
Abstract
Background
Sampling of saliva for diagnosing Plasmodium falciparum infections is a safe, non-invasive alternative to sampling of blood. However, the use of saliva presents a challenge because lower concentrations of parasite DNA are present in saliva compared to peripheral blood. Therefore, a sensitive method is needed for detection of parasite DNA in saliva. This study utilized two recently reported “ultra-sensitive” PCR assays based on detection of the P. falciparum mitochondrial cox3 gene and the multi-copy nuclear varATS gene. The ultra-sensitive assays have an advantage over standard 18S rRNA gene-based PCR assay as they target genes with higher copy numbers per parasite genome. Stored saliva DNA samples from 60 Cameroonian individuals with infections previously confirmed by 18S rRNA gene PCR in peripheral blood were tested with assays targeting the cox3 and varATS genes.
Results
Overall, the standard 18S rRNA gene-based PCR assay detected P. falciparum DNA in 62% of the stored saliva DNA samples, whereas 77 and 68% of the samples were positive with assays that target the cox3 and varATS genes, respectively. Interestingly, the ultra-sensitive assays detected more P. falciparum infections in stored saliva samples than were originally detected by thick-film microscopy (41/60 = 68%). When stratified by number of parasites in the blood, the cox3 assay successfully detected more than 90% of infections using saliva when individuals had > 1000 parasites/μl of peripheral blood, but sensitivity was reduced at submicroscopic parasitemia levels. Bands on electrophoresis gels were distinct for the cox3 assay, whereas faint or non-specific bands were sometimes observed for varATS and 18S rRNA that made interpretation of results difficult. Assays could be completed in 3.5 and 3 h for the cox3 and varATS assays, respectively, whereas the 18S rRNA gene assays required at least 7 h.
Conclusions
This study demonstrates that a PCR assay targeting the cox3 gene detected P. falciparum DNA in more saliva samples than primers for the 18S rRNA gene. Non-invasive collection of saliva in combination with the proposed cox3 primer-based PCR assay could potentially enhance routine testing of P. falciparum during disease surveillance, monitoring, and evaluation of interventions for malaria elimination.
Similar content being viewed by others


Background
Early and accurate diagnosis of Plasmodium falciparum (Pf) infections is crucial for treatment and monitoring of malaria transmission. Thick-film microscopy (TFM) has traditionally been the gold standard for the identification of Pf from peripheral blood. A trained microscopist can typically detect 50–100 parasites/μl of blood in Giemsa’s solution-stained blood smear [1], but individuals with fewer parasites may be erroneously diagnosed as parasite-free. Although people with submicroscopic infections are usually not ill, they are capable of transmitting sexual-stage parasites to mosquitoes [2]. Submicroscopic infections can be detected by highly sensitive molecular assays, such as nested PCR (nPCR) and loop-mediated isothermal amplification (LAMP) [3]. With the use of nPCR and LAMP, the limit of detection is estimated to be 0.1–10 parasites/μl of blood and can be lower still with qRT-PCR [4]. Current diagnostic and molecular methods have an inherent problem; they require collection of peripheral blood. Routine blood collection methods are invasive procedures that are painful to patients, require trained phlebotomists, and involve the use of needles or lancets. Improper handling of sharps exposes healthcare workers and the patient to blood-borne pathogens, while disposal of used sharps is hazardous for waste management in malaria-endemic countries [5]. Repetitive sampling of blood from the same person, e.g., in mass studies monitoring malaria elimination, often results in low compliance.
Non-blood samples, e.g., saliva and urine, can serve as non-invasive and safe alternatives to blood samples. Plasmodium DNA has been detected in saliva and urine [6], however, at 600 and 2500 times lower levels, respectively [7], than those present in peripheral blood. Despite the reduced amount of parasite DNA in saliva, quantitative studies show a positive correlation between parasite numbers detected in peripheral blood by microscopy and parasite DNA in saliva [7, 8], which supports the use of saliva as an alternative for blood.
The traditional molecular assay used for Pf detection targets a specific region in the 18S ribosomal RNA (18S rRNA) gene [9] that has 4–8 copies per Pf genome. Recently, primers to multi-copy gene targets have been reported to have improved sensitivity. One is the mitochondrial cytochrome c oxidase III (cox3) gene, with 20 to 150 copies per Pf genome. Because cox3 is a mitochondrial gene, it is less likely to undergo immune pressure and genetic variation. The other gene is the var gene acidic terminal sequence (varATS), ~ 59 copies/Pf genome [4, 10, 11]. Cunha et al. reported detection of submicroscopic infections in the blood by qPCR using varATS primers that were missed by 18S rRNA gene primers. Since conventional instrumentation of qPCR can be difficult to maintain in resource poor countries where malaria is transmitted, a conventional PCR method might be more useful.
A recent study was conducted in Cameroon using 18S rRNA gene PCR to detect Pf DNA in the saliva of 222 fever patients. The study was able to detect Pf DNA in saliva in 95% [95% CI 85–99] of 53 subjects who were peripheral blood smear positive by microscopy and 82% [95% CI 72–90] of 78 blood samples positive by PCR [8]. The current study hypothesized that the use of these “ultra-sensitive” primers to detect malaria DNA in saliva would increase the number of Pf cases detected compared to the use of the traditional 18S rRNA gene PCR.
Methods
Sample selection
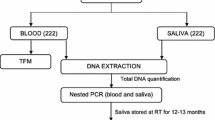
Paired blood and saliva samples were collected in 2015 from febrile patients in Cameroon as previously described [8]. Saliva were collected using the OMNIgene®ORAL (OM-501) kits (Genotek, Ottawa, Canada). In the original study, DNA was isolated from whole blood and saliva using a DNA purification kit (Macherey Nagel, Duren, Germany) and stored at − 20 °C until used. In the current study, 60 stored archival saliva DNA samples were available from subjects who originally tested peripheral blood positive by PCR using 18S rRNA gene PCR. As a positive control, DNA isolated from laboratory-cultured Pf of the 3D7 strain was serially diluted five-folds from 3.74 to 4 × 10−7 ng/μl of parasite DNA. Negative controls were saliva samples collected from three U.S. individuals who were malaria naïve, and a non-template control with nuclease-free PCR-grade water was substituted for the DNA template.
Nested PCR amplification of 18S rRNA
In the original study, the following protocol was used. One PCR reaction consisted of 12.5 μl of 2x Go Taq Green (Promega, Madison, USA), 1 μl each of 10 μM forward and reverse primers (IDT, IA, USA), 5.5 μl of nuclease-free water, and 5 μl of DNA template, with a total of 25 μl. The standard protocols for the cycling conditions were followed [9]. For the initial amplification targeting the 18S rRNA gene, forward and reverse primers, rPLU5 (5′-CCTGTTGTTGCCTTAAACTTC-3′) and rPLU6 (5′-TTAAAATTGTTGCAGTTAAAACG-3′) were used. Five microliters of the amplified product was used for the second round of amplification with forward and reverse primers, rFAL-1 (5′-TTAAACTGGTTTGGGAAAACCAAATATATT-3′) and rFAL-2 (5′-ACACAATGAACTCAATCATGACTACCCGTC) [9]. The expected product size was 205 base pairs (bp). In the current study, the above protocol was employed, except 5 μl of DNA was used (instead of 2 μl) to allow for direct comparison among the primers.
Nested PCR amplification of mitochondrial cytochrome C oxidase III (cox3)
For the initial amplification targeting cox3, forward and reverse primers, MtU.F (5′-CTCGCCATTTGATAGCGGTTAACC-3′) and MtU.R (5′-CCTGTTATCCCCGGCGAACCTTC-3′) were used with standard protocols for the cycling conditions [10]. Five microliters of the amplified product, diluted at 1:50, was used for the second amplification with forward and reverse primers MtNst_falF (5′-GAACACAATTGTCTATTCGTACAATTATTC-3′) and MtNst_falR (5′-CTTCTACCGAATGGTTTATAAATTCTTTC-3′). The expected product size was 201 bp.
PCR amplification of varATS
For the amplification targeting varATS, forward varATS primer (5′-CCCATACACAACCAAYTGGA-3′) and reverse varATS primer (5′-TTCGCACATATCTCTATGTCTATCT-3′) were used with standard protocols for the cycling conditions [4]. The expected product size was 65 bp.
Gel electrophoresis
Ten microliters of the PCR products were run on a 1.5% agarose gel (Sigma, Fisher, USA) stained with ethidium bromide (Sigma-Aldrich, USA) for 45 min at 100 V. Products were visualized under a UV lamp. A 100-bp ladder (exACTGene Ladder, Fisher) was used as a reference. The presence of a band at the expected size for each primer set was determined positive.
Statistical data analysis
Data analyses were conducted using Microsoft Excel and Prism version 7.04 (Graph Pad Software Inc.). The 95% confidence interval was calculated using the modified Wald method.
Results
Comparison of the limit of detection of the three primers
DNA isolated from P. falciparum cultured in vitro that contained from 3.74 to 4 × 10−7 ng/μl of parasite DNA was serially diluted fivefold and used as the positive control in the assays. The lower limit of detection (i.e., presence of a band following gel electrophoresis) using the 18S rRNA primers reached 1 × 10−5 ng/μl; cox3 primer was 4 × 10−7 ng/μl; and varATS primer was 2 × 10−6 ng/μl. It is important to point out that both the 18S rRNA and cox3 assays are nested PCRs, where 5 μl of the nest 1 produce is used in nest 2 of the 18S rRNA assay, but 5 μl of a 1:50 dilution of the nest 1 was used in nest 2 of the cox3 assay. The varATS assay was a single-step PCR assay. Thus, the cox3 assay was the most sensitive assay, with the varATS having a lower limit of detection similar to that of the nest-PCR 18S rRNA method.
Detection of Pf DNA in saliva using 18S rRNA primers, cox3, and varATS
Overall, the PCR assay using standard 18S rRNA primers detected Pf DNA in 62% [95% CI 49–73] of the stored saliva DNA samples, whereas 77% [95% CI 64–86] and 68% [95% CI 56–79] of the samples were positive with assays using cox3 and varATS primers, respectively (Table 1). Although the differences were not significant based on 95% CI, the cox3 primers detected more submicroscopic infections compared to the 18S rRNA and varATS (47% vs. 21 and 26%, respectively) and had the highest efficiency for malaria detection (Table 2). Interestingly, the ultra-sensitive primers detected Pf in more stored saliva DNA samples than were originally detected by TFM (41/60 = 68%) and thus were more efficient (Table 2). When stratified by number of parasites in the blood, the cox3 assay detected over 90% of infections using saliva DNA when individuals had > 1000 parasites/μl in their peripheral blood (Table 1). For individuals with > 10,000 parasites/μl in peripheral blood, both cox3 and varATS assays successfully detected 100% of the infections, but the sensitivity was reduced at submicroscopic parasitemia levels. US control samples were negative for all primer sets (Additional file 1: Table S1).
Advantages and disadvantages of each assay
Cox3 PCR consistently showed the brightest and sharpest bands on the gel, whereas the 18S rRNA PCR often produced smeared and non-specific bands with a different size from the expected size (Fig. 1). Interpretation of the gel was also difficult for varATS PCR, as bands were constantly faint and resulted in over 10% of “unclear” samples (Additional file 1: Table S1). The ultra-sensitive primers-based PCR had another advantage over traditional PCR with much shorter turnaround times than 18S rRNA, ~ 3.5 and ~ 3 h for cox3 and varATS, respectively, compared to ~ 7 h for 18S rRNA (Table 3).

Representative photographs of gel electrophoresis for 18S rRNA, cox3, and varATS primers using six saliva DNA samples (sample nos. 10–15). Sample nos. 10 and 11 were positive with 18S rRNA, since distinct bands of the correct size were visible; sample nos. 10 through 13 were positive with cox3; and sample nos. 10 and 11 were positive with varATS primers, but sample nos. 12–15 were difficult to score. The gel pictures show brighter and more distinct bands for cox3 than 18s rRNA and varATS
Discussion
Non-invasive and sensitive tests for malaria have many applications. This study compared the use of three PCR-based assays to detect Pf DNA in saliva. Saliva samples have multiple advantages over blood samples, e.g., no pain for patients, minimal training required for healthcare workers, reduced transmission of blood-borne pathogens, and contaminated sharp wastes. The WHO reported that 37.6% of hepatitis B, 39% of hepatitis C, and 4.4% of HIV/AIDS prevalence in health workers worldwide are due to needle-stick injury [12]. Saliva tests have been shown to be convenient and safe for cancer, HIV, HCV, HPV, and recently in the field of malaria by detecting HRP-2, lactate dehydrogenase, and Pf DNA [6, 13,14,15,16,17,18].
In the current study, two sets of primers were evaluated, namely primers to the P. falciparum genes for cox3 and varATS, which were first described in 2015. Following its initial report, the cox3 PCR has been used in epidemiological studies [19, 20]. Since the assay is a nested PCR, cox3 PCR has the benefit of identifying the presence or absence of Plasmodium species in the blood after a single round of PCR amplification. The mitochondrial genome is highly conserved in Plasmodium species [21]. Plasmodium detection using another mitochondrial gene, cytochrome b gene (cytb), in saliva and urine has been reported in symptomatic patients [6]. However, a more recent study demonstrated that cytb-based PCR has lower sensitivity than cox3-based assays [10].
In a resource- and personnel-limited setting, clear and easy interpretation of results is extremely important and the presence of a single distinct band in gel electrophoresis-based assays help assure accurate diagnosis. The traditional 18S rRNA gene-based PCR frequently amplified non-specific bands (Fig. 1), a phenomenon also been reported by others [10, 22]. The extra bands could possibly be due to cross reactivity between human and parasite small subunit rRNA [10]. In contrast, cox3 primers target a gene specific to Plasmodium, thereby eliminating the risk of non-specific amplification of human DNA. VarATS PCR uses a single amplification and produces a small-sized product, making it difficult for researchers to be confident in distinguishing between the true product and non-specific bands on the gel. On the other hand, varATS primers have the advantage of being used in a single-step PCR, thereby significantly reducing the turnaround time and workload required for nPCR.
The process of how parasite DNA enters the human salivary glands and circulates in the saliva remains unclear. Further investigation is also required to determine whether Pf DNA in the saliva is present only during an active Pf infection or if it persists after parasites are cleared from the blood. An inherent problem with rapid diagnostic test based on detection of parasite histidine-rich protein-two (HRP-2) is the persistence of the antigen in the peripheral blood after parasite clearance [23,24,25].
The project has several limitations. The DNA samples were collected in 2015 and DNA was isolated and stored at − 20 °C until re-evaluated in 2017. The saliva samples were collected in 2015 and DNA degradation was likely to have occurred, as the detection of positive saliva samples was higher in the original study than that when archival saliva DNA was used in 2017 with the 18S rRNA primers. DNA degradation in stored samples is not unexpected, as power outages frequently occur in Cameroon. Whether DNA degradation took place is not clear, but it is likely that ultra-sensitive primers would perform even better using fresh saliva samples.
In future studies, a larger number of fresh saliva and blood should be collected and tested with 18S rRNA, cox3, and varATS primers to establish the true sensitivity of each PCR-based approach. In addition, future studies are needed to determine if other species of malaria (e.g., P. vivax, P. malariae, P. ovale) can also be diagnosed using saliva and if the amount of Plasmodial DNA in saliva reflects the level of parasitemia in the peripheral blood.
Conclusions
The present study found the PCR assay based on detection of the mitochondrial cox3 gene to be better than the traditional 18s rRNA assay for detecting Pf DNA in stored DNA samples extracted. In addition, PCR using cox3 primers provided bright and distinct band following gel electrophoresis and the assay could be completed in ~ 3.5 h. The proposed combination of saliva and PCR using cox3 primers may provide a new tool for use in diagnosis, epidemiological studies, and monitoring interventions for malaria control.
Abbreviations
- 18S rRNA:
-
18S small subunit ribosomal RNA
- AIDS:
-
Acquired Immune Deficiency Syndrome
- bp:
-
Base pair
- cox3 :
-
Mitochondrial cytochrome c oxidase III
- Cytb:
-
Cytochrome b gene
- HIV:
-
Human immunodeficiency virus
- HPV:
-
Human papillomavirus
- HRP-2:
-
Histidine-rich protein 2
- LAMP:
-
Loop-mediated isothermal amplification
- nPCR:
-
Nested polymerase chain reaction
- Pf :
-
Plasmodium falciparum or P. falciparum
- qPCR:
-
Qualitative PCR
- qRT-PCR:
-
Quantitative reverse-transcription PCR
- TFM:
-
Thick-film microscopy
- varATS :
-
var gene acidic terminal sequence
- WHO:
-
World Health Organization
References
Wongsrichanalai C, Barcus MJ, Muth S, Sutamihardja A, Wernsdorfer WH. A review of malaria diagnostic tools: microscopy and rapid diagnostic test (RDT). Am J Trop Med Hyg. 2007;77(6 Suppl):119–27.
Karl S, Gurarie D, Zimmerman PA, King CH, St Pierre TG, Davis TM. A sub-microscopic gametocyte reservoir can sustain malaria transmission. PLoS One. 2011;6(6):e20805.
Britton S, Cheng Q, McCarthy JS. Novel molecular diagnostic tools for malaria elimination: a review of options from the point of view of high-throughput and applicability in resource limited settings. Malar J. 2016;15:88.
Hofmann N, Mwingira F, Shekalaghe S, Robinson LJ, Mueller I, Felger I. Ultra-sensitive detection of Plasmodium falciparum by amplification of multi-copy subtelomeric targets. PLoS Med. 2015;12(3):e1001788.
Haylamicheal ID, Desalegne SA. A review of legal framework applicable for the management of healthcare waste and current management practices in Ethiopia. Waste Manag Res. 2012;30(6):607–18.
Putaporntip C, Buppan P, Jongwutiwes S. Improved performance with saliva and urine as alternative DNA sources for malaria diagnosis by mitochondrial DNA-based PCR assays. Clin Microbiol Infect. 2011;17(10):1484–91.
Nwakanma DC, Gomez-Escobar N, Walther M, Crozier S, Dubovsky F, Malkin E, Locke E, Conway DJ. Quantitative detection of Plasmodium falciparum DNA in saliva, blood, and urine. J Infect Dis. 2009;199(11):1567–74.
Mfuh KO, Tassi Yunga S, Esemu LF, Bekindaka ON, Yonga J, Djontu JC, Mbakop CD, Taylor DW, Nerurkar VR, Leke RGF. Detection of Plasmodium falciparum DNA in saliva samples stored at room temperature: potential for a non-invasive saliva-based diagnostic test for malaria. Malar J. 2017;16(1):434.
Snounou G, Viriyakosol S, Jarra W, Thaithong S, Brown KN. Identification of the four human malaria parasite species in field samples by the polymerase chain reaction and detection of a high prevalence of mixed infections. Mol Biochem Parasitol. 1993;58(2):283–92.
Isozumi R, Fukui M, Kaneko A, Chan CW, Kawamoto F, Kimura M. Improved detection of malaria cases in island settings of Vanuatu and Kenya by PCR that targets the Plasmodium mitochondrial cytochrome c oxidase III (cox3) gene. Parasitol Int. 2015;64(3):304–8.
Cunha MG, Medina TS, Oliveira SG, Marinho AN, Povoa MM, Ribeiro-dos-Santos AK. Development of a polymerase chain reaction (PCR) method based on amplification of mitochondrial DNA to detect Plasmodium falciparum and Plasmodium vivax. Acta Trop. 2009;111(1):35–8.
Organization WH: World health report 2002. 2002.
Zachary D, Mwenge L, Muyoyeta M, Shanaube K, Schaap A, Bond V, Kosloff B, de Haas P, Ayles H. Field comparison of OraQuick ADVANCE rapid HIV-1/2 antibody test and two blood-based rapid HIV antibody tests in Zambia. BMC Infect Dis. 2012;12:183.
SahebJamee M, Boorghani M, Ghaffari SR, AtarbashiMoghadam F, Keyhani A. Human papillomavirus in saliva of patients with oral squamous cell carcinoma. Med Oral Patol Oral Cir Bucal. 2009;14(10):e525–8.
Wilson NO, Adjei AA, Anderson W, Baidoo S, Stiles JK. Detection of Plasmodium falciparum histidine-rich protein II in saliva of malaria patients. Am J Trop Med Hyg. 2008;78(5):733–5.
Gbotosho GO, Happi CT, Folarin O, Keyamo O, Sowunmi A, Oduola AM. Rapid detection of lactate dehydrogenase and genotyping of Plasmodium falciparum in saliva of children with acute uncomplicated malaria. Am J Trop Med Hyg. 2010;83(3):496–501.
Buppan P, Putaporntip C, Pattanawong U, Seethamchai S, Jongwutiwes S. Comparative detection of Plasmodium vivax and Plasmodium falciparum DNA in saliva and urine samples from symptomatic malaria patients in a low endemic area. Malar J. 2010;9:72.
Lee SR, Kardos KW, Schiff E, Berne CA, Mounzer K, Banks AT, Tatum HA, Friel TJ, Demicco MP, Lee WM, et al. Evaluation of a new, rapid test for detecting HCV infection, suitable for use with blood or oral fluid. J Virol Methods. 2011;172(1–2):27–31.
Chan CW, Iata H, Yaviong J, Kalkoa M, Yamar S, Taleo G, Isozumi R, Fukui M, Aoyama F, Pomer A, et al. Surveillance for malaria outbreak on malaria-eliminating islands in Tafea Province, Vanuatu after tropical cyclone Pam in 2015. Epidemiol Infect. 2017;145(1):41–5.
Idris ZM, Chan CW, Kongere J, Gitaka J, Logedi J, Omar A, Obonyo C, Machini BK, Isozumi R, Teramoto I, et al. High and heterogeneous prevalence of asymptomatic and sub-microscopic malaria infections on islands in Lake Victoria, Kenya. Sci Rep. 2016;6:36958.
Hikosaka K, Watanabe Y, Kobayashi F, Waki S, Kita K, Tanabe K. Highly conserved gene arrangement of the mitochondrial genomes of 23 Plasmodium species. Parasitol Int. 2011;60(2):175–80.
Kimura M, Kaneko O, Liu Q, Zhou M, Kawamoto D, Wataya Y, Otani S, Yamaguchi Y, Tanabe K. Identification of the four species of human malaria parasites by nested PCR that targets variant sequences in the small subunit rRNA gene. Parasitol Int. 1997;46(2):91–5.
Grandesso F, Nabasumba C, Nyehangane D, Page AL, Bastard M, De Smet M, Boum Y, Etard JF. Performance and time to become negative after treatment of three malaria rapid diagnostic tests in low and high malaria transmission settings. Malar J. 2016;15(1):496.
Kilauzi AL, Mulumba JG, Magafu MG, Matchaba-Hove R, Tapera R, Magafu NS, Tamfum JJ. SD Bioline malaria antigen Pf (HRP-2/pLHD) for assessing efficacy of artemisinin combination therapy against Plasmodium falciparum in pediatric patients in the Democratic Republic of the Congo. Pan Afr Med J. 2015;22:304.
Kattenberg JH, Tahita CM, Versteeg IA, Tinto H, Traore-Coulibaly M, Schallig HD, Mens PF. Antigen persistence of rapid diagnostic tests in pregnant women in Nanoro, Burkina Faso, and the implications for the diagnosis of malaria in pregnancy. Tropical Med Int Health. 2012;17(5):550–7.
Acknowledgements
We thank the researchers and the laboratory staff at the Biotechnology Centre, University of Yaoundé I, Cameroon, for collecting, processing, and archiving the samples. Our sincere appreciation is sent to all the participants in the study, as this study could not have been carried out without their participation. The OMNIgene®ORAL (OM-501) kits were kindly donated by DNA Genotek, Ottawa, Canada.
Funding
This research was supported by grants T37MD008636 (Minority Health and Health Disparities International Research Training Program, NIMHD), D43TW009074 (Global Infectious Diseases, FIC), P30GM114737 (Centers of Biomedical Research Excellence, NIGMS, NIH), and 5R21AI 105286-02 (NIAID, NIH).
Availability of data and materials
Comparative data for the three PCR assays are provided in Additional file 1: Table S1.
Author information
Authors and Affiliations
Contributions
LEF and YML conceived the project and designed the experiments. RC provided ideas on the cox3 primers. STY and KM designed and carried out the original project in 2015 and collected samples with BO and LEF. JA, BT, BO, CN, LEF, and YML carried out the experiments. JA, YML, and DWT analyzed the data. RFGL supervised the collection of the samples. YML, JA, RC, VRN, and DWT wrote the manuscript. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Blood and saliva samples used were deidentified, archival samples collected in 2015. The current study was exempt from human subject research by the Committee on Human Studies, University of Hawaii at Manoa (Protocol Number: 2017-00395).
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. Raw data are provided in the Table. The results have been sorted by parasitemia that was determined by thick-film microscopy. Shaded blocks indicate where a difference between the three PCR assays evaluated was seen. (DOCX 26 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lloyd, Y.M., Esemu, L.F., Antallan, J. et al. PCR-based detection of Plasmodium falciparum in saliva using mitochondrial cox3 and varATS primers. Trop Med Health 46, 22 (2018). https://doi.org/10.1186/s41182-018-0100-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41182-018-0100-2