, Lai E, Bendale G, Berry A, et al. Enhancing plasticity in central networks improves motor and sensory recovery after nerve damage. Nat Commun. 2019;10(1):5782.
Oliveira Júnior JO, Portella Junior CS, Cohen CP. Inflammatory mediators of neuropathic pain. Rev Dor. 2016;17:35–42.
Matsuda M, Huh Y, Ji RR. Roles of inflammation, neurogenic inflammation, and neuroinflammation in pain. J Anesth. 2019;33(1):131–9.
Davidson S, Copits BA, Zhang J, Page G, Ghetti A, Gereau RW IV. Human sensory neurons: membrane properties and sensitization by inflammatory mediators. Pain. 2014;155(9):1861–70.
Legrain V, Iannetti GD, Plaghki L, Mouraux A. The pain matrix reloaded: a salience detection system for the body. Prog Neurobiol. 2011;93(1):111–24.
Gold MS, Gebhart GF. Nociceptor sensitization in pain pathogenesis. Nat Med. 2010;16(11):1248–57.
Jensen TS, Finnerup NB. Allodynia and hyperalgesia in neuropathic pain: clinical manifestations and mechanisms. Lancet Neurol. 2014;13(9):924–35.
Woolf CJ. Pain amplification—a perspective on the how, why, when, and where of central sensitization. J Appl Biobehav Res. 2018;23(2):e12124.
Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10(9):895–926.
Nijs J, Malfliet A, Ickmans K, Baert I, Meeus M. Treatment of central sensitization in patients with ‘unexplained’ chronic pain: an update. Expert Opin Pharmacother. 2014;15(12):1671–83.
Pace MC, Passavanti MB, De Nardis L, Bosco F, Sansone P, Pota V, et al. Nociceptor plasticity: a closer look. J Cell Physiol. 2018;233(4):2824–38.
Nijs J, Leysen L, Vanlauwe J, Logghe T, Ickmans K, Polli A, et al. Treatment of central sensitization in patients with chronic pain: time for change? Expert Opin Pharmacother. 2019;20(16):1961–70.
Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg 2+ of NMDA responses in spinal cord neurones. Nature. 1984;309(5965):261–3.
Liu XG, Sandkühler J. Long-term potentiation of C-fiber-evoked potentials in the rat spinal dorsal horn is prevented by spinal N-methyl-D-aspartic acid receptor blockage. Neurosci Lett. 1995;191(1–2):43–6.
Yang HW, Hu XD, Zhang HM, Xin WJ, Li MT, Zhang T, et al. Roles of CaMKII, PKA, and PKC in the induction and maintenance of LTP of C-fiber-evoked field potentials in rat spinal dorsal horn. J Neurophysiol. 2004;91(3):1122–33.
Hu NW, Zhang HM, Hu XD, Li MT, Zhang T, Zhou LJ, et al. Protein synthesis inhibition blocks the late-phase LTP of C-fiber evoked field potentials in rat spinal dorsal horn. J Neurophysiol. 2003;89(5):2354–9.
Kuner R. The plastic spinal cord: functional and structural plasticity in the transition from acute to chronic pain. Neuroforum. 2017;23(3):137–43.
May A. Chronic pain may change the structure of the brain. Pain. 2008;137(1):7–15.
Mansour AR, Farmer MA, Baliki MN, Apkarian AV. Chronic pain: the role of learning and brain plasticity. Restor Neurol Neurosci. 2014;32(1):129–39.
Kuner R, Flor H. Structural plasticity and reorganisation in chronic pain. Nat Rev Neurosci. 2017;18(1):20–30.
Gustin SM, Peck CC, Cheney LB, Macey PM, Murray GM, Henderson LA. Pain and plasticity: is chronic pain always associated with somatosensory cortex activity and reorganization? J Neurosci. 2012;32(43):14874–84.
Mekhail NA, Cheng J, Narouze S, Kapural L, Mekhail MN, Deer T. Clinical applications of neurostimulation: forty years later. Pain Pract. 2010;10(2):103–12.
Kaye AD, Ridgell S, Alpaugh ES, Mouhaffel A, Kaye AJ, Cornett EM, et al. Peripheral nerve stimulation: a review of techniques and clinical efficacy. Pain Ther. 2021. https://doi.org/10.1007/s40122-021-00298-1.
Moisset X, Lanteri-Minet M, Fontaine D. Neurostimulation methods in the treatment of chronic pain. J Neural Transm. 2020;127(4):673–86.
Melzack R, Wall PD. Pain mechanisms: a new theory. Science. 1965;150(3699):971–9.
Heijmans L, Joosten EA. Mechanisms and mode of action of spinal cord stimulation in chronic neuropathic pain. Postgrad Med. 2020;132(sup3):17–21.
Deer TR, Jain S, Hunter C, Chakravarthy K. Neurostimulation for intractable chronic pain. Brain Sci. 2019;9(2):23.
Kapural L, Yu C, Doust MW, Gliner BE, Vallejo R, Sitzman BT, et al. Novel 10-kHz high-frequency therapy (HF10 therapy) is superior to traditional low-frequency spinal cord stimulation for the treatment of chronic back and leg pain: the SENZA-RCT randomized controlled trial. Anesthesiology. 2015;123(4):851–60.
Kapural L, Yu C, Doust MW, Gliner BE, Vallejo R, Sitzman BT, et al. Comparison of 10-kHz high-frequency and traditional low-frequency spinal cord stimulation for the treatment of chronic back and leg pain: 24-month results from a multicenter, randomized, controlled pivotal trial. Neurosurgery. 2016;79(5):667–77.
Deer T, Slavin KV, Amirdelfan K, North RB, Burton AW, Yearwood TL, et al. Success using neuromodulation with BURST (SUNBURST) study: results from a prospective, randomized controlled trial using a novel burst waveform. Neuromodulation. 2018;21(1):56–66.
Demartini L, Terranova G, Innamorato MA, Dario A, Sofia M, Angelini C, et al. Comparison of tonic vs. burst spinal cord stimulation during trial period. Neuromodulation. 2019;22(3):327–32.
De Ridder D, Lenders MW, De Vos CC, Dijkstra-Scholten C, Wolters R, Vancamp T, et al. A 2-center comparative study on tonic versus burst spinal cord stimulation: amount of responders and amount of pain suppression. Clin J Pain. 2015;31(5):433–7.
Sayed D, Salmon J, Khan TW, Sack AM, Braun T, Barnard A, et al. Retrospective analysis of real-world outcomes of 10 kHz SCS in patients with upper limb and neck pain. J Pain Res. 2020;13:1441–8.
Sayed D, Foster J, Nairizi A, Sills S, Miller A. 10 kHz high-frequency spinal cord stimulation for chronic thoracic pain: a multicenter case series and a guide for optimal anatomic lead placement. Pain Phys. 2020;23(4):E369–76.
Tate JL, Stauss T, Li S, Rotte A, Subbaroyan J. A prospective, multi-center, clinical trial of a 10-kHz spinal cord stimulation system in the treatment of chronic pelvic pain. Pain Pract. 2021;21(1):45–53.
Gupta M, Scowcroft J, Kloster D, Guirguis M, Carlson J, McJunkin T, et al. 10-kHz spinal cord stimulation for chronic postsurgical pain: results from a 12-month prospective, multicenter study. Pain Pract. 2020;20(8):908–18.
Salmon J. High-frequency spinal cord stimulation at 10 kHz for widespread pain: a retrospective survey of outcomes from combined cervical and thoracic electrode placements. Postgrad Med. 2019;131(3):230–8.
De Groote S, Goudman L, Peeters R, Linderoth B, Vanschuerbeek P, Sunaert S, et al. Magnetic resonance imaging exploration of the human brain during 10 kHz spinal cord stimulation for failed back surgery syndrome: a resting state functional magnetic resonance imaging study. Neuromodulation. 2020;23(1):46–55.
De Groote S, Goudman L, Van Schuerbeek P, Peeters R, Sunaert S, Linderoth B, et al. Effects of spinal cord stimulation on voxel-based brain morphometry in patients with failed back surgery syndrome. Clin Neurophysiol. 2020;131(11):2578–87.
Sandkühler J, Liu X. Induction of long-term potentiation at spinal synapses by noxious stimulation or nerve injury. Eur J Neurosci. 1998;10(7):2476–80.
Svendsen F, Tjølsen A, Gjerstad J, Hole K. Long term potentiation of single WDR neurons in spinalized rats. Brain Res. 1999;816(2):487–92.
Rygh LJ, Svendsen F, Hole K, Tjølsen A. Natural noxious stimulation can induce long-term increase of spinal nociceptive responses. Pain. 1999;82(3):305–10.
Wallin J, Fiskå A, Tjølsen A, Linderoth B, Hole K. Spinal cord stimulation inhibits long-term potentiation of spinal wide dynamic range neurons. Brain Res. 2003;973(1):39–43.
Cui JG, O’Connor WT, Ungerstedt U, Linderoth B, Meyerson BA. Spinal cord stimulation attenuates augmented dorsal horn release of excitatory amino acids in mononeuropathy via a GABAergic mechanism. Pain. 1997;73(1):87–95.
Yakhnitsa V, Linderoth B, Meyerson BA. Spinal cord stimulation attenuates dorsal horn neuronal hyperexcitability in a rat model of mononeuropathy. Pain. 1999;79(2–3):223–33.
Tilley DM, Lietz CB, Cedeno DL, Kelley CA, Li L, Vallejo R. Proteomic modulation in the dorsal spinal cord following spinal cord stimulation therapy in an in vivo neuropathic pain model. Neuromodulation. 2021;24(1):22–32.
Liao WT, Tseng CC, Wu CH, Lin CR. Early high-frequency spinal cord stimulation treatment inhibited the activation of spinal mitogen-activated protein kinases and ameliorated spared nerve injury-induced neuropathic pain in rats. Neurosci Lett. 2020;721:134763.
Shinoda M, Fujita S, Sugawara S, Asano S, Koyama R, Fujiwara S, et al. Suppression of superficial microglial activation by spinal cord stimulation attenuates neuropathic pain following sciatic nerve injury in rats. Int J Mol Sci. 2020;21(7):2390.
Shu B, He SQ, Guan Y. Spinal cord stimulation enhances microglial activation in the spinal cord of nerve-injured rats. Neurosci Bull. 2020;36(12):1441–53.
Chen G, Zhang YQ, Qadri YJ, Serhan CN, Ji RR. Microglia in pain: detrimental and protective roles in pathogenesis and resolution of pain. Neuron. 2018;100(6):1292–311.
Liao WT, Tseng CC, Chia WT, Lin CR. High-frequency spinal cord stimulation treatment attenuates the increase in spinal glutamate release and spinal miniature excitatory postsynaptic currents in rats with spared nerve injury-induced neuropathic pain. Brain Res Bull. 2020;164:307–13.
Polania R, Nitsche MA, Ruff CC. Studying and modifying brain function with non-invasive brain stimulation. Nat Neurosci. 2018;21(2):174–87.
Terranova C, Rizzo V, Cacciola A, Chillemi G, Calamuneri A, Milardi D, et al. Is there a future for non-invasive brain stimulation as a therapeutic tool? Front Neurol. 2019;9:1146.
Cruccu G, Aziz TZ, Garcia-Larrea L, Hansson P, Jensen TS, Lefaucheur JP, et al. EFNS guidelines on neurostimulation therapy for neuropathic pain. Eur J Neurol. 2007;14(9):952–70.
Cruccu G, Garcia-Larrea L, Hansson P, Keindl M, Lefaucheur JP, Paulus W, et al. EAN guidelines on central neurostimulation therapy in chronic pain conditions. Eur J Neurol. 2016;23(10):1489–99.
Saavedra LC, Mendonca M, Fregni F. Role of the primary motor cortex in the maintenance and treatment of pain in fibromyalgia. Med Hypotheses. 2014;83(3):332–6.
Seminowicz DA, Moayedi M. The dorsolateral prefrontal cortex in acute and chronic pain. J Pain. 2017;18(9):1027–35.
Quartarone A, Bagnato S, Rizzo V, Morgante F, Sant’Angelo A, Battaglia F, et al. Distinct changes in cortical and spinal excitability following high-frequency repetitive TMS to the human motor cortex. Exp Brain Res. 2005;161(1):114–24.
Seewoo BJ, Feindel KW, Etherington SJ, Rodger J. Frequency-specific effects of low-intensity rTMS can persist for up to 2 weeks post-stimulation: a longitudinal rs-fMRI/MRS study in rats. Brain Stimul. 2019;12(6):1526–36.
Yoo WK, You SH, Ko MH, Kim ST, Park CH, Park JW, et al. High frequency rTMS modulation of the sensorimotor networks: behavioral changes and fMRI correlates. Neuroimage. 2008;39(4):1886–95.
Tik M, Hoffmann A, Sladky R, Tomova L, Hummer A, de Lara LN, et al. Towards understanding rTMS mechanism of action: stimulation of the DLPFC causes network-specific increase in functional connectivity. Neuroimage. 2017;162:289–96.
Thomson AC, Kenis G, Tielens S, De Graaf TA, Schuhmann T, Rutten BP, et al. Transcranial magnetic stimulation-induced plasticity mechanisms: TMS-Related gene expression and morphology changes in a human neuron-like cell model. Front Mol Neurosci. 2020;13:528396.
Wang HY, Crupi D, Liu J, Stucky A, Cruciata G, Di Rocco A, et al. Repetitive transcranial magnetic stimulation enhances BDNF–TrkB signaling in both brain and lymphocyte. J Neurosci. 2011;31(30):11044–54.
Thickbroom GW, Byrnes ML, Edwards DJ, Mastaglia FL. Repetitive paired-pulse TMS at I-wave periodicity markedly increases corticospinal excitability: a new technique for modulating synaptic plasticity. Clin Neurophysiol. 2006;117(1):61–6.
Tsuji T, Rothwell JC. Long lasting effects of rTMS and associated peripheral sensory input on MEPs, SEPs and transcortical reflex excitability in humans. J Physiol. 2002;540(1):367–76.
Shang Y, Wang X, Shang X, Zhang H, Liu Z, Yin T, et al. Repetitive transcranial magnetic stimulation effectively facilitates spatial cognition and synaptic plasticity associated with increasing the levels of BDNF and synaptic proteins in Wistar rats. Neurobiol Learn Mem. 2016;134:369–78.
Takeuchi N, Izumi SI. Maladaptive plasticity for motor recovery after stroke: mechanisms and approaches. Neural Plast. 2012;2012:359728.
Ellaway PH, Vásquez N, Craggs M. Induction of central nervous system plasticity by repetitive transcranial magnetic stimulation to promote sensorimotor recovery in incomplete spinal cord injury. Front Integr Neurosci. 2014;8:42.
Cywiak C, Ashbaugh RC, Metto AC, Udpa L, Qian C, Gilad AA, et al. Non-invasive neuromodulation using rTMS and the electromagnetic-perceptive gene (EPG) facilitates plasticity after nerve injury. Brain Stimul. 2020;13(6):1774–83.
Li X, Qi G, Yu C, Lian G, Zheng H, Wu S, et al. Cortical plasticity is correlated with cognitive improvement in Alzheimer’s disease patients after rTMS treatment. Brain Stimul. 2021;14(3):503–10.
Andrade DC, Borges I, Bravo GL, Bolognini N, Fregni F. Therapeutic time window of noninvasive brain stimulation for pain treatment: inhibition of maladaptive plasticity with early intervention. Expert Rev Med Devices. 2013;10(3):339–52.
Che X, Cash RF, Luo X, Luo H, Lu X, Xu F, et al. High-frequency rTMS over the dorsolateral prefrontal cortex on chronic and provoked pain: a systematic review and meta-analysis. Brain Stimul. 2021;14(5):1135–46.
Taylor JJ, Borckardt JJ, George MS. Endogenous opioids mediate left dorsolateral prefrontal cortex rTMS-induced analgesia. Pain. 2012;153(6):1219–25.
Mori N, Hosomi K, Nishi A, Oshino S, Kishima H, Saitoh Y. Analgesic effects of repetitive transcranial magnetic stimulation at different stimulus parameters for neuropathic pain: a randomized study. Neuromodulation. 2021. https://doi.org/10.1111/ner.13328.
Rao N, Chen YT, Ramirez R, Tran J, Li S, Parikh PJ. Time-course of pain threshold after continuous theta burst stimulation of primary somatosensory cortex in pain-free subjects. Neurosci Lett. 2020;722:134760.
Nitsche MA, Seeber A, Frommann K, Klein CC, Rochford C, Nitsche MS, et al. Modulating parameters of excitability during and after transcranial direct current stimulation of the human motor cortex. J Physiol. 2005;568(1):291–303.
Fritsch B, Reis J, Martinowich K, Schambra HM, Ji Y, Cohen LG, Lu B. Direct current stimulation promotes BDNF-dependent synaptic plasticity: potential implications for motor learning. Neuron. 2010;66(2):198–204.
Podda MV, Cocco S, Mastrodonato A, Fusco S, Leone L, Barbati SA, et al. Anodal transcranial direct current stimulation boosts synaptic plasticity and memory in mice via epigenetic regulation of Bdnf expression. Sci Rep. 2016;6:22180.
Wu YJ, Lin CC, Yeh CM, Chien ME, Tsao MC, Tseng P, et al. Repeated transcranial direct current stimulation improves cognitive dysfunction and synaptic plasticity deficit in the prefrontal cortex of streptozotocin-induced diabetic rats. Brain Stimul. 2017;10(6):1079–87.
Rroji O, van Kuyck K, Nuttin B, Wenderoth N. Anodal tDCS over the primary motor cortex facilitates long-term memory formation reflecting use-dependent plasticity. PLoS ONE. 2015;10(5):e0127270.
Goodwill AM, Reynolds J, Daly RM, Kidgell DJ. Formation of cortical plasticity in older adults following tDCS and motor training. Front Aging Neurosci. 2013;5:87.
Agboada D, Mosayebi-Samani M, Kuo MF, Nitsche MA. Induction of long-term potentiation-like plasticity in the primary motor cortex with repeated anodal transcranial direct current stimulation–better effects with intensified protocols? Brain Stimul. 2020;13(4):987–97.
David MC, Moraes AA, Costa ML, Franco CI. Transcranial direct current stimulation in the modulation of neuropathic pain: a systematic review. Neurol Res. 2018;40(7):557–65.
Cerrahoğlu Şirin T, Aksu S, Hasirci Bayir BR, Ulukan Ç, Karamürsel S, Kurt A, et al. Is allodynia a determinant factor in the effectiveness of transcranial direct current stimulation in the prophylaxis of migraine? Neuromodulation. 2021;24(5):899–909.
Pollonini L, Miao H, Ahn H. Longitudinal effect of transcranial direct current stimulation on knee osteoarthritis patients measured by functional infrared spectroscopy: a pilot study. Neurophotonics. 2020;7(2):025004.
Hughes SW, Ward G, Strutton PH. Anodal transcranial direct current stimulation over the primary motor cortex attenuates capsaicin-induced dynamic mechanical allodynia and mechanical pain sensitivity in humans. Eur J Pain. 2020;24(6):1130–7.
Kold S, Graven-Nielsen T. Effect of anodal high-definition transcranial direct current stimulation on the pain sensitivity in a healthy population: a double-blind, sham-controlled study. Pain. 2021;162(6):1659–68.
Carvalho VG, de Almeida RL, Boechat-Barros R. Motor cortical excitability behavior in chronic spinal cord injury neuropathic pain individuals submitted to transcranial direct current stimulation. Spinal Cord Ser Cases. 2020;6(1):101.
DosSantos MF, Love TM, Martikainen IK, Nascimento TD, Fregni F, Cummiford C, et al. Immediate effects of tDCS on the μ-opioid system of a chronic pain patient. Front Psychiatry. 2012;3:93.
Khedr EM, Omran EA, Ismail NM, El-Hammady DH, Goma SH, Kotb H, et al. Effects of transcranial direct current stimulation on pain, mood and serum endorphin level in the treatment of fibromyalgia: a double blinded, randomized clinical trial. Brain Stimul. 2017;10(5):893–901.
Heimrath K, Brechmann A, Blobel-Lüer R, Stadler J, Budinger E, Zaehle T. Transcranial direct current stimulation (tDCS) over the auditory cortex modulates GABA and glutamate: a 7 T MR-spectroscopy study. Sci Rep. 2020;10(1):20111.
Fonteneau C, Redoute J, Haesebaert F, Le Bars D, Costes N, Suaud-Chagny MF, et al. Frontal transcranial direct current stimulation induces dopamine release in the ventral striatum in human. Cereb Cortex. 2018;28(7):2636–46.
Melo L, Mosayebi-Samani M, Ghanavati E, Nitsche MA, Kuo MF. Dosage-dependent impact of acute serotonin enhancement on transcranial direct current stimulation effects. Int J Neuropsychopharmacol. 2021. https://doi.org/10.1093/ijnp/pyab035.
Adelhöfer N, Mückschel M, Teufert B, Ziemssen T, Beste C. Anodal tDCS affects neuromodulatory effects of the norepinephrine system on superior frontal theta activity during response inhibition. Brain Struct Funct. 2019;224(3):1291–300.
Bazzari AH, Parri HR. Neuromodulators and long-term synaptic plasticity in learning and memory: a steered-glutamatergic perspective. Brain Sci. 2019;9(11):300.
Knotkova H, Nitsche MA, Cruciani RA. Putative physiological mechanisms underlying tDCS analgesic effects. Front Hum Neurosci. 2013;7:628.
Satava RM. Emerging medical applications of virtual reality: a surgeon’s perspective. Artif Intell Med. 1994;6(4):281–8.
Viñas-Diz S, Sobrido-Prieto M. Virtual reality for therapeutic purposes in stroke: a systematic review. Neurologia. 2016;31(4):255–77.
Silva TD, Fontes AM, Oliveira-Furlan BS, Roque TT, Lima AI, Souza BM, et al. Effect of combined therapy of virtual reality and transcranial direct current stimulation in children and adolescents with cerebral palsy: a study protocol for a triple-blinded randomized controlled crossover trial. Front Neurol. 2020;11:953.
Kothgassner OD, Goreis A, Kafka JX, Van Eickels RL, Plener PL, Felnhofer A. Virtual reality exposure therapy for posttraumatic stress disorder (PTSD): a meta-analysis. Eur J Psychotraumatol. 2019;10(1):1654782.
Sengkeh MY, Chayati N. Audiovisual virtual reality distraction in reduction of pain and anxiety intention in post-operative patients: a review study. Open Access Maced J Med Sci. 2021;9(F):76–80.
Botella C, Fernández-Álvarez J, Guillén V, García-Palacios A, Baños R. Recent progress in virtual reality exposure therapy for phobias: a systematic review. Curr Psychiatry Rep. 2017;19(7):42.
Ahmadpour N, Randall H, Choksi H, Gao A, Vaughan C, Poronnik P. Virtual Reality interventions for acute and chronic pain management. Int J Biochem Cell Biol. 2019;114:105568.
Osumi M, Inomata K, Inoue Y, Otake Y, Morioka S, Sumitani M. Characteristics of phantom limb pain alleviated with virtual reality rehabilitation. Pain Med. 2019;20(5):1038–46.
McCracken LM. “Attention” to pain in persons with chronic pain: a behavioral approach. Behav Ther. 1997;28(2):271–84.
Bantick SJ, Wise RG, Ploghaus A, Clare S, Smith SM, Tracey I. Imaging how attention modulates pain in humans using functional MRI. Brain. 2002;125(2):310–9.
Schreiber KL, Campbell C, Martel MO, Greenbaum S, Wasan AD, Borsook D, et al. Distraction analgesia in chronic pain patients: the impact of catastrophizing. Anesthesiology. 2014;121(6):1292–301.
Eccleston C, Crombez G. Pain demands attention: a cognitive–affective model of the interruptive function of pain. Psychol Bull. 1999;125(3):356–66.
Villemure C, Bushnell CM. Cognitive modulation of pain: how do attention and emotion influence pain processing? Pain. 2002;95(3):195–9.
Klossika I, Flor H, Kamping S, Bleichhardt G, Trautmann N, Treede RD, et al. Emotional modulation of pain: a clinical perspective. Pain. 2006;124(3):264–8.
Wiech K, Ploner M, Tracey I. Neurocognitive aspects of pain perception. Trends Cogn Sci. 2008;12(8):306–13.
Peters ML. Emotional and cognitive influences on pain experience. Mod Trends Pharmacopsychiatry. 2015;30:138–52.
Jones T, Moore T, Choo J. The impact of virtual reality on chronic pain. PLoS ONE. 2016;11(12):e0167523.
Mallari B, Spaeth EK, Goh H, Boyd BS. Virtual reality as an analgesic for acute and chronic pain in adults: a systematic review and meta-analysis. J Pain Res. 2019;12:2053–85.
Smith V, Warty RR, Sursas JA, Payne O, Nair A, Krishnan S, et al. The effectiveness of virtual reality in managing acute pain and anxiety for medical inpatients: systematic review. J Med Internet Res. 2020;22(11):e17980.
Pourmand A, Davis S, Marchak A, Whiteside T, Sikka N. Virtual reality as a clinical tool for pain management. Curr Pain Headache Rep. 2018;22(8):53.
Botella C, Garcia-Palacios A, Vizcaíno Y, Herrero R, Baños RM, Belmonte MA. Virtual reality in the treatment of fibromyalgia: a pilot study. Cyberpsychol Behav Soc Netw. 2013;16(3):215–23.
Shiri S, Feintuch U, Weiss N, Pustilnik A, Geffen T, Kay B, et al. A virtual reality system combined with biofeedback for treating pediatric chronic headache—a pilot study. Pain Med. 2013;14(5):621–7.
Mehesz E, Karoui H, Strutton PH, Hughes SW. Exposure to an immersive virtual reality environment can modulate perceptual correlates of endogenous analgesia and central sensitization in healthy volunteers. J Pain. 2021;22(6):707–14.
Orakpo N, Vieux U, Castro-Nuñez C. Case report: virtual reality neurofeedback therapy as a novel modality for sustained analgesia in centralized pain syndromes. Front Psychiatry. 2021;12:660105.
Hughes SW, Zhao H, Auvinet EJ, Strutton PH. Attenuation of capsaicin-induced ongoing pain and secondary hyperalgesia during exposure to an immersive virtual reality environment. Pain Rep. 2019;4(6):e790.
Spiegel BM. Virtual medicine: how virtual reality is easing pain, calming nerves and improving health. Med J Aust. 2018;209(6):245–7.
Collins KL, Russell HG, Schumacher PJ, Robinson-Freeman KE, O’Conor EC, Gibney KD, et al. A review of current theories and treatments for phantom limb pain. J Clin Invest. 2018;128(6):2168–76.
Fuchs X, Diers M, Trojan J, Kirsch P, Milde C, Bekrater-Bodmann R, et al. Phantom limb pain after unilateral arm amputation is associated with decreased heat pain thresholds in the face. Eur J Pain. 2021. https://doi.org/10.1002/ejp.1842.
Vase L, Svensson P, Nikolajsen L, Arendt-Nielsen L, Jensen TS. The effects of menthol on cold allodynia and wind-up-like pain in upper limb amputees with different levels of phantom limb pain. Neurosci Lett. 2013;534:52–7.
Vase L, Nikolajsen L, Christensen B, Egsgaard LL, Arendt-Nielsen L, Svensson P, et al. Cognitive-emotional sensitization contributes to wind-up-like pain in phantom limb pain patients. Pain. 2011;152(1):157–62.
Makin TR, Flor H. Brain (re) organisation following amputation: Implications for phantom limb pain. Neuroimage. 2020;218:116943.
Herrador Colmenero L, Perez Marmol JM, Martí-García C, Querol Zaldivar MD, Tapia Haro RM, Castro Sánchez AM, et al. Effectiveness of mirror therapy, motor imagery, and virtual feedback on phantom limb pain following amputation: a systematic review. Prosthet Orthot Int. 2018;42(3):288–98.
Matamala-Gomez M, Donegan T, Bottiroli S, Sandrini G, Sanchez-Vives MV, Tassorelli C. Immersive virtual reality and virtual embodiment for pain relief. Front Hum Neurosci. 2019;13:279.
Zanini A, Montalti M, Caola B, Leadbetter A, Martini M. Pain during illusory own arm movement: a study in immersive virtual reality. Eur Med J. 2017;2(2):90–7.
Tong X, Wang X, Cai Y, Gromala D, Williamson O, Fan B, et al. “I dreamed of my hands and arms moving again”: a case series investigating the effect of immersive virtual reality on phantom limb pain alleviation. Front Neurol. 2020;11:876.
Chau B, Phelan I, Ta P, Humbert S, Hata J, Tran D. Immersive virtual reality therapy with myoelectric control for treatment-resistant phantom limb pain: case report. Innov Clin Neurosci. 2017;14(7–8):3–7.
Ambron E, Miller A, Kuchenbecker KJ, Buxbaum LJ, Coslett H. Immersive low-cost virtual reality treatment for phantom limb pain: evidence from two cases. Front Neurol. 2018;9:67.
Ortiz-Catalan M, Guðmundsdóttir RA, Kristoffersen MB, Zepeda-Echavarria A, Caine-Winterberger K, Kulbacka-Ortiz K, et al. Phantom motor execution facilitated by machine learning and augmented reality as treatment for phantom limb pain: a single group, clinical trial in patients with chronic intractable phantom limb pain. Lancet. 2016;388(10062):2885–94.
Meints SM, Mawla I, Napadow V, Kong J, Gerber J, Chan ST, et al. The relationship between catastrophizing and altered pain sensitivity in patients with chronic low back pain. Pain. 2019;160(4):833–43.
Terry EL, Tanner JJ, Cardoso JS, Sibille KT, Lai S, Deshpande H, et al. Associations of pain catastrophizing with pain-related brain structure in individuals with or at risk for knee osteoarthritis: sociodemographic considerations. Brain Imaging Behav. 2021;15(4):1769–77.
Christensen KS, O’Sullivan K, Palsson TS. Conditioned pain modulation efficiency is associated with pain catastrophizing in patients with chronic low back pain. Clin J Pain. 2020;36(11):825–32.
Toledo T, Lannon E, Kuhn B, Hellman N, Sturycz C, Palit S, et al. State catastrophizing is associated with facilitation of spinal nociception during conditioned pain modulation (CPM). J Pain. 2018;19(3):S15.
Bazzari FH, Bazzari AH. Orofacial neuropathic pain: a pharmacological approach. SA Pharm J. 2019;86(4):23–8.
Davis CE, Stockstill JW, Stanley WD, Wu Q. Pain-related worry in patients with chronic orofacial pain. J Am Dent Assoc. 2014;145(7):722–30.
Dinan JE, Hargitai IA, Watson N, Smith A, Schmidt JE. Pain catastrophising in the oro-facial pain population. J Oral Rehabil. 2021;48(6):643–53.
Bernardy K, Klose P, Busch AJ, Choy EH, Häuser W. Cognitive behavioural therapies for fibromyalgia. Cochrane Database Syst Rev. 2013;9:CD009796.
Hilton L, Hempel S, Ewing BA, Apaydin E, Xenakis L, Newberry S, et al. Mindfulness meditation for chronic pain: systematic review and meta-analysis. Ann Behav Med. 2017;51(2):199–213.
Yu L, Norton S, McCracken LM. Change in “self-as-context” (“perspective-taking”) occurs in acceptance and commitment therapy for people with chronic pain and is associated with improved functioning. J Pain. 2017;18(6):664–72.
Pardos-Gascón EM, Narambuena L, Leal-Costa C, Van-der Hofstadt-Román CJ. Differential efficacy between cognitive-behavioral therapy and mindfulness-based therapies for chronic pain: systematic review. Int J Clin Health Psychol. 2021;21(1):100197.
Salomons TV, Moayedi M, Erpelding N, Davis KD. A brief cognitive-behavioural intervention for pain reduces secondary hyperalgesia. Pain. 2014;155(8):1446–52.
Lazaridou A, Kim J, Cahalan CM, Loggia ML, Franceschelli O, Berna C, et al. Effects of cognitive-behavioral therapy (CBT) on brain connectivity supporting catastrophizing in fibromyalgia. Clin J Pain. 2017;33(3):215–21.
Kang D, McAuley JH, Kassem MS, Gatt JM, Gustin SM. What does the grey matter decrease in the medial prefrontal cortex reflect in people with chronic pain? Eur J Pain. 2019;23(2):203–19.
Seminowicz DA, Shpaner M, Keaser ML, Krauthamer GM, Mantegna J, Dumas JA, et al. Cognitive-behavioral therapy increases prefrontal cortex gray matter in patients with chronic pain. J Pain. 2013;14(12):1573–84.
Jensen KB, Kosek E, Wicksell R, Kemani M, Olsson G, Merle JV, et al. Cognitive Behavioral Therapy increases pain-evoked activation of the prefrontal cortex in patients with fibromyalgeia. Pain. 2012;153(7):1495–503.
Shpaner M, Kelly C, Lieberman G, Perelman H, Davis M, Keefe FJ, et al. Unlearning chronic pain: a randomized controlled trial to investigate changes in intrinsic brain connectivity following cognitive behavioral therapy. Neuroimage Clin. 2014;5:365–76.
Yoshino A, Okamoto Y, Okada G, Takamura M, Ichikawa N, Shibasaki C, et al. Changes in resting-state brain networks after cognitive–behavioral therapy for chronic pain. Psychol Med. 2018;48(7):1148–56.
Nascimento SS, Oliveira LR, DeSantana JM. Correlations between brain changes and pain management after cognitive and meditative therapies: a systematic review of neuroimaging studies. Complement Ther Med. 2018;39:137–45.
Zeidan F, Emerson NM, Farris SR, Ray JN, Jung Y, McHaffie JG, et al. Mindfulness meditation-based pain relief employs different neural mechanisms than placebo and sham mindfulness meditation-induced analgesia. J Neurosci. 2015;35(46):15307–25.
Aytur SA, Ray KL, Meier SK, Campbell J, Gendron B, Waller N, et al. Neural mechanisms of acceptance and commitment therapy for chronic pain: a network-based fMRI approach. Front Hum Neurosci. 2021;15:587018.
Meier SK, Ray KL, Waller NC, Gendron BC, Aytur SA, Robin DA. Network analysis of induced neural plasticity post-acceptance and commitment therapy for chronic pain. Brain Sci. 2020;11(1):10.
Hanley AW, Gililland J, Garland EL. To be mindful of the breath or pain: comparing two brief preoperative mindfulness techniques for total joint arthroplasty patients. J Consult Clin Psychol. 2021;89(7):590–600.
Buvanendran A, Sremac AC, Merriman PA, Della Valle CJ, Burns JW, McCarthy RJ. Preoperative cognitive–behavioral therapy for reducing pain catastrophizing and improving pain outcomes after total knee replacement: a randomized clinical trial. Reg Anesth Pain Med. 2021;46(4):313–21.
Hadlandsmyth K, Conrad M, Steffensmeier KS, Van Tiem J, Obrecht A, Cullen JJ, Vander Weg MW. Enhancing the biopsychosocial approach to perioperative care: a pilot randomized trial of the perioperative pain self-management (PePS) intervention. Ann Surg. 2020. https://doi.org/10.1097/SLA.0000000000004671.
Ambrose KR, Golightly YM. Physical exercise as non-pharmacological treatment of chronic pain: why and when. Best Pract Res Clin Rheumatol. 2015;29(1):120–30.
Santos RD, Galdino G. Endogenous systems involved in exercise-induced analgesia. J Physiol Pharmacol. 2018;69(1):3–13.
Nijs J, Kosek E, Van Oosterwijck J, Meeus M. Dysfunctional endogenous analgesia during exercise in patients with chronic pain: to exercise or not to exercise? Pain Phys. 2012;15(3 Suppl):ES205–13.
Sluka KA, Law LF, Bement MH. Exercise-induced pain and analgesia? Underlying mechanisms and clinical translation. Pain. 2018;159(Suppl 1):S91–7.
Holm M, Wernbom M, Schrøder HM, Arendt-Nielsen L, Skou ST. Strength training in addition to neuromuscular exercise and education in individuals with knee osteoarthritis the effects on pain and sensitization. Eur J Pain. 2021;25(9):1898–911.
Thorlund JB, Roos EM, Goro P, Ljungcrantz EG, Grønne DT, Skou ST. Patients use fewer analgesics following supervised exercise therapy and patient education: an observational study of 16 499 patients with knee or hip osteoarthritis. Br J Sports Med. 2021;55(12):670–5.
Bruehl S, Burns JW, Koltyn K, Gupta R, Buvanendran A, Edwards D, et al. Are endogenous opioid mechanisms involved in the effects of aerobic exercise training on chronic low back pain? A randomized controlled trial. Pain. 2020;161(12):2887–97.
Bid DD, Soni N, Yadav A, Rathod P. A study on central sensitization in chronic non-specific low back pain. Indian J Physiother Occup Ther. 2017;11(4):165–75.
Meeus M, Hermans L, Ickmans K, Struyf F, Van Cauwenbergh D, Bronckaerts L, et al. Endogenous pain modulation in response to exercise in patients with rheumatoid arthritis, patients with chronic fatigue syndrome and comorbid fibromyalgia, and healthy controls: a double-blind randomized controlled trial. Pain Pract. 2015;15(2):98–106.
Mayer TG, Neblett R, Cohen H, Howard KJ, Choi YH, Williams MJ, et al. The development and psychometric validation of the central sensitization inventory. Pain Pract. 2012;12(4):276–85.
Neblett R, Hartzell MM, Mayer TG, Cohen H, Gatchel RJ. Establishing clinically relevant severity levels for the central sensitization inventory. Pain Pract. 2017;17(2):166–75.
Neblett R, Hartzell MM, Cohen H, Mayer TG, Williams M, Choi Y, et al. Ability of the central sensitization inventory to identify central sensitivity syndromes in an outpatient chronic pain sample. Clin J Pain. 2015;31(4):323–32.
Meeker TJ, Jupudi R, Lenz FA, Greenspan JD. New developments in non-invasive brain stimulation in chronic pain. Curr Phys Med Rehabil Rep. 2020;8(3):280–92.
Leung A, Shirvalkar P, Chen R, Kuluva J, Vaninetti M, Bermudes R, Expert Consensus Panel, et al. Transcranial magnetic stimulation for pain, headache, and comorbid depression: INS-NANS expert consensus panel review and recommendation. Neuromodulation. 2020;23(3):267–90.
U.S. Food and Drug Administration. Senza Spinal Cord Stimulation System—P130022/S039. 2021. https://www.fda.gov/medical-devices/recently-approved-devices/senza-spinal-cord-stimulation-system-p130022s039. Accessed 16 Dec 2021.
Tieppo Francio V, Polston KF, Murphy MT, Hagedorn JM, Sayed D. Management of chronic and neuropathic pain with 10 kHz spinal cord stimulation technology: summary of findings from preclinical and clinical studies. Biomedicines. 2021;9(6):644.
National Institute for Health and Care Excellence. Senza spinal cord stimulation system for delivering HF10 therapy to treat chronic neuropathic pain. Medical technologies guidance [MTG41]. 2019. https://www.nice.org.uk/guidance/mtg41/chapter/1-Recommendations. Accessed 16 Dec 2021.
Skelly AC, Chou R, Dettori JR, Turner JA, Friedly JL, Rundell SD, et al. Noninvasive nonpharmacological treatment for chronic pain: a systematic review. Rockville (MD): Agency for Healthcare Research and Quality (US). 2018.
National Institute for Health and Care Excellence. Chronic pain (primary and secondary) in over 16s: assessment of all chronic pain and management of chronic primary pain. 2021. https://www.nice.org.uk/guidance/ng193/chapter/Recommendations. Accessed 16 Dec 2021.
Acknowledgements
Not applicable.
Funding
None.
Author information
Authors and Affiliations
Contributions
Conceptualization: AHB and FHB; Literature Search: AHB and FHB; Manuscript Synthesis: AHB and FHB; Manuscript Review and Editing: AHB and FHB. Both authors read and approved the final manuscript.
Authors’ information
Amjad H. Bazzari, Ph.D., Assistant Professor in Neurophysiology & Translational Neuroscience, Faculty of Medicine, Arab American University, 240 Jenin, 13 Zababdeh, Palestine. Firas H. Bazzari, Ph.D., Assistant Professor in Neuropharmacology, Faculty of Pharmacy, Arab American University, 240 Jenin, 13 Zababdeh, Palestine.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors report no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bazzari, A.H., Bazzari, F.H. Advances in targeting central sensitization and brain plasticity in chronic pain. Egypt J Neurol Psychiatry Neurosurg 58, 38 (2022). https://doi.org/10.1186/s41983-022-00472-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41983-022-00472-y