
Abstract
Primary liver cancers are rare in children, and the most common type is hepatoblastoma (HB), an embryonal tumor with histological features that resemble different stages of liver cell differentiation. However, mainly because of its rarity, molecular data on HB tumorigenesis remain scarce. This article reviews the current knowledge regarding genetic and epigenetic alterations reported in HB cases, with emphasis on the recent findings of next-generation sequencing studies.
Similar content being viewed by others

Background
Cancer cells are characterized by the acquisition of several capacities during the stages of tumorigenesis [1], including maintenance of proliferation signaling, inhibition of growth suppressors, resistance to cell death, induction of angiogenesis, activation of invasive and metastatic pathways, ability to evade immunological destruction and modification of cellular metabolism. These features are the outcome of the progressive accumulation of somatic mutations in key genes, a process that generally requires long periods of time [1]. In tumors of adults, most mutations triggering tumorigenesis are often due to DNA repair dysfunction due to aging as well as exposure to environmental factors such as cigarette smoke, chemicals and ultraviolet radiation.
Although neoplasia is rare in children, it is the leading cause of death among young people in developed countries. For example, in the United States, 15,789 children and adolescents (up to 19 years old) were diagnosed with cancer in 2014, and 1,960 died from the disease [2]. It is estimated that approximately 10% of all tumors in children are driven by inherited mutations, with a portion being associated with known genetic syndromes, such as Li-Fraumeni and Beckwith-Wiedemann, which confer an increased risk of developing certain types of tumors [2].
Embryonal tumor, a subtype of pediatric cancer, is defined mainly by its presumptive origin from partially undifferentiated primordial cells that have undergone genetic mutations [3]. The major hypothesis of origin is based on the occurrence of signaling errors that prevent both cell maturation and differentiation during embryonic development. These molecular defects during organogenesis could lead to either malformation of the organ or development of an embryonal tumor.
This article reviews the current knowledge regarding the major genetic and epigenetic alterations that occur in the genome of hepatoblastomas (HBs), an embryonal liver tumor. We discuss novel genomic data for HB, with emphasis on the recent findings of next-generation sequencing and their implications for HB tumorigenesis.
Main text
Prevalence and clinical features of hepatoblastomas
Primary liver cancers are rare in children. HB is the most common type, representing approximately 1% of all cancers in this age group [4]. According to a US study [5] carried out between 2002 and 2008, approximately 10 of every one million children under one year of age are affected by HB. The diagnosis is based primarily on ultrasound and CT scan following detection of an increased abdominal mass; symptoms include anorexia, weight loss and pain [6]. An elevated level of α-fetoprotein (AFP), a major component of the carcinoembryonic proteome [7], is considered a HB marker as well as a prognostic factor [8].
Regarding histological features, HBs can be highly variable both within and between cases with epithelial and stromal components. The epithelial component can be embryonal or fetal, the stromal component is composed of connective tissue and even heterologous elements such as bone, cartilage and skeletal muscle are usually found. HBs are classified according to their components. Mixed stromal and epithelial subtype is the most common (approximately 44% of cases). Among pure epithelial, a combination of embryonal and fetal type epithelia can also be found in different proportions. Purely fetal HBs account for 31% of the cases, and embryonal type accounts for 19% of the tumors. Macrotrabecular tumors, which are infrequent and need to be distinguished from hepatocellular carcinoma (HCCs), and small cell undifferentiated type, account for only 3% of HBs, each [9]. An important aspect is the differential diagnosis between HBs occurring in elder children (>5 years old) and HCCs. In these cases, the presence of a stromal component, which is exclusively found on HBs, is a major feature to distinguish HCCs from HBs.
Between 1975 and 2010, childhood cancer mortality decreased by more than 50% [10], and this statistic is also valid for HB, because most cases are responsive to chemotherapy, with reductions in both tumor mass and AFP serum levels. Indeed, preoperative chemotherapy can completely eradicate metastatic pulmonary disease and multinodular liver disease. A relevant milestone in HB was the risk stratification of patients that allowed differential treatments.
Historically, four independent groups proposed trials for HB treatment: the International Childhood Liver Tumor Strategy Group (SIOPEL); the Children’s Oncology Group (COG), and its derived groups: the Children’s Cancer Group (CCG) and the Pediatric Oncology Group (POG); the German Society for Pediatric Oncology and Hematology (GPOH); and the Japanese Study Group for Pediatric Liver Tumors (JPLT) [11]. All groups contributed to HB management collecting data that have been used to associate clinical characteristics with different disease outcomes. Some characteristics that predict for worse outcomes, the ‘poor’ prognostic factors, have been identified across groups, such as metastatic disease at enrollment [12,13,14,15]; however, some risk factors achieved statistical significance in certain group studies, while remaining non-significant in others [12,13,14,15].
A review published in 2014 [16] summarizes the studies carried out by SIOPEL trials, which in 1990 established the preoperative chemotherapy for all cases, and introduced radiology-based staging called PRETEXT (PRETreatment EXTent of disease). This first trial was called SIOPEL 1 and collected data about 194 patients during four years, investigating the role of the cisplatin/doxorubicin combination chemotherapy [17]. The PRETEXT is based on pretreatment imaging with ultrasound and CT scans and/or magnetic resonance imaging, and describes the site and size of the tumor, invasion of the vessels and distant spread; the system identifies four PRETEXT stages (I–IV), which reflect the number of liver sections that are affected by the tumor [13, 18]. SIOPEL 1 data also showed that two risk groups could be distinguished: the standard risk patients, with resectable tumors and no metastases, and the high-risk patients, with either unresectable tumors and/or metastasis, or low AFP. Following, SIOPEL 2 (1994–1998) introduced a patient stratification for treatment based on the two risk categories; standard risk patients were treated with cisplatin alone to avoid the cardiotoxicity associated with doxorubicin, whereas high-risk patients were treated with the combination of cisplatin/doxorubin/carboplatin [19]. Other SIOPEL trials have occurred [20, 21], and current trial is SIOPEL 6, with standard risk patients [22].
The progress in the genomic research leads to the development of individualized strategies for the management of many types of cancer, based on specific biomarkers. However, in very rare tumors, like HB, the identification of biomarkers with impact on prognosis and treatment remains very challenging. Besides AFP levels, the main risk factors significantly associated with HB are clinical features. The Children’s Hepatic tumors International Collaboration (CHIC) was launched to address the challenge of the development of biologic markers, and the identification of reliable prognostic risk factors for tailoring treatment, in very rare tumors, like hepatoblastomas. A centralized online platform merged data from eight completed hepatoblastoma trials with a total of 1,605 cases treated between 1988 and 2008 [23]. Using the resulting database, increased risk was detected for adverse disease outcome for PRETEXT IV tumors, macrovascular venous or portal involvement, contiguous extrahepatic disease, primary tumor multifocality, and tumor rupture. Risk factors associated with the worst outcome were higher age (≥8 years), low AFP (<100 ng/ml), and metastatic disease. The novel prognostic factors that were identified, as well as the established factors, will be used to develop a future common global risk stratification system.
Environmental and genetic risk factors
Although the etiology of embryonal tumors has not been fully clarified, evidence supports the hypothesis of failure in the normal process of differentiation during fetal or postnatal development [3, 24]. Several studies have investigated the role of environmental factors during pregnancy that could be causally related to HB development [5], including pre-eclampsia [25], maternal overweight condition during pregnancy [26, 27], and treatment for infertility [26]. However, the only factor consistently associated with increased HB risk is low weight at birth [5] and prematurity [23] followed by tobacco consumption during pregnancy; the two latter may also be related to low weigh at birth [5].
In adult tumors, the current simplified hypothesis of cancer origin is accumulation of somatic mutations acquired over time [28]. In contrast, the development of pediatric tumors occurs over brief periods, suggesting the existence of either genetic predisposition or highly penetrant somatic mutations in undifferentiated cells [29]. Indeed, some genetic syndromes present an increased predisposition for HB development due to the presence of germline mutations. These syndromic patients correspond to approximately 15% of all HB cases [30]. Familial adenomatous polyposis (FAP), a cancer predisposition syndrome caused by germline mutations in the APC gene, is associated with HB in children 0-4 years of age [30, 31], with an estimated risk of HB development in FAP patients of 0.42% [32]. A study on 93 patients who developed HB published in 2005 identified eight (8.6%) cases in families with early-onset colon cancer or multiple colon polyps, features that suggest that FAP and APC germline mutations were present in these patients [33]. Interestingly, there was an apparent over representation of mutations in the 5’ region of APC in these cases. Beckwith-Wiedemann syndrome is an overgrowth syndrome associated with predisposition to the development of embryonal tumors, including Wilm’s tumor, neuroblastoma, adrenocortical carcinoma and HB [34, 35]. The relative risk of developing HB in this syndrome is 2–3 times higher than in the general population, and the underlying molecular defect is related to dysfunction of the imprinting mechanism at the 11p15.5 region, which contains the H19 and IGF2 genes [36]. The role of H19 and IGF2 in sporadic HBs (not related to genetic syndromes) has been investigated in some studies that seek to compare expression of these genes between HBs and normal liver [37, 38]. Two other syndromes of growth disorder that were already associated with HB are Sotos, caused by mutations or deletions of the NSD1 gene, located at 5q35 [39], and Simpson-Golabi-Behmel, caused by mutations in the GPC3 gene, located at Xq26 [40].
Interestingly, a few cases of HB have been reported in children with trisomy of chromosome 18 (Edwards Syndrome) [41,42,43,44,45]. Given the rarity of the two conditions, such findings suggest an etiologic association rather than a random occurrence.
Genomic alterations
Cytogenetic findings
Whole-chromosome aneuploidy is a common feature of HBs; the most common gains affect chromosomes 2, 8, and 20 [16], whereas the most frequent genomic loss is of chromosome 18 [16]. It is still not well established whether HB tumorigenesis is driven by changes in the copy number of genes mapped to these aneuploidies or whether such chromosomal changes already reflect mitotic errors inherent to HB development [46,47,48].
Structural chromosomal rearrangements have also been observed in HBs, such as the recurrent translocation der (4) t (1;4) (q12;q34) in three cases described by Schneider et al. in 1997 [49]. At least one focal rearrangement, 2q24 gain, is a relatively frequent copy number alteration in HB genomes, correlating with tumor aggressiveness. Our group [50] has recently evaluated the pattern of chromosomal gains and losses in samples of sporadic HBs using array-CGH, and we delineated a critical region of 10 Mb at 2q24, which harbors several genes. Among these genes, DAPL1 and ERMN exhibited overexpression in HBs as compared to control livers, suggesting their potential relevance to this type of tumor.
Genetic findings and next-generation sequencing studies
Pediatric tumors harbor an average number of non-synonymous mutations that is generally lower than the number detected in adult solid tumors, with approximately only 9.6 mutations per sample [28]. The proposed explanation for such a small number of mutations is based on two main assumptions. Firstly, pediatric tumors probably arise early from undifferentiated precursor cells, leading to tumor development in a short period [51]. Secondly, mutations in pediatric tumors would be more penetrant than those arising in adult tumors [52].
In 1999, Koch and co-workers [53] studied a group of 52 tumors, reporting a recurrent mutation in CTNNB1, currently the most frequent mutation reported for HBs. The CTNNB1 gene, located at 3p21, encodes the cytosolic protein β-catenin, plays an important role in cell adhesion and communication [53], and is also involved in Wnt signaling [54]. Wnt signaling is closely related to embryonic development, promoting cell growth and division (proliferation) and acting in cell differentiation. The CTNNB1 mutations reported in HB result in the absence of β-catenin degradation and migration to the nucleus, with activation of the canonical Wnt pathway in events not related to embryonic development [55].
The AXIN1 and AXIN2 genes, downstream components of the Wnt pathway that also participate in the degradation of β-catenin, were reported to be mutated in some cases of HB [56,57,58]. In addition, mutations were described in the PIK3CA gene, and in growth factors of this signaling pathway, such as IGF2, showing altered expression in this tumor type [59].
Next-generation sequencing (NGS) has greatly accelerated the identification of driver genes in cancer, including those related to HB tumorigenesis. Overall, the exome studies performed to date have revealed a very low frequency of somatic mutations in HBs (<5 mutations per tumor) even compared to other pediatric tumors [60].
Jia et al. published [51] exome sequencing data from six HB samples, identifying 24 non-synonymous somatic mutations affecting 21 different genes. Among these mutations, they described a novel alteration in CTNNB1 (G512V), two different mutations in a proposed novel HB gene, CAPRIN2 (R968H/S969), and nucleotide changes in Ubiquitin ligase complex-related genes (SPOP, KLHL22, TRPC4AP and RNF169). In this work, the protein level of β-catenin was found to be increased in the nucleus of HB cells overexpressing CAPRIN2, both wild-type and mutant forms, indicating activation of the Wnt pathway. The authors also employed a shRNA-mediated loss-of-function screening strategy to identify potential cancer-associated genes, performing proliferation assays with HepG2 and HUH-6 cell lines. Knock-down of SPOP, OR5I1 and CDC20B significantly promoted proliferation in both cell lines, suggesting that although CAPRIN2 may be an oncogene, SHOP, OR5I1 and CDC20B could represent novel tumor suppressors in HB.
Three other HB exome sequencing studies [60,61,62] have been published. The first study on HBs was conducted by Eichenmuller et al. [60], who investigated 15 samples of HB and three cases of an aggressive subtype of HB (HCC-like) that affects children over 5 years of age, and exhibits clinical and histopathological features that resemble hepatocellular carcinoma [63]. The authors described a relatively simple genome for HBs, with only ~2.9 mutations per tumor, particularly affecting CTNNB1 (12/15 cases) and NFE2L2 (2/15 cases) genes. NFE2L2 encodes a protein recognized by the complex KEAP1/CUL3, which is active in proteasomal degradation. The identified NFE2L2 mutations alter the structure of the protein, which is not recognized by the KEAP1/CUL3 complex and thus accumulates in the cytoplasm. Immunohistochemical analysis confirmed this effect, showing that NFE2L2 overexpression occurs in association with vascular invasion and metastasis. In addition to CTNNB1 and NFE2L2 mutations, the TERT promoter was found to be mutated in this tumor cohort, and deletions were detected in RAD17 and TP53.
Fujita et al. [61] reported a patient that had been with osteopathia striata, cranial sclerosis, and HB. A nonsense heterozygous mutation was identified in WTX (c.1045C > T p.Glu349), a tumor suppressor gene related to Wilm’s tumor. The somatic WTX mutation identified in the HB was also present in peripheral blood DNA, indicating that a germline mutation was responsible for the syndromic signs, and probably associated with the HB development in this case.
A third study by Kosaki et al. [62] involved HB exome sequencing for a patient diagnosed with Simpson-Golabi-Behmel syndrome. The authors detected a CTNNB1 somatic mutation (p.Ile35Ser), previously reported in HBs and other cancer types [44]. The patient was also carrier of a germline mutation in GPC3, a gene already known to cause Simpson-Golabi-Behmel syndrome. Changes in GPC3 expression have been associated with several forms of cancer. In particular, this gene is overexpressed in hepatocellular carcinoma [64], representing an important diagnostic biomarker.
Epigenetics
DNA methylation is a major epigenetic control mechanism of gene expression that involves the addition of a covalent methyl radical to the cytosine carbon 5 via the action of DNA methyltransferase enzymes (DNMTs) [65, 66]. The methylation patterns in differentiated cells are generally stable and inheritable during development. However, at least two events of overall methylation pattern reprogramming occur through removal of these epigenetic marks followed by new methylation [67] in both embryogenesis and gametogenesis. This reprogramming seems to have a crucial role in establishing differentiation potential of cells, as well as specific expression patterns in different tissues [68]. Aberrant DNA methylation patterns have been associated with several diseases, including cancer [69]. The anomalous methylation pattern often detected in tumor samples [70,71,72] are characterized by global hypomethylation, mainly affecting repetitive sequences, and specific promoter hypermethylation in early stages of tumorigenesis and throughout cancer progression [73], as an alternative mechanism for tumor suppressor gene inactivation. To date, HBs have remained largely unexplored regarding DNA methylation profile, both in coding and repetitive sequences. Methylation studies have identified hypermethylation in several gene promoters, including MT1G, that showed a significant correlation with poor prognosis [73]; RASSF1A, which plays a role as a tumor suppressor [74], and SOCS1, CASP8, SFRP1, APC, HHIP and IGFBP3 [73, 75, 76]. Additionally, a high frequency of H19 gene inactivation has been reported in HBs, including promoter hypermethylation [77].
A recent study [78] of HB evaluated the level of methylation of 33 differentially methylated regions subjected to imprinting. Compared to control livers, hypomethylation at specific loci was detected both in tumors and in the adjacent normal tissues, suggesting that hypomethylation may precede neoplastic growth. However, hypermethylation of INPP5, RB1 and GNASXL was observed as exclusive tumor events. This work also analyzed the level of methylation of the repetitive sequence LINE-1, which is usually hypomethylated in solid tumors. Except for a very low level hypomethylation in the first CpG of LINE-1, no significant differences were observed between HBs, adjacent normal tissues and normal livers. The authors suggested that the absence of LINE-1 hypomethylation could reflect a distinct mechanism for the development of embryonal tumors.
Comprehensive methylation studies of HB genomes are still required to clarify a possible link between HB tumorigenesis and changes in methylation.
Conclusions
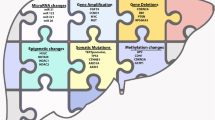
The box below summarizes the main genetic findings already disclosed for HB (Fig. 1). The paucity of genetic alterations can be explained using the most accepted model of the origin of embryonal tumors, which suggests that oncogenesis probably occurs in yet undifferentiated cells. Thus, fewer mutational events would be required in comparison to adult solid tumors, thereby exhibiting a type of “shortcut” for tumorigenesis.

Major genetic and epigenetic findings in hepatoblastomas
Some current studies are focusing on strategies that could highlight common underlying events for both organ development and tumorigenesis. Such research lines seek to strengthen the hypothesis that embryonal tumors have an intrauterine/perinatal origin. However, full characterization of the mutational burden of HBs is warranted to define how these genetic and epigenetics changes may disrupt development and influence tumorigenesis.
Abbreviations
- AFP:
-
α-fetoprotein
- CCG:
-
Children’s cancer group
- CHIC:
-
Children’s hepatic tumors international collaboration
- COG:
-
Children’s oncology group
- DNMTs:
-
DNA methyltransferase enzymes
- FAP:
-
Familial adenomatous polyposis
- GPOH:
-
German society for pediatric oncology and haematology
- HB:
-
Hepatoblastoma
- HCC:
-
Hepatocellular carcinoma
- JPLT:
-
Japanese study group for pediatric liver tumors
- NGS:
-
Next-generation sequencing
- POG:
-
Pediatric oncology group
- PRETEXT:
-
Pretreatment extent of disease
- SIOPEL:
-
Société internationale d’oncologie pédiatrique—epithelial liver tumor study group.
References
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Ward E, Desantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics. Cancer J Clin. 2014;64(2):83–103.
Maris JM, Denny CT. Focus on embryonal malignancies. Cancer Cell. 2002;2:447–50.
Stiller CA, Pritchard J, Steliarova-Foucher E. Liver cancer in European children: incidence and survival, 1978–1997. Report from the automated childhood cancer information system project. Eur J Cancer. 2006;42:2115–123.
Spector LG, Birch J. The epidemiology of hepatoblastoma. Pediatr Blood Cancer. 2012;59(5):776–9.
Exelby PR, Filler RM, Grosfeld JL. Liver tumors in children in the particular reference to hepatoblastoma and hepatocellular carcinoma: American academy of pediatrics surgical section survey. J Pediatr Surg. 1975;10:329–37.
Wu JT, knight JA. Alpha-fetoprotein: its use in clinical medicine ASCP check sample. Clin Chem. 1987;27:1.
Van Tornout JM, Buckley JD, Quinn JJ, et al. Timing and magnitude of decline in alpha-fetoprotein levels in treated children with unresectable or metastatic hepatoblastoma are predictors of outcome: a report from the Children’s cancer group. J Clin Oncol. 1997;15:1190–7.
Terrada D, Alaggio R, Dávila MT. Towards an international pediatric liver tumor consensus classification: proceedings of the Los Angeles COG liver tumors symposium. Mod Pathol. 2014;27:472–91.
Smith MA, Altekruse SF, Adamson PC, et al. Declining childhood and adolescent cancer mortality. Cancer. 2014;16:2497–506.
Perilongo G, Malogolowkin M, Feusner J. Hepatoblastoma clinical research: lessons learned and future challenges. Pediatr Blood Cancer. 2012;59:818e21.
Fuchs J, Rydzynski J, von Schweinitz D, Bode U, Hecker H, Weinel P, et al. Pretreatment prognostic factors and treatment results in children with hepatoblastoma. A report of the German cooperative paediatric liver tumor study HB-94. Cancer. 2002;95:172e82.
Aronson DC, Schnater JM, Staalman CR, Weverling GJ, Plaschkes J, Perilongo G, et al. Predictive value of the pretreatment extent of disease system in hepatoblastoma: results from the international society of pediatric oncology liver tumor study group SIOPEL-1 study. J Clin Oncol. 2005;23:1245e52.
Meyers RL, Rowland JR, Krailo M, Chen Z, Katzenstein HM, Malogolowkin MH. Pretreatment prognostic factors in hepatoblastoma: a report of the Children’s oncology group. Pediatr Blood Cancer. 2009;53:1016e22.
Maibach R, Roebuck D, Brugieres L, Capra M, Brock P, Dall’Igna P, et al. Prognostic stratification for children with hepatoblastoma: the SIOPEL experience. Eur J Cancer. 2012;48:1543e9.
Czauderna P, Lopez-Terrada D, Hiyama E, et al. Hepatoblastoma state of the art: pathology, genetics, risk stratification, and chemotherapy. Curr Opin Pediatr. 2014;26:19–28.
Guglielmi M, Perilongo G, Cecchetto G, Rondelli R, Lanino E, Siracusa F, et al. Rationale and results of the international society of pediatric oncology (SIOP) Italian pilot study on childhood hepatoma: surgical resection d’emblee or after primary chemotherapy? J Surg Oncol Suppl. 1993;3:122–6.
Roebuck DJ, Aronson D, Clapuyt P, Czauderna P, de Ville de Goyet J, Gauthier F, et al. International childhood liver tumor strategy group 2005 PRETEXT: a revised staging system for primary malignant liver tumors of childhood developed by the SIOPEL group. Pediatr Radiol. 2007;37:123–32.
Perilongo G, Shafford E, Maibach R, et al. Risk adapted treatment for childhood hepatoblastoma: final report of the second study of the internal society of pediatric oncology, SIOPEL 2. Eur J Cancer. 2004;40:411–21.
Zsiros J, Maibach R, Shafford E, et al. Successful treatment of childhood high-risk hepatoblastoma with dose-intensive multiagent chemotherapy and surgery: final results of the SIOPEL-3HR study. J Clin Oncol. 2010;28:2584–90.
Zsiros J, Brugieres L, Brock P, Roebuck D, Maibach R, Zimmermann A, Childs M, Pariente D, Laithier V, Otte JB, Branchereau S, Aronson D, Rangaswami A, Ronghe M, Casanova M, Sullivan M, Morland B, Czauderna P, Perilongo G. Dose-dense cisplatin-based chemotherapy and surgery for children with high-risk hepatoblastoma (SIOPEL-4): a prospective, single-arm, feasibility study. International childhood liver Tumours strategy group (SIOPEL). Lancet Oncol. 2013;14(9):834–42.
Perilongo G, Maibach R, Shafford E, et al. Cisplatin versus cisplatin plus doxorubicin for standard risk hepatoblastoma. N Engl J Med. 2009;361:1662–70.
Meyers RL, Maibach R, Hiyama E, Häberle B, Krailo M, Rangaswami A, Aronson DC, Malogolowkin MH, Perilongo G, von Schweinitz D, Ansari M, Lopez-Terrada D, Tanaka Y, Alaggio R, Leuschner I, Hishiki T, Schmid I, Watanabe K, Yoshimura K, Feng Y, Rinaldi E, Saraceno D, Derosa M, Czauderna P. Risk-stratified staging in paediatric hepatoblastoma: a unified analysis from the Children’s hepatic tumors international collaboration. Lancet Oncol. 2016;S1470–2045(16):30598–8.
Maschietto M, de Camargo B, Brentani H, Grundy P, Sredni ST, Torres C, Mota LD, Cunha IW, Patrão DF, Costa CM, Soares FA, Brentani RR, Carraro DM. Molecular profiling of isolated histological components of wilms tumor implicates a common role for the Wnt signaling pathway in kidney and tumor development. Oncology. 2008;75(1-2):81–91.
Ansell P, Mitchell CD, Roman E. Relationships between perinatal and maternal characteristics and hepatoblastoma: a report from the UKCCS. Eur J Cancer. 2005;41:741–48.
Mclaughlin CC, Baptiste MS, Schymura MJ. Maternal and infant birth characteristics and hepatoblastoma. Am J Epidemiol. 2006;163:818–28.
Pu CL, Guo CB, Jin XQ. Retrospective analysis of maternal and infant birth features of hepatoblastoma patients. Zhonghua Gan Zang Bing Za Zhi. 2009;17:459–61.
Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science. 2013;339:1546–58.
Davenport KP, Blanco FC, Sandler AD. Pediatric malignancies: neuroblastoma, Wilm’s tumor, hepatoblastoma, rhabdomyosarcoma, and sacroccygeal teratoma. Surg Clin North Am. 2012;92:745–67.
Kingston JE, Herbert A, Draper GJ, et al. Association between hepatoblastoma and polyposis coli. Arch Dis Child. 1983;58:959–62.
Garber JE, Li FP, Kingston JE, et al. Hepatoblastoma and familial adenomatous polyposis. J Natl Cancer Inst. 1988;80:1626–628.
Hughes LJ, Michels VV. Risk of hepatoblastoma in familial adenomatous polyposis. Am J Med Genet. 1992;43:1023–25.
Hirschman BA, Pollock BH, Tomlinson GE. The spectrum of APC mutations in children with Hepatoblastoma from familial adenomatous polyposis kindreds. J Pediatr. 2005;147:263–66.
Beckwith J. Macroglossia, Omphalocele, adrenal cytomegaly, gigantism and hyperplastic visceromegaly. Birth Defects. 1969;5:188–96.
Wiedemann H. Complexe malformatif familial avec hernie ombilicale et macroglossie— “un syndrome nouveau?”. J Genet Hum. 1964;13:223–32.
Bliek J, Gicquel C, Maas S. Epigenotyping as a tool for the prediction of tumor risk and tumor type in patients with beckwith-Wiedemann syndrome (BWS). J Pediatr. 2004;145:796–99.
Fukuzawa R, Umezawa A, Ochi K, et al. High frequency of inactivation of the imprinted H19 gene in “sporadic” hepatoblastoma. Int J Cancer. 1999;82:490–97.
Ross JA, Rakloff GA, Davies SM. H19 and IFG-2 allele-specific expression in hepatoblastoma. Br J Cancer. 2000;82:753–56.
Kato M, Takita J, Takahashi K, et al. Hepatoblastoma in a patient with Sotos syndrome. J Pediatr. 2009;155:937–39.
Toretsky JA, Zitomersky NL, Eskenazi AE, et al. Glypican-3 expression in wilms tumor and hepatoblastoma. J Pedatr Hematol Oncol. 2001;23:496–99.
Dasouki M, Barr M. Trisomy 18 and hepatic neoplasia. Am J Med Genet. 1987;7:203–05.
Mamlok V, Nichols M, Lockhart L, et al. Trisomy 18 and hepatoblastoma. Am J Med Genet. 1989;33:125–26.
Teraguchi M, Nogi S, Ikemoto Y, et al. Multiple hepatoblastomas associated with trisomy 18 in a 3-year-old girl. Pediatr Hematol Oncol. 1997;14:463–67.
Bove K, Soukup S, Ballard E, et al. Hepatoblastoma in a child with trisomy 18: Cytogenetics, liver anomalies and literature review. Pediatr Pathol Lab Med. 1996;16:253–62.
Maruyama K, Ikeda H, Koizumi T. Hepatoblastoma associated with trisomy 18 syndrome: a case report and a review of the literature. Pediatr Int. 2001;43:302–5.
Soukup SW, Lampkin BL. Trisomy 2 and 20 in two hepatoblastomas. Genes Chromosomes Cancer. 1991;3:231–34.
Tonk VS, Wilson KS, Timmons CF, et al. Trisomy 2, trisomy 20, and del (17p) as sole chromosomal abnormalities in three cases of hepatoblastoma. Genes Chromosomes Cancer. 1994;11:199–202.
Tomlinson GE, Douglass EC, Pollock BH, et al. Cytogenetic analysis of a large series of hepatoblastoma: numerical aberrations with recurring translocations involving 1q12-21. Genes Chromosomes Cancer. 2005;44:177–84.
Schneider NR, Cooley LD, Finegold MJ, et al. The first recurring chromosome translocation in hepatoblastoma: der (4) t (1;4) (q12;q34). Genes Chromosomes Cancer. 1997;19:291–94.
Rodrigues TC, Fidalgo F, da Costa CML, Ferreira EN, da Cunha IW, Carraro DM, Krepischi ACV, Rosenberg C. Upregulated genes at 2q24 gain as candidate oncogenes in hepatoblastomas. Future Oncol. 2014;10(15):2449–57.
Jia D, Dong R, Jing Y, Xu D, Wang Q, Chen L, Li Q, Huang Y, Zhang Y, Zhang Z, Liu L, Zheng S, Xia Q, Wang H, Dong K, He X. Exome sequencing of hepatoblastoma reveals novel mutations and cancer genes in the Wnt pathway and ubiquitin ligase complex. Hepatology. 2014;60(5):1686–96.
Tomasetti C, Vogelstein B, Parmigiani G. Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci U S A. 2013;110(6):1999–2004.
Koch A, Denkhaus D, Albrecht S, et al. Childhood hepatoblastomas frequently carry a mutated degradation targeting box of the beta-catenin gene. Cancer Res. 1999;59:26973.
Udatsu Y, Kusafuka T, Kuroda S, et al. High frequency of beta-catenin mutations in hepatoblastoma. Pediatr Surg Int. 2001;17:508–12.
Park WS, Oh RR, Park JY, et al. Nuclear localization of beta-catenin is an important prognostic factor in hepatoblastoma. J Pathol. 2001;193:83–490.
Koch A, Weber N, Waha A, et al. Mutations and elevated transcriptional activity of conductin (AXIN2) in hepatoblastomas. J Pathol. 2004;204:546–54.
Miao J, Kusafuka T, Udatsu Y, et al. A. Sequence variants of the Axin gene in hepatoblastoma. Hepatol. Res. 2003;25:174–9.
Taniguchi K, Roberts LR, Aderca IN, et al. Mutational spectrum of beta-catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene. 2002;21:4863–71.
Hartmann W, Kuchler J, Koch A, et al. Activation of phosphatidylinositol-3’kinase/AKT signaling is essential in hepatoblastoma survival. Clin Cancer Res. 2009;15:4538–45.
Eichenmuller M, Trippel F, Kreuder M, Beck A, Schwarzmayr T, Haberle B, et al. The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J Hepatol. 2014;61:1312–20.
Fujita, A., Ochi, N.; Fujimaki, H. et al. A novel WTX mutation in a female patient with osteopathia striata with cranial sclerosis and hepatoblastoma. Am J Med Genet A 2014.
Kosaki R, Takenouchi T, Takeda N, Kagami M, Nakabayashi K, Hata K, Kosaki K. Somatic CTNNB1 mutation in hepatoblastoma from a patient with Simpson-Golabi-Behmel syndrome and germline GPC3 mutation. Am J Med Genet A. 2014;164A(4):993–7. doi:10.1002/ajmg.a.36364.
Prokurat A, Kluge P, Kosciesza A, et al. Transitional liver cell tumors (TLCT) in older children and adolescents: a novel group of aggressive hepatic tumors expressing betacatenin. MedPediatr Oncol. 2002;39:510–8.
Midorikawa Y, Ishikawa S, Iwanari H, et al. Glypican-3, overexpressed in hepatocellular carcinoma, modulates FGF2 and BMP-7 signaling. Int J Cancer. 2003;103:455–65.
Branco MR, Ficz G, REIK W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat Rev Genet. 2012;13:7–13.
Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92.
Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–20.
Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–93.
Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610.
Ponder BA. Cancer genetics. Nature. 2001;411:336–41.
Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol. 2010;11:607–20.
Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7:21–33.
Sakamoto LH, De Camargo B, Cajaiba M, Soares FA, Vettore AL. MT1G hypermethylation: a potential prognostic marker for hepatoblastoma. Pediatr Res. 2010;67:387–93.
Honda S, Miyagi H, Suzuki H, Minato M, Haruta M, Kaneko Y, Hatanaka KC, Hiyama E, Kamijo T, Okada T, Taketomi A. RASSF1A methylation indicates a poor prognosis in hepatoblastoma patients. Pediatr Surg Int. 2013;29:1147–52.
Regel I, Eichenmuller M, Joppien S, Liebl J, Haberle B, Muller-Hocker J, Vollmar A, Von SD, Kappler R. IGFBP3 impedes aggressive growth of pediatric liver cancer and is epigenetically silenced in vascular invasive and metastatic tumors. Mol Cancer. 2012;11:9.
Tomlinson GE, Kappler R. Genetics and epigenetics of hepatoblastoma. Pediatr Blood Cancer. 2012;59:785–92.
Honda S, Arai Y, Haruta M, Sasaki F, Ohira M, Yamaoka H, Horie H, Nakagawara A, Hiyama E, Todo S, Kaneko Y. Loss of imprinting of IGF2 correlates with hypermethylation of the H19 differentially methylated region in hepatoblastoma. Br J Cancer. 2008;99:1891–99.
Rumbajan JM, Maeda T, Souzaki R, Mitsui K, Higashimoto K, Nakabayashi K, Yatsuki H, Nishioka K, Harada R, Aoki S, Kohashi K, Oda Y, Hata K, Saji T, Taguchi T, Tajiri T, Soejima H, Joh K. Comprehensive analyses of imprinted differentially methylated regions reveal epigenetic and genetic characteristics in hepatoblastoma. BMC Cancer. 2013;13:608.
Acknowledgements
Not applicable.
Funding
Not applicable.
Availability of data and materials
Not applicable.
Authors’ contributions
TFMA is a PhD student and contributed to the research and organization of all information used in the text also contributed formatting and article design; TN is master’s degree student and contributed to the research and organization of all information used in the text; CMLC is a pediatrist and contributed to the review and improvement of clinical and epidemiological data presented in the text; CR is a biologist and contributed to the improvement of information on next Generation sequencing, as well as revision of the text; IWC is a pathologist and contributed to the improvement of information related to pathology; ACVK is biologist and contributed to the research and organization of all information used in the text, formatting, design and revising it, and is the corresponding author. All authors read and approved the final manuscript.
Authors’ information
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Aguiar, T.F.M., Carneiro, T.N., da Costa, C.M.L. et al. The genetic and epigenetic landscapes of hepatoblastomas. Appl Cancer Res 37, 20 (2017). https://doi.org/10.1186/s41241-017-0021-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41241-017-0021-0