
Abstract
Background
There is conflicting data as to the prevalence of seizures and the relevance of epileptiform EEGs in autistic children. With the focus primarily on aetiology in autism research, very little emphasis has been placed on comorbid conditions in recent years. This imbalance must be brought to attention, as the comorbidities may harbour the prospect for effective treatment options.
Objectives
To determine the prevalence and characteristics of seizures in autistic children and to develop a clinical profile of the autistic child with seizures.
Method
Clinical, EEG, and MRI data of 86 autistic children between the ages of 3 and 13 years old, spanning a 10-year period, from 1 January 2008 until 1 January 2018, were analysed retrospectively.
Results
The prevalence of epilepsy was 23% with a 95% confidence interval (14.8, 33.6). There were 17 males and 3 females. There was no correlation with a history of regression and sleep hygiene.
Conclusion
Seizures are common in autistic children (23%) and have a much higher prevalence than in the general childhood population (0.5–1.0%). Semiology can be subtle, mimicking autistic behaviour. EEG and MRI evidence is often lacking, making the diagnosis of epilepsy difficult. If left unrestrained, the seizures frequently evolve into intractable epilepsy. Greater awareness is imperative in order to prevent early cognitive and behavioural decline. This is one of only a few such studies to have come out of Africa. More studies from different geographical locations are urgently required.
Similar content being viewed by others


Introduction
Autism has reached epidemic proportions, and there is no obvious explanation for this increase [1]. Researchers are scrambling for a reliable aetiology, and the comorbid conditions of autism are largely being overlooked. This is to the detriment of the child with autism, as some of these conditions may be preventable to a certain degree, or at least harbour valid targets for current treatment options. Of great interest is that comorbidities such as cognitive impairment and epilepsy may share a common and dysfunctional neurobiological substrate with autism, leading to the possibility that improvement in one domain may result in more widespread benefit in neurodevelopment. In order for this to happen, certain circuits must be shared by the respective conditions.
If it is considered that the mesial temporal lobes form the main site of epileptogenesis in the brain [2], and that they also constitute the amygdala and hippocampus, which are pivotal to the creation of fear processing, social cognition, and behaviour [3], as well as memory formation and storage [4], then it becomes entirely plausible that epilepsy could be producing the autistic phenotype through the early destruction of this region, called the salience network, and possibly even in utero [5].
Whereas the prevalence of epilepsy in the general childhood population is known and relatively stable (0.5–1.0%), the prevalence of epilepsy in autism remains largely undetermined [6] and currently ranges between 5 and 38%, depending on the selection criteria adopted for each cohort [7, 8]. Similarly, the global prevalence of autism also varies according to geographical distribution, and if a composite of data from both rural and urban areas is taken into account, it is estimated to be 62/10,000 (although many researchers estimate this value to be much higher) [9]. Due to this alarming increase in cases of autism in the world today, and the fact that unregulated seizures seem to accompany the dysfunctional neural circuitry, greater emphasis should be placed on addressing what is both known and treatable.
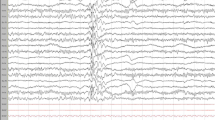
However, temporal lobe epilepsy is often difficult to diagnose. In making a concrete diagnosis of epilepsy, a patient’s history, clinical examinations, EEG (electroencephalogram), and MRI (magnetic resonance imaging) findings all come together to support the supposition that there is abnormal and ongoing electrical activity in the brain [10].
Certain characteristics specific to the temporal lobe complicate seizure surveillance. A child with temporal lobe seizures may present with sudden and unprovoked temper tantrums [11], or intermittent psychotic episodes [12], which can mimic routine autistic behaviour. Seizures arising within the confines of the temporal lobe may involve visual pathways and result in brief staring episodes [13]. This can be masked by expected anti-social behaviour, as autistic children are known to avoid visual contact and close interpersonal contact. Children with temporal lobe epilepsy often experience seizures at night, which makes their detection difficult and greatly limits parental reporting of seizures [14]. Sleep is a potent driver of epileptogenesis. Melatonin lowers the physiological levels of dopamine during sleep, and dopamine counteracts epilepsy by elevating the seizure threshold [15].
Even the anatomy of the temporal lobes impedes the diagnostic process. As the medial temporal lobes diverge inwardly from the exterior of the brain, discharges arising from these regions are often not detected by the more superficially placed cortical leads applied during EEG [16]. This frequently results in a normal EEG, a misdiagnosis, and a false sense of security. Additionally, the MRIs of the brain often appear normal, even though prolonged epileptogenesis may have occurred [17]. All of these factors make confirmation of the diagnosis of epilepsy elusive and leave both the clinician and the parents of the child in doubt. This can result in delayed treatment and the deterioration in cognition, speech, and behaviour due to unrestrained epileptogenesis.
The dilemma facing clinicians is that seizure semiology in autism, and especially in children, has not been accurately validated [18]. Autism, epilepsy, and cognitive impairment may be comorbid conditions arising from the same defective neurobiological substrate [19]. As a result, clinical presentation, duration, and frequency of seizures in the autistic child may differ from those in normal children. However, all seizure types have previously been reported in children with autism, and autism has not been linked to any particular seizure semiology [20]. The seizure semiology in autism should be accurately characterized in order to facilitate the early detection and prevention of seizure escalation in these already vulnerable children [21].
Lastly, clinical biomarkers are urgently required to identify the autistic child who is at risk of developing epilepsy [22]. By determining the seizure semiology and clinical profile of the epileptic, epileptogenesis can be slowed and possibly even prevented [23]. The fact that seizures may alter and disrupt both cognition and behaviour in the child with autism, then this study of the association between autism and epilepsy, becomes important for both children at risk and their parents [24].
The aim of the study is to raise awareness of the high prevalence of epilepsy in autistic children and to demonstrate the complexities and difficulties of its diagnosis and management in these children.
Methodology
This was a retrospective study that involved 86 children (64 males/22 females) with autism who were followed up at a tertiary level paediatric neurology clinic at Steve Biko Academic Hospital, in the Republic of South Africa, from January 2008 to January 2018. Children were diagnosed with autism based on criteria in the DSM-5 (Diagnostic and Statistical Manual-5) and therefore displayed fixed deficits in both social communication and interaction across multiple domains, as well as restricted interests and repetitive patterns of behaviour in activities of daily living [25].
Clinical records relating to the medical history and clinical examination of each child, together with EEG data that had been collected over a 10-year period, were retrospectively analysed in order to determine the prevalence and characteristics of the seizures in these children. EEG data comprised a mixture of 30 min recordings performed during both sleep and wakefulness. EEGs were done by using Niyon Koden EEG-90100/9200 machines from Nihon Kohden Corporation. Japan.
Inclusion criteria
Children who had been diagnosed with autism according to the DSM-5 between the ages of 3 and 13 years old, who had previously displayed fixed deficits in social (non-verbal) and verbal communication, together with restricted interests in activities of daily living, and a preoccupation with purposeless, repetitive behaviours, were included.
Exclusion criteria
The exclusion criteria are as follows:
-
1)
Cases of “syndromic autism”, such as neurocutaneous syndromes (tuberous sclerosis) and other known genetic causes of autism (Angelman syndrome/Rett syndrome).
-
2)
Those with a history of neonatal encephalopathy (Apgar score below 7/cord blood pH below 7.2).
-
3)
Those with microcephaly, cerebral palsy, dysmorphism, or laboratory evidence of a congenital infection.
In the children who were later found to have seizures, four additional clinical parameters were assessed in order to create a clinical profile of the autistic child most at risk of developing epilepsy, namely (1) the level of intellectual disability, (2) history of language and behavioural regression as a young child, (3) the presence of motor stereotypies, and (4) the presence of sleep disturbances, as indicated merely by parental reporting, and without the implementation of formal sleep studies or polysomnography (PSG).
A formal visual-motor assessment (Beery-Buktenica Test) was conducted on each child with epilepsy in order to ascertain their intellectual quotient (IQ) [26]. A Beery Test was performed on the children with epilepsy in order to find a correlation between level of cognition and the presence of epilepsy. The Beery-Buktenica Developmental Test of Visual-Motor Integration (Beery Test) evaluates activities of daily living, such as being able to get dressed without assistance, eating and drinking with a cup and spoon, and going to the toilet unsupervised [27]. These measures of executive function are incorporated into the assessment and may be used to determine IQ [28].
Consent for the study was received from the Human Research Ethics Committee of the University of Pretoria.
Statistical methods
Data was be captured, cleaned, and analysed in Stata: Release 11 (StataCorp. 2009. Statistical Software. College Station, TX, USA: StataCorp LP). Categorical variables were represented as absolute values and percentages. Non-normally distributed numerical variables were represented as medians and interquartile ranges. Normally distributed numerical data were represented as mean and standard deviation. The Wilcoxon rank sum test was used to test differences between numerical variables, and Fisher’s exact test was used to test for differences between categorical variables. A p value of less than 0.05 was used to denote statistical significance.
Sampling and sample size
This retrospective study made use of opportunistic sampling to obtain the required sample size. Opportunistic sampling was used as the prevalence of epilepsy is low (between 0.5 and 1%), and it would be the best sampling method to reach the required sample size.
For the proposed cross sectional study, using the formula n = Z2P (1 − P)/d2 where n denotes the sample size, Z denotes the statistic corresponding to the level of confidence, P denotes the expected prevalence, and d denotes the level of precision with d = 0.05 and prevalence of 0.01, a total of 16 participants with epilepsy would be required.
Results
The prevalence of epilepsy in the autistic children was 23% (17 boys and 3 girls). The median age of the children with epilepsy was 7 years (IQR 5.5 to 9): 7 years (IQR 6 to 8) for the male children and 6 years (IQR 5 to 12) for the female children (p value 0.96).
Staring episodes, lasting either seconds or a few minutes, occurred in 85% of the children with epilepsy, and they were always the first manifestation in the series of ictal events. Lateral version of the eyes occurred in 60% of the children, and a few also displayed recurrent blinking of the eyelids at the onset of a seizure. Nineteen patients (95%) presented with at least one of these ocular signs during the initial stages of the seizure. In 55% of the patients with epilepsy, seizures would only occur at night during sleep.
Generalized tonic-clonic seizures were observed in 11 children (55%). Only two children displayed focal semiology involving the limbs on one side of the body. Although seizure frequency could only be determined in 15 of the 20 children with epilepsy, due to either absent guardians at follow-up visits or unreliable record keeping, 60% of the patients with adequately maintained seizure charts would only have seizures on 2 or 3 days over a 1- to 6-month period. The relative infrequency of seizures may complicate the detection of seizures in autism.
Although the age of seizure onset could not be determined in all cases, eight of the epileptic children developed seizures during infancy, and five of these had seizure-onset within the first 6 months of life. Epilepsy had been confirmed in 11 children before the age of 3 years, at which time autism is often first diagnosed.
All seizure types were noted, and no specific seizure type could be reliably linked to autism. In toddlers, seizures would manifest in the form of apnoeic episodes, breath-holding spells, and sudden unprovoked temper tantrums. Drop attacks (atonic seizures) involving the head, neck, and shoulders and occurring in clusters were often accompanied by other seizure types, similar to that which occurs in other epileptic encephalopathies. However, not all seizure types were atypical, and one child presented with typical absence epilepsy. Unusual semiology was also noted, as evidenced by a child with gelastic seizures (recurrent bouts of impersonal laughter) that occurred routinely in the early hours of the morning, and another who experienced a very rare form of epilepsy, namely cursive seizures, that resulted in the child to getting up abruptly and running out of the house.
Five children (25%) endured recurrent episodes of status epilepticus (seizures lasting longer than 20 min). Four of these patients with recurrent status epilepticus had persistently normal EEGs (Table 1).
The majority of the EEGs (55%) demonstrated a focal, as opposed to a generalized epileptiform dysfunction. In five EEGs, the epileptiform focus was situated in the frontal lobes. The seizures originated from the temporal lobe in three children and from the occipital lobe in another. Two patients had multifocal epileptiform dysfunction. Only two of the EEGs (10%) revealed a generalized epileptiform dysfunction.
The EEGs of three children revealed a non-specific dysfunction which may have been epileptiform but which did not demonstrate typical spike-wave discharges. These were in the form of runs of either theta slow-wave or beta fast-wave activity in the frontal or temporal regions, which may have preceded epileptiform discharges. One EEG showed the absence of regular interictal alpha wave rhythms during wakefulness in one hemisphere, which may indicate the presence of an underlying epileptiform dysfunction. In four children, the EEG was normal on more than one occasion, in the presence of recurrent and unprovoked clinical seizures. There was no statistically significant association between EEG findings and clinical semiology (OR 0.29 to 40.95, p value 0.33). Whereas a significant proportion of the EEGs were focal, most clinical seizures were generalized.
All of the children with epilepsy (100%) had intellectual disability (ID) with an IQ below 80 (Table 2).
Of great importance is that 45% of these had severe (IQ 20–40) to profound (IQ < 20) ID. The three girls in the group all had severe to profound intellectual disability with an IQ below 40 (Table 3).
Females used a significantly higher number of drugs (4, IQR 3 to 4) than the males (1, IQR 1 to 3) (p value 0.03) (Table 4). Two thirds of the children who had been placed on two or more anti-epileptic drugs had severe to profound intellectual disability.
A history of regression was an unreliable marker of epilepsy. Only a single child had a history of language or developmental regression. With regard to the presence of motor stereotypies as a possible marker of epilepsy, 12 children (60%) with epilepsy demonstrated recurrent, purposeless, or stereotypical behaviour. Six of them displayed primarily hand-flapping, and another six demonstrated recurrent rocking of either the head or torso and a preoccupation with turning around in circles. All of those who engaged in rocking behaviour had a level of cognition that was severely to profoundly impaired (IQ below 40).
Half of the children with epilepsy experienced sleep disturbances at night (either in the form of delayed sleep-onset or recurrent nocturnal awakenings), which was well within the expected limits experienced by autistic children without epilepsy. Sleep disturbances therefore could not be used as a reliable marker of epilepsy.
Discussion
Over the last few years, there has been a surge in interest within the field of autism research regarding the rising incidence of autism within the childhood population [29]. It is widely postulated that a continuous interaction exists between genetic and epigenetic [30] or environmental factors which play out on a second-by-second basis. With all this focus on aetiology, the comorbidities linked with autism have been progressively overlooked. Regular reporting of different patient cohorts is becoming more and more necessary in determining the natural history and developmental characteristics of the disorder [31].
The prevalence of epilepsy in autism is extremely high (23% in this study). The seizures in autism are subtle, nocturnal, and infrequent. Although staring represents a complex partial seizure, the significance of this type of seizure in autism is not known, and this is relevant when choosing anti-convulsant treatment [32]. They often occur in infancy and can simulate autistic behaviour. This implies that they are unrecognized and underreported. In fact, the relative frequency of the various types of seizure disorders in autism is unknown [33], and there has been surprisingly little study of the relationship between staring episodes and temporal lobe epilepsy in children in general [34]. Very few studies are available that attempt to create a clinical profile for the child with autism who may be having occult seizures late at night. Further studies are needed to determine modifiable risk factors for non-febrile seizures in autism [35]. If left untreated, the seizures can accelerate epileptogenesis, resulting in cognitive deficits and worsening behaviour. Early diagnosis and intervention are therefore critical.
The epilepsy in autism is also severe. In many children, the epilepsy becomes intractable, and in most cases, two or more anti-epileptic drugs are used to control the seizures. The epilepsy is dangerous; a significant number of children present with recurrent episodes of status epilepticus, which has an ominous mortality rate attached to it [36, 37]. Herein lies the paradox when dealing with epilepsy in autism; it appears early and often remains concealed during the early stages of development.
Any seizure type can present in autism, and the same seems to apply to the EEG [38]. To complicate matters, the EEG often does not correlate with the seizure semiology. The MRI scans are often reported as being normal, and parenchymal lesions are not responsible for the epilepsy in the vast majority of cases [39]. The resulting paucity of auxiliary evidence creates doubt in the mind of the attending physician as to if epilepsy is indeed present or not. Similarly, a history of developmental regression is not a reliable marker of epilepsy in autism [40].
It has also been shown that a similar developmental trajectory exists between autistic children who underwent a period of regression and those who did not [41]. There are studies that state that epilepsy is associated with autistic regression in children [42, 43]. Other studies contradict this and state that epilepsy is a comorbidity of autism and does not contribute to the initiation of a period of loss in previously acquired milestones [44]. Despite the occurrence of clinical seizures, the presence or absence of developmental regression has no predictive value or influence on the epileptiform nature of these EEGs [45]. The truth probably lies somewhere in between, and epilepsy plays a minor but not negligible role in autistic regression [46]. The neurobiological basis of regression needs to be investigated and characterized, as there still may be a minor but non-negligible association with epilepsy.
The prevalence and nature of stereotypical behaviour may be an “indirect” marker of epilepsy in autism. The more robust the clinical presentation of a particular comorbidity, namely autism, epilepsy, and mental retardation, within an individual, the greater the likelihood will be that stereotypical behaviour will occur [47]. Hand and finger stereotypies, and in particular hand flapping, seem to occur more often in higher-functioning children, whereas self-stimulatory behaviour (in the form of head nodding, body rocking, and, in some cases, episodes of hyperventilation) appears to be more prevalent in lower-functioning children [48].
As the nature and presence of motor stereotypical behaviour can reflect the level of cognition, and as lower levels of cognition are more associated with epilepsy, self-stimulatory behaviour will be more likely to occur in severely cognitively impaired children with epilepsy. Epilepsy can also mimic stereotypical behaviour and aggravate existing motor stereotypies [49]. Motor stereotypies are also associated with the severity of progression in autistic individuals, beyond that predicted by social communication measures alone [50], although symptomatology in autism may change with age [51]. Therefore, further studies are required that can elucidate reliable and quantitative measurements of autistic symptoms [52].
The ratio of males to females in the total cohort of children with autism was 3.75:1, which is in keeping with international estimates [53]. All of the females who had epilepsy had an IQ below 40 and were severely to profoundly cognitively impaired. Each one of them had intractable epilepsy and was on at least three or more anti-epileptics. The above factors seem to indicate that epilepsy severity is directly proportional to the level of intellectual disability. There is a need for a detailed neuropsychiatric assessment in all children who present with the combination of epilepsy, learning disability, and autistic behaviour [54]. The risk of epilepsy is proportionate to the degree of underlying brain dysfunction [55].
Each female had definitive epileptiform spikes or sharp waves on her EEG and had been referred for MRI. Of critical importance is that all of them had their first seizures during infancy. In two of these children, seizure onset was below the age of 6 months. There was also a strong correlation between early seizure onset and the later development of recurrent episodes of status epilepticus. Therefore, female gender could be a useful marker for epilepsy. Remarkably little research has focused on the reasons for this disparity in gender. It remains to be seen whether or not the female brain is vulnerable to the effects of epileptogenesis in autism. Better understanding of this gender difference could lead to major advancements in the prevention of the autistic phenotype in both males and females [56].
This study aims to explore a possible association between autism and epilepsy. Such an undertaking is important for a number of reasons. Very few systematic investigations that explore the link between autism and epilepsy have been done. Danielsson et al. conducted the largest and longest prospective population-based study to establish the prevalence and characteristics of epilepsy in autism. They followed up 120 autistic individuals (diagnosed in childhood) to determine how many of them would develop epilepsy in childhood and how many would undergo remission in adulthood [57]. The landmark study was published in Epilepsia and highlighted the need for similar studies from different geographical locations, in order to “neutralize” the effect which the environment may exert on susceptible individuals.
Conclusion
Brain wiring is abnormal in autism. Cognitive impairment and epilepsy share the same dysfunction in messaging. Epilepsy, therefore, is extremely common in autism. Not only does it delay neurodevelopment but it worsens behaviour. This has a huge impact not only on the individual but also on the whole family. If left unchecked, the seizures become intractable, and numerous anti-epileptics are utilized, thereby increasing the side-effect profile. As these seizures can mimic autistic behaviour, a greater awareness must be fostered throughout the autism community. This is one of only a few such studies to have come out of Africa. While the world rushes to identify modifiable risk factors in autism, the significance of epilepsy should not be overlooked.
Study limitations
Due to the fact that autism, epilepsy, and mental retardation overlap, the results of the study may reflect the selection bias of the group. It was difficult to ascertain which had arisen first, autism or epilepsy in two children. This is due to the fact that the epilepsy in both of these children started in infancy. However, the parents noticed from an early age that the pervasive symptoms were dominant and occurred alongside the epilepsy, and not as a consequence thereof.
Autistic regression is determined by three factors, namely language regression or a loss of communication skills, loss of social skills, and onset of restricted and repetitive behaviour. Parents only reported language regression; however, the loss of social skills and the development of motor stereotypies are as important when assessing regression. The prevalence of regression and its association with epilepsy may have been significantly higher had all three aspects been considered.
Availability of data and materials
The datasets generated and/or analysed during the current study are not publicly available due institutional rules but are available from the corresponding author on reasonable request.
Abbreviations
- DSM-5:
-
Diagnostic and Statistical Manual-5
- EEG:
-
Electroencephalogram
- ID:
-
Intellectual disability
- IQ:
-
Intellectual quotient
- MRI:
-
Magnetic resonance imaging
References
Leonard H, Glasson E, Nassar N, Whitehouse A, Bebbington A, Bourke J, et al. Autism and intellectual disability are differentially related to sociodemographic background at birth. PLoS One. 6(3):e17875.
Ben-Ari Y, Dudek FE. Primary and secondary mechanisms of epileptogenesis in the temporal lobe: there is a before and an after. Epilepsy Curr. 2010;10(5):118–25.
Olson IR, McCoy D, Klobusicky E, Ross LA. Social cognition and the anterior temporal lobes: a review and theoretical framework. Soc Cogn Affect Neurosci. 2013;8(2):123–33.
Alvarez P, Squire LR. Memory consolidation and the medial temporal lobe: a simple network model. Proc Natl Acad Sci U S A. 1994;91:7041–5.
Green SA, Hernandez L, Bookheimer SY, Dapretto M. Salience network connectivity in autism is related to brain and behavioural markers of sensory over-responsivity. J Am Acad Child Adolesc Psychiatry. 2016;55(7):618–26.
Evangelista E, Benar C, Bonini F, Carron R, Colombet B, Regis J, et al. Does the thalamo-cortical synchrony play a role in seizure termination? Front Neurol. 2015;6:192.
Besag FMC. The relationship between epilepsy and autism: a continuing debate. Acta Paediatr. 2009;98(4):618–20.
Berg AT, Plioplys S. Epilepsy and autism: is there a special relationship? Epilepsy Behav. 2012;23(3):193–8.
Elsabbagh M, Divan G, Koh Y. Global prevalence of autism and other pervasive developmental disorders. Autism Res. 2012;5(3):160–70.
Engel J Jr. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE Task Force on classification and terminology. Epilepsia. 2001;42(6):796–803.
Brodie MJ, Besag F, Ettinger AB, Mula M, Gobbi G, Comai S, et al. Epilepsy, antiepileptic drugs, and aggression: an evidence-based review. Pharmacol Rev. 2016;68(3):563–602.
Beletsky V, Mirsattari SM. Epilepsy, mental health disorder, or both? Epilepsy Res Treat. 2012;2012:163731.
Tufenkjian K, Luders HO. Seizure semiology: its value and limitations in localizing the epileptogenic zone. J Clin Neurol. 2012;8(4):243–50.
Bernasconi A, Andermann F, Cendes F, Dubeau F, Andermann E, Olivier A. Nocturnal temporal lobe epilepsy. Neurology. 1998;50:1772–7.
Starr MS. The role of dopamine in epilepsy. Synapse. 1996;22(2):159–94.
Javidan M. Electroencephalography in mesial temporal lobe epilepsy: a review. Epilepsy Res Treat. 2012;2012:637430.
Jeste SS. The neurology of autism spectrum disorders. Curr Opin Neurol. 2011;24(2):132–9.
El Achkar CM, Spence SJ. Clinical characteristics of children and young adults with co-occurring autism spectrum disorder and epilepsy. Epilepsy Behav. 2015;47:183–90.
Sansa G, Carlson C, Doyle W, Weiner HL, Bluvstein J, Barr W, et al. Medically refractory epilepsy in autism. Epilepsia. 2011;52(6):1071–5.
Camfield P, Camfield C. Incidence, prevalence and aetiology of seizures and epilepsy in children. Epileptic Disord. 2015;17(2):117–23.
Bombardieri R, Pinci M, Moavero R, Cerminara C, Curatolo P. Early control of seizures improves long-term outcome in children with tuberous sclerosis complex. Eur J Paediatr Neurol. 2010;14:146–9.
Esler AN, Ruble LA. DSM-5 diagnostic criteria for autism spectrum disorder with implications for school psychologists. Int J Sch Educ Psychol. 2015;3(1):1–15.
Solomon M, Ozonoff S, Cummings N, Cameron Carter MD. Cognitive control in autism spectrum disorders. Int J Dev Neurosci. 2008;26(2):239–47.
Solomon M, Ozonoff SJ, Ursu S, Ravizza S, Cummings N, Ly S, et al. The neural substrates of cognitive control deficits in autism spectrum disorders. Neuropsychologia. 2009;47(12):2515–26.
Sortor JM, Kulp MT. Are the results of the Beery-Buktenica Developmental Test of Visual-Motor Integration and its subsets related to achievement test scores? Optom Vis Sci. 2003;80(11):758–63.
Dickerson Mayes S, Calhoun SL. Analysis of WISC-III, Stanford-Binet: IV and Academic Achievement Test Scores in children with autism. J Autism Dev Disord. 2003;33(3):329–41.
Green RR, Bigler ED, Froehlich A, Prigge MB, Travers BG, Cariello AN, et al. Beery VMI performance in autism spectrum disorder. Child Neuropsychol. 2016;22(7):795–817.
Frye RE. Prevalence, significance, and clinical characteristics of seizures, epilepsy and subclinical electrical activity in autism. N Am J Med Sci. 2015;8(3):113–22.
Rutter M. Incidence of autism spectrum disorders: changes over time and their meaning. Acta Paediatr. 2005;94(1):2–15.
Persico AM, Bourgeron T. Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci. 2006;29(7):349–58.
Head AM, McGillivray JA, Stokes MA. Gender differences in emotionality and sociability in children with autism spectrum disorders. Mol Autism. 2014;5:19.
Gillberg C. The treatment of epilepsy in autism. J Autism Dev Disord. 1991;21(1):61–77.
Olsson I, Steffenburg S, Gillberg C. Epilepsy in autism and autistic like conditions: a population-based study. Arch Neurol. 1988;45:666–8.
Ott D, Caplan R, Guthrie D, Siddarth P, Komo S, Shields WD, et al. Measures of psychopathology in children with complex partial seizures and primary generalized epilepsy with absence. J Am Acad Child Adolesc Psychiatry. 2001;40(8):907–14.
McCue LM, Flick LH, Twyman KA, Xian H, Conturo TE. Prevalence of non-febrile seizures in children with idiopathic autism spectrum disorder and their unaffected siblings: a retrospective cohort study. BMC Neurol. 2016;16:245.
DeLorenzo RJ, Hauser WA, Towne AR, Boggs JG, Pellock JM, Penberthy L, et al. A prospective population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology. 1996;46(4):1029–35.
Hauser WA. Status epilepticus: epidemiologic considerations. Neurology. 1990;40(5):9–13.
Canitano R. Epilepsy in autism spectrum disorders. Eur Child Adolesc Psychiatry. 2007;16(1):61–6.
Ming X, O’Connor M, Bandari S, He W, Brewer G. The utility of MRI in children with autism spectrum disorder. Autism Open Access. 2016;6(1):1–4.
Tuchman RF, Rapin I. Regression in pervasive developmental disorders: seizures and epileptiform electroencephalographic correlates. Pediatrics. 1997;99:560–6.
Jones LA, Campbell JM. Clinical characteristics associated with language regression for children with autism spectrum disorders. J Autism Dev Disord. 2010;40(1):54–62.
Giannotti F, Cortesi F, Cerquiglini A. An investigation of sleep characteristics, EEG abnormalities and epilepsy in developmentally regressed and non- regressed children with autism. J Autism Dev Disord. 2008;38:1888–97.
Hrdlicka M, Komarek V, Propper L, Kulisek R, Zumrova A, Faladova L, et al. Not EEG abnormalities but epilepsy is associated with autistic regression and mental functioning in childhood autism. Eur Child Adolesc Psychiatry. 2004;13:209–13.
Canitano R, Luchetti A, Zappella M. Epilepsy, electroencephalographic abnormalities, and regression in children with autism. J Child Neurol. 2005;20:27–31.
Chez MG, Chang M, Krasne V, Coughlan C, Kominsky M, Schwartz A. Frequency of epileptiform EEG abnormalities in a sequential screening of autistic patients with no known clinical epilepsy from 1995 to 2005. Epilepsy Behav. 2006;8(1):267–71.
Rapin I. Autistic regression and disintegrative disorder: how important is the role of epilepsy? Semin Pediatr Neurol. 1995;2(4):278–85.
Smith KRM, Matson JL. Behavior problems: differences among intellectually disabled adults with co-morbid autism spectrum disorders and epilepsy. Res Dev Disabil. 2010;31(5):1062–9.
Goldman S, Wang C, Salgado MW, Greene PE, Kim M, Rapin I. Motor stereotypies in children with autism and other developmental disorders. Dev Med Child Neurol. 2009;51:30–8.
Deonna T, Roulet E. Evaluating a possible contributing or causal role of epilepsy. Epilepsia. 2006;47(2):79–82.
Ghanizadeh A. Clinical approach to motor stereotypies in autistic children. Iran J Pediatr. 2010;20(2):149–59.
Baranek GT. Autism during infancy: a retrospective video analysis of sensory- motor and social behaviors at 9-12 months of age. J Autism Dev Disord. 1999;29(3):213–24.
Viscidi EW, Johnson AL, Spence SJ, Buka SL, Morrow EM, Triche EW. The association between epilepsy and autism symptoms and maladaptive behaviors in children with autism spectrum disorder. Autism. 2014;18(8):996–1006.
Baker SM, Milivojevich A. Gender differences among children with autism spectrum disorder: differential symptom patterns. Glob Adv Health Med. 2013;2(6):8–18.
Hagberg B, Hagberg G, Lewerth A, Lindberg U. Mild mental retardation in Swedish school children. II. Etiologic and pathogenetic aspects. Acta Paediatr Scand. 1981;70:445–52.
Steffenburg S, Steffenburg U, Gillberg C. Autism spectrum disorders in children with active epilepsy and learning disability: comorbidity, pre-and perinatal background, and seizure characteristics. Dev Med Child Neurol. 2003;45:724–30.
Halladay AK, Bishop S, Constantino JN, Daniels AM, Koenig K, Palmer K, et al. Sex and gender differences in autism spectrum disorder: summarizing evidence gaps and identifying emerging areas of priority. Mol Autism. 2015;6:36.
Danielsson S, Gillberg IC, Billstedt E, Gillberg C, Olsson I. Epilepsy in young adults with autism: a prospective population-based follow-up study of 120 individuals diagnosed in childhood. Epilepsia. 2005;46(6):918–23.
Acknowledgements
I would like to formally thank the following colleagues for their contributions towards this article:
1) Professor Robin J. Green, my supervisor.
2) Mrs. Henriette Dippenaar, from the Department of Occupational Therapy, who assisted me in assessing the Beery tests, which were used to determine the IQs (intellectual coefficients) of each patient.
3) Miss Itumeleng Phele, from the Department of Neurophysiology, who made the EEG (electroencephalogram) data available.
4) The mothers of the children in the study, who made this research possible, so that others may benefit from it.
Funding
Not applicable
Author information
Authors and Affiliations
Contributions
LGV is responsible for the conception of the work, analysing and interpreting the data, approving the final revision of the work, and communicating with the journal during the manuscript submission, peer review, and publication process. GRJ is the supervisor of the research, giving guidance and support throughout. OSAS provided the statistical analysis and guidance. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent from the CEO of the hospital to review hospital records was obtained. Written informed consent from the mothers of all patients to perform the Beery-Buktenica test was also obtained.
Ethics reference no: 170/2018.
The Faculty of Health Sciences Research Ethics Committee approved ethics on 25 April 2018.
Contact: Dr. R Sommers; Deputy Chairperson of the Faculty of Health Sciences Research Ethics Committee, University of Pretoria.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lamb, G.V., Green, R.J. & Olorunju, S. Tracking epilepsy and autism. Egypt J Neurol Psychiatry Neurosurg 55, 55 (2019). https://doi.org/10.1186/s41983-019-0103-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41983-019-0103-x