
Abstract
Background
Multidrug resistance-associated protein 1 (MRP1), an energy-dependent efflux pump, is expressed widely in various tissues and contributes to many physiological and pathophysiological processes. 6-Bromo-7-[11C]methylpurine ([11C]7m6BP) is expected to be useful for the assessment of MRP1 activity in the human brain and lungs. However, the radiochemical yield (RCY) in the synthesis of [11C]7m6BP was low, limiting its clinical application, because the methylation of the precursor with [11C]CH3I provided primarily the undesired isomer, 6-bromo-9-[11C]methylpurine ([11C]9m6BP). To increase the RCY of [11C]7m6BP, we investigated conditions for improving the [11C]7m6BP/[11C]9m6BP selectivity of the methylation reaction.
Results
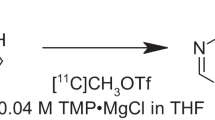
[11C]7m6BP was manually synthesized via the methylation of 6-bromopurine with [11C]CH3I in various solvents and at different temperatures in the presence of potassium carbonate for 5 min. Several less polar solvents, including tetrahydrofuran (THF), 2-methyltetrahydrofuran (2-MeTHF), and ethyl acetate (AcOEt) improved the [11C]7m6BP/[11C]9m6BP selectivity from 1:1 to 2:1, compared with the conventionally used solvents for the alkylation of 6-halopurines, acetone, acetonitrile, and N,N-dimethylformamide. However, a higher temperature (140 °C or 180 °C) was needed to progress the 11C-methylation in the less polar solvents, and the manual conditions could not be directly translated to an automated synthesis. [11C]Methyl triflate ([11C]CH3OTf) was thus used as a methylating agent to increase the conversion at a lower temperature. The 11C-methylation using [11C]CH3OTf at 100 °C proceeded efficiently in THF, 2-MeTHF, and AcOEt with maintenance of the improved selectivity. Starting from 28 to 34 GBq [11C]CO2, [11C]7m6BP was produced with 2.3–2.6 GBq for THF, 2.7–3.3 GBq for AcOEt, and 2.8–3.9 GBq for 2-MeTHF at approximately 30 min after the end of bombardment (n = 3 per solvent). The isolated RCYs (decay corrected) for THF, 2-MeTHF, and AcOEt were 24–28%, 29–35%, and 22–31% (n = 3), respectively.
Conclusions
The use of THF, 2-MeTHF, and AcOEt improved the [11C]7m6BP/[11C]9m6BP selectivity in the methylation reaction, and the improved method provided [11C]7m6BP with sufficient radioactivity for clinical use.
Similar content being viewed by others
Background
Multidrug resistance-associated protein 1 (MRP1) is a member of the adenosine triphosphate (ATP)-binding cassette superfamily of transporters and pumps various compounds, including therapeutic agents and physiological substances, out of cells using the energy of ATP hydrolysis (Bakos and Homolya 2007; Cole et al. 1992). This protein is widely expressed in normal tissues, including the brain, lung, heart, and kidney (Bakos and Homolya 2007; Flens et al. 1996). In addition to conferring multidrug resistance (Leslie et al. 2005; Löscher and Potschka 2005), changes in MRP1 activity or expression are associated with brain and lung diseases (Krohn et al. 2011, 2015; Qosa et al. 2015; van der Deen et al. 2006). Studies have also reported that MRP1 protects the heart against chronic doxorubicin-induced cardiotoxicity and protects intestinal epithelial cells against inflammation-induced apoptotic cell death (Blokzijl et al. 2008; Zhang et al. 2015). Thus, the noninvasive measurement of MRP1 activity in organs of interest would help to elucidate the pathogenesis and diagnoses of diseases.
6-Bromo-7-[11C]methylpurine ([11C]7m6BP) has been used to measure MRP1 activity in the brain and lungs of rodents (Krohn et al. 2019; Mairinger et al. 2020; Okamura et al. 2009b, 2013, 2020; Zoufal et al. 2019, 2020), and [11C]7m6BP is thus expected to be useful for assessment of the MRP1 activity in the tissues of human. Although a recent report has suggested a limited sensitivity of [11C]7m6BP for measuring MRP1 activity in the brain of a mouse species (Okamura et al. 2020), if the efflux process via MRP1 is the rate-limiting step in the human brain, it may be possible to assess the MRP1 activity in the brain. However, the low yield of the current synthetic method remains a problem, limiting the clinical use of [11C]7m6BP. In the conventional method using alkyl halides and bases, the alkylation of 6-chloropurine or 6-bromopurine results in a mixture of N7- and N9-alkylated purines, and the N9-isomers are predominantly produced (Galante et al. 2014; Hanna et al. 1994; Montgomery and Temple 1961; Tuncbilek et al. 2009; Zhang et al. 2012). Consistent with this production of isomers, the 11C-methylation of 6-bromopurine has resulted in a low radiochemical yield of [11C]7m6BP (Okamura et al. 2009b; Zoufal et al. 2019). In the present study, we investigated conditions for improving the [11C]7m6BP/[11C]9m6BP selectivity in this methylation reaction.
Methods
Chemicals
6-Bromopurine was purchased from Sigma-Aldrich/Merck (St. Louis, MO, USA). 6-Bromo-9-methylpurine and 6-bromo-7-methylpurine were synthesized as described previously (Okamura et al. 2009a, b). Chloroform (CHCl3, dehydrated) and super dehydrated solvents—acetone (ACT), acetonitrile (MeCN), ethyl acetate (AcOEt), N,N-dimethylformamide (DMF), tetrahydrofuran (THF), 1,4-dioxane (1,4-DO), diisopropyl ether (iPr2O), toluene (Tol), and dichloromethane (DCM)—were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). Methyl acetate (AcOMe, anhydrous) and 1,3-dioxane (1,3-DO) were purchased from Sigma-Aldrich/Merck. Methyl propionate (MP, special grade) was purchased from FUJIFILM Wako Pure Chemical Corporation. 2-Methyltetrahydrofuran (2-MeTHF, dehydrated) was purchased from KANTO CHEMICAL Co., INC (Tokyo, Japan). Aqueous 57% hydrogen iodide (HI) solution was purchased from FUJIFILM Wako Pure Chemical Corporation. Potassium Carbonate (K2CO3, guaranteed reagent) was purchased from FUJIFILM Wako Pure Chemical Corporation and was ground in a mortar into a fine powder, which was used for manual and automated syntheses. A 0.05–0.08 M solution of lithium aluminum hydride (LAH) in THF was prepared by diluting a 1.0 M solution of LAH in THF, which was purchased from Sigma-Aldrich/Merck.
Production of [11C]carbon dioxide ([11C]CO2)
[11C]CO2 was produced by the 14N(p,α)11C nuclear reaction in an atmosphere of nitrogen gas containing 0.01% oxygen with 18 MeV protons using the CYPRIS HM-18 cyclotron (Sumitomo Heavy Industry, Tokyo, Japan).
Production of [11C]CH3I
[11C]CH3I was prepared from [11C]CO2 as described previously, except for the volumes of the solutions of LAH in THF and 57% HI (Kikuchi et al. 2013). The volumes of the LAH in THF and 57% HI solutions used were both 100 µL. [11C]CH3I was transferred using a N2 gas stream with a flow rate of 50 mL/min and collected in MeCN (ca. 2 mL) in a glass vial.
General conditions for 11C-methylation (manual synthesis)
The following solvents were investigated in this reaction: ACT; MeCN; DMF; AcOMe; AcOEt; MP; THF; 1,3-DO; 2-MeTHF; 1,4-DO; iPr2O; CHCl3; Tol; and DCM. The [11C]CH3I solution (< 80 MBq, 10 μL) was added to a mixture of 6-bromopurine (5.1 mg, 25.6 µmol), K2CO3 (3.6 mg, 26.0 µmol), and each solvent (400 μL) in a 1.6-mL glass vial. The vial was sealed with a screw cap, and the temperature was then held at 100, 140, or 180 °C for 5 min. At the end of the reaction, the reaction mixture was cooled on ice, 200 μL of methanol was added, and the resulting mixture was then analyzed by both high-performance liquid chromatography (HPLC) and thin-layer chromatography (TLC).
Radiochemical yields from [11C]CH3I
All radiochemical yields (RCYs) were determined by radio-HPLC analysis and radio-TLC of the crude product. HPLC was used for the separation of [11C]CH3I and the methylated products including the N7-isomer ([11C]7m6BP) and the N9-isomer (6-bromo-9-[11C]methylpurine; [11C]9m6BP). Because [11C]7m6BP and [11C]9m6BP could not be separated by the HPLC conditions used here, TLC was used for the separation of these compounds. RCYs based on [11C]CH3I were calculated as follows: RCYHPLC multiplied by RCYTLC, where RCYHPLC and RCYTLC show the proportion of the 11C-methylated products determined by HPLC and that of [11C]7m6BP determined by TLC, respectively.
HPLC analysis
The HPLC system consisted of a JASCO PU-2089 plus pump (JASCO Corporation, Tokyo, Japan), a multiwavelength detector (MD-2015 plus, JASCO Corporation), and a sensitive positron detector (Ohyo Koken Kogyo, Co., Ltd. Tokyo, Japan) for radioactivity detection. Data acquisition and interpretation were performed with ChromNAV (version 1.18.03, JASCO Corporation). The RCYs were calculated after the correction of the radiochromatograms for decay. A COSMOSIL 5C18-MS-II column (150 × 4.6 mm; Nacalai Tesque, Kyoto, Japan) and a 5C18-MS-II guard column (10 × 4.6 mm) were used for the RCY analyses with a mobile phase consisting of a mixture of water and MeCN. The peaks corresponding to the 11C-labeled compounds in the radiochromatogram were determined based on the UV absorptions of the corresponding nonradioactive compounds. The retention times of the 11C-methylated products ([11C]7m6BP and the N9-isomer) and [11C]CH3I (mobile phase: water/MeCN, 40/60; flow rate: 1 mL/min) were approximately 2 and 4 min, respectively.
TLC analysis
TLC was conducted on glass-backed silica gel TLC plates (silica gel 60 F254; Merck Ltd., Tokyo, Japan), and unlabeled 7m6BP was pre-spotted on the TLC plates. The reaction solvents were used, except for DMF, and each reaction mixture (2 µL) was co-spotted with unlabeled 7m6BP. The TLC plates were developed with AcOEt/EtOH (9/1, v/v). When DMF was used as the reaction solvent, the reaction mixture was further diluted with methanol (four-fold dilution, in total six-fold dilution). One µL of the reaction mixture was co-spotted with unlabeled 7m6BP, and the TLC plates were developed with CHCl3/AcOEt (9/1, v/v). After drying, the plates were developed with AcOEt/EtOH (9/1, v/v) again. The air-dried TLC plates were covered with a 5 μm-thick film and placed in a cassette in contact with a phosphor imaging plate. Radioactivity on the TLC plate was quantified with an imaging plate reader (BAS-5000, FUJIFILM Corporation, Tokyo, Japan). The fraction of radioactivity on the TLC plates and a typical image of a TLC plate were shown in Additional file 1: Table S1 and Fig. S2, respectively.
Automated synthesis of 6-bromo-7-[11C]methylpurine using [11C]methyl triflate
Production of [11C]methyl triflate
[11C]CH3I was produced as described above. [11C]Methyl triflate ([11C]CH3OTf) was prepared from [11C]CH3I as described previously (Jewett 1992). In brief, [11C]CH3OTf was produced by passing [11C]CH3I through a glass column containing silver triflate-impregnated graphitized carbon (200–300 mg) at 180 °C with a N2 flow of 50 mL/min.
11C-methylation using a synthetic apparatus
Automated radiochemical synthesis was performed using a system built in-house (Additional file 1: Fig. S1). 6-Bromopurine (2 mg, 10.0 µmol), K2CO3 (6.4 mg, 26.0 µmol), and solvent (400 μL) were added into a 1.6-mL glass vial. The mixture was vortexted and sonicated at room temperature (20–23 °C) for approximately 20 s, to form a suspension. The suspension was drawn up into a 1-mL syringe with a 20G needle and then injected into a reaction vessel for the automated synthesis. When DMSO was used, the reaction conditions including the reaction temperature, the reaction time, and the solvent volume were according to a previous report (Zoufal et al. 2019).
[11C]CH3OTf was trapped in the precursor mixture at room temperature, and the reaction vessel was heated at 100 °C for 5 min. The reaction vessel was cooled to room temperature, 1 mL of AcOEt was added, and the radioactive mixture was transferred into a COSMOSIL 5SL-II column (10 ID × 250 mm; Nacalai Tesque). The column was eluted with AcOEt/EtOH (93:7, v/v) at a flow rate of 5.0 mL/min. The radioactive fraction corresponding to the desired product (retention time: approximately 10–12 min) was collected in a rotary evaporator flask containing 50 μL of 25% ascorbic acid injection, evaporated in vacuo, and dissolved in 5 mL of saline. The total synthesis time, after 10 min of proton bombardment with a beam current of 20 µA, was 29–32 min. The identification and radiochemical purity (RCP) of [11C]7m6BP obtained after the formulation were determined by HPLC using an authentic sample of 7m6BP. HPLC was performed on a COSMOSIL 5C18-AR-II column (4.6 I.D. × 250 mm; Nacalai Tesque) and a 5C18-AR-II guard column (10 × 4.6 mm) with a mobile phase of H2O/methanol (75:25, v/v) at a flow rate of 0.9 mL/min. The molar activity (Am) was determined by comparing the assayed radioactivity to the mass associated with the 7m6BP UV peak at 288 nm.
Results
Effect of the solvent on the RCY of [11C]7m6BP (manual synthesis)
When ACT, MeCN, or DMF was used for the 11C-methylation of 6-bromopurine, the reaction proceeded efficiently at 100 °C, and unreacted [11C]CH3I was not observed. However, the product ratio of [11C]7m6BP/[11C]9m6BP was low (Table 1, entries 1–3), and the RCYs of [11C]7m6BP were 24–26%. When iPr2O, CHCl3, Tol, or DCM was used, a considerable amount of unreacted [11C]CH3I was observed, and the reaction scarcely proceeded even at high temperature (180 °C) (Table 1, entries 25–34). These results are likely to be because of the poor solubility of 6-bromopurine in these solvents. While 1,4-DO and 2-MeTHF improved the [11C]7m6BP/[11C]9m6BP selectivity to 2:1, the methylation did not proceed efficiently (Table 1, entries 19–24). The RCYs of [11C]7m6BP were thus low even at high temperature (180 °C), and were lower than those using ACT, MeCN, or DMF. The use of AcOMe, AcOEt, MP, or THF gave a [11C]7m6BP/[11C]9m6BP selectivity of > 1:1 (Table 1, entries 4–15). Although 32–76% of unreacted [11C]CH3I was observed at 100 °C, the reaction proceeded smoothly at higher temperatures (140 °C and 180 °C), and the amount of unreacted [11C]CH3I was reduced to 0.97–7.5%. Furthermore, 1,3-DO increased the selectivity compared with ACT, MeCN, and DMF; however, the ratio of [11C]7m6BP/[11C]9m6BP was still < 1:1 (Table 1, entries 16–18).
Effect of the solvent on the isolated RCY of [11C]7m6BP (automated synthesis)
From the results of the manual synthesis, the conditions in which the reaction was performed in THF at 140 °C (Table 1, entry 14) appeared to be appropriate for the production of [11C]7m6BP. However, such reaction conditions that generated a high pressure were not suitable for [11C]7m6BP production using the apparatus for the automated synthesis. In the automated synthesis, [11C]7m6BP was therefore synthesized using [11C]CH3OTf and THF at a lower temperature (100 °C), and the tracer was successfully obtained under these conditions (Table 2 and Fig. 1). The reactivity of 6-bromopurine with [11C]CH3I at 100 °C in AcOEt was much lower than in THF, and 58% of [11C]CH3I was unreacted (Table 1, entry 7). However, the reaction with [11C]CH3OTf in the automated synthesis proceeded efficiently, and the selectivity remained almost unchanged (Table 2 and Fig. 2). The isolated RCY of [11C]7m6BP in AcOEt was comparable to that in THF (Table 2). The use of 2-MeTHF and [11C]CH3OTf improved the low reactivity with a similar [11C]7m6BP/[11C]9m6BP selectivity (Table 2 and Fig. 2). By contrast, the reaction using 1,4-DO and [11C]CH3OTf was unsuccessful, and a peak of an unknown 11C-labeled by-product of comparable intensity to [11C]7m6BP and [11C]9m6BP was observed (Fig. 2), which was probably a decomposed product derived from unreacted [11C]CH3OTf. When ACT or DMSO was used as the solvent, the isolated RCYs of [11C]7m6BP were low (Table 2). Taken together, these results indicated that the use of THF, 2-MeTHF, or AcOEt increased the isolated RCY of [11C]7m6BP. Starting from 28 to 34 GBq [11C]CO2, [11C]7m6BP was produced with 2.3–2.6 GBq using THF, 2.7–3.3 GBq using AcOEt, and 2.8–3.9 GBq using 2-MeTHF at approximately 30 min after the end of bombardment (EOB). The isolated RCYs (decay corrected) for THF, 2-MeTHF, and AcOEt were 24–28%, 29–35%, and 22–31% (n = 3), respectively. The radiochemical purity of [11C]7m6BP was higher than 95% up to 60 min after being formulated (Table 2), indicating the radiochemical stability of the compound for the duration of at least one positron emission tomography scan.

Representative analytical chromatograms of isolated [11C]7m6BP. UV-chromatogram of the authentic sample of 7m6BP (A). UV-chromatogram (B) and radiochromatogram (C) of isolated [11C]7m6BP immediately after synthesis. The UV absorbance was measured at 288 nm. The radiochromatogram was corrected for baseline noise and decay. The synthesis of [11C]7m6BP was performed using [11C]CH3OTf in THF at 100 °C

Typical chromatograms of semi-preparative HPLC for the reaction mixture. The peaks of N7 and N9 show [11C]7m6BP and [11C]9m6BP, respectively. ACT, DMSO, THF, 2-MeTHF, 1,4-DO, and AcOEt were used as reaction solvents
Discussion
In the present study, we investigated conditions for improving the [11C]7m6BP/[11C]9m6BP of selectivity in the methylation reaction in the synthesis of [11C]7m6BP. The methylation of the precursor with [11C]CH3I in ACT, MeCN, or DMF provided primarily [11C]9m6BP (Table 1, entries 1–3), and the reaction in iPr2O, CHCl3, Tol, or DCM scarcely proceeded (entries 25–34). Although 1,4-DO and 2-MeTHF improved the [11C]7m6BP/[11C]9m6BP selectivity to 2:1, the methylation did not proceed efficiently (entries 19–24). The use of less polar solvents—AcOMe, AcOEt, MP, THF, and 1,3-DO—appeared promising for increasing the RCY of [11C]7m6BP at 140 °C or 180 °C. From the results of the manual synthesis, we selected the conditions using THF at 140 °C (entry 14), which might increase the isolated RCY of [11C]7m6BP, compared with previous methods for the synthesis of [11C]7m6BP (Okamura et al. 2009b; Zoufal et al. 2019). In a preliminary examination, however, a considerable loss of radioactivity in the reaction vial during the automated synthesis was observed. This loss resulted from the passing of [11C]CH3I through a solenoid valve before the precursor was methylated, probably because of the high pressure caused by heating THF (boiling point: 66 °C) at 140 °C. The temperature parameter from the manual synthesis could thus not be translated directly to the automated synthesis. While a lower temperature was required for the automated synthesis, a higher temperature was needed for the efficient methylation of 6-bromopurine (Table 1). The methylation of 6-bromopurine with [11C]CH3I in THF at 100 °C proceeded moderately (Table 1, entry 13), but the methylation at 80 °C scarcely proceeded (data not shown). The [11C]7m6BP/[11C]9m6BP selectivity was almost constant from 100 to 180 °C (Table 1, entries 13–15). In the automated synthesis, the temperature was thus changed from 140 to 100 °C, and [11C]CH3OTf replaced [11C]CH3I as the methylating agent to compensate for the reactivity loss. [11C]7m6BP was successfully obtained under these conditions, and the selectivity of [11C]7m6BP/[11C]9m6BP in THF in the automated synthesis (Table 2) was consistent with that in the manual synthesis (Table 1, entry 13).
In addition to THF, the 11C-methylation at 100 °C proceeded efficiently in 2-MeTHF and AcOEt, while maintaining the improved selectivity (Table 2 and Fig. 2). By contrast, when ACT was used as the solvent, the isolated RCYs of [11C]7m6BP were 6.6–11%, which were a little higher than those in a previous study using ACT and [11C]CH3I (4–9%) (Okamura et al. 2009b). This increase may be because there were differences in the reactivity between [11C]CH3OTf and [11C]CH3I but the selectivity was not changed. The [11C]7m6BP/[11C]9m6BP selectivity was similar in the automated synthesis to that in the manual synthesis (Tables 1 and 2). When DMSO was used as the solvent, the radiochemical purity of [11C]7m6BP (two out of three runs) did not achieve 95%, because of contamination with the N9-isomer. Aside from the radiochemical purity, the isolated RCY of [11C]7m6BP in DMSO was still low, which was consistent with a previous study (Zoufal et al. 2019).
Chen et al. have reported that treating 6-bromopurine with an alkylmagnesium reagent, which was then reacted with CH3I at 25 °C for 20 h, gave the N7-isomer (Chen et al. 2016). If 6-bromopurine can be reacted with [11C]CH3OTf or [11C]CH3I at a lower temperature (room temperature) and for a longer time, [11C]7m6BP might be formed more selectively. However, such a long reaction time is impractical for the radiosynthesis of [11C]7m6BP. Although our conditions did not provide only [11C]7m6BP, the reaction can potentially produce sufficient [11C]7m6BP for clinical use (Table 2).
The present study showed that the solvents, THF, 2-MeTFH, and AcOEt, increased the [11C]7m6BP/[11C]9m6BP selectivity in the methylation of 6-bromopurine, compared with the traditionally used solvents, ACT, MeCN, DMF, and DMSO. The mechanism of the reaction remains unknown; however, the solvent and temperature might affect the relative populations of the tautomers (6-bromo-7H-purine and 6-bromo-9H-purine). To the best of our knowledge, no reports are available on the determination of tautomerism in 6-bromopurine in any solvent. For 6-chloropurine, the population of the 7H tautomer has been reported to be 8–22% at 303 K (30 °C) in DMSO (Seckarova et al. 2004), which probably led to the low yield of 6-chloro-7-alkylpurine (Montgomery and Temple 1961). The population of the 7H tautomer of 6-bromopurine is thus assumed to be comparable to, or higher than, that of the 9H tautomer in the less polar solvents, such as THF, 2-MeTFH, and AcOEt, whereas the 9H tautomer may be the dominant tautomeric form in more polar solvents.
Conclusions
The use of 2-MeTHF, THF, and AcOEt improved the [11C]7m6BP/[11C]9m6BP selectivity in the methylation reaction. The reaction conditions for providing [11C]7m6BP with sufficient radioactivity for clinical use are the use of 2-MeTHF (THF or AcOEt), [11C]CH3OTf as a methylating agent, a reaction temperature of 100 °C, and a reaction time of 5 min.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- 1,3-DO:
-
1,3-Dioxane
- 1,4-DO:
-
1,4-Dioxane
- 2-MeTHF:
-
2-Methyltetrahydrofuran
- [11C]7m6BP:
-
6-Bromo-7-[11C]methylpurine
- [11C]9m6BP:
-
6-Bromo-9-[11C]methylpurine
- ACT:
-
Acetone
- AcOEt:
-
Ethyl acetate
- AcOMe:
-
Methyl acetate
- DCM:
-
Dichloromethane
- DMF:
-
N,N-Dimethylformamide
- EOB:
-
End of bombardment
- EOS:
-
End of synthesis
- HI:
-
Hydrogen iodide
- HPLC:
-
High-performance liquid chromatography
- iPr2O:
-
Diisopropyl ether
- MeCN:
-
Acetonitrile
- MP:
-
Methyl propionate
- MRP1:
-
Multidrug resistance-associated protein 1
- LAH:
-
Lithium aluminum hydride
- RCP:
-
Radiochemical purity
- RCY:
-
Radiochemical yield
- THF:
-
Tetrahydrofuran
- TLC:
-
Thin-layer chromatography
- Tol:
-
Toluene
- UV:
-
Ultraviolet
References
Bakos É, Homolya L. Portrait of multifaceted transporter, the multidrug resistance-associated protein 1 (MRP1/ABCC1). Pflügers Arch. 2007;453:621–41.
Blokzijl H, van Steenpaal A, Vander Borght S, et al. Up-regulation and cytoprotective role of epithelial multidrug resistance-associated protein 1 in inflammatory bowel disease. J Biol Chem. 2008;283:35630–7.
Chen S, Graceffa RF, Boezio AA. Direct, regioselective N-alkylation of 1,3-azoles. Org Lett. 2016;18:16–9.
Cole SP, Bhardwaj G, Gerlach JH, et al. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science. 1992;258:1650–4.
Flens MJ, Zaman GJ, van der Valk P, et al. Tissue distribution of the multidrug resistance protein. Am J Pathol. 1996;148:1237–47.
Galante E, Okamura T, Sander K, et al. Development of purine-derived 18F-labeled pro-drug tracers for imaging of MRP1 activity with PET. J Med Chem. 2014;57:1023–32.
Hanna NB, Bhattacharya BK, Robins RK, et al. Sulfinosine congeners: synthesis and antitumor-activity in mice of certain N9-alkylpurines and purine ribonucleosides. J Med Chem. 1994;37:177–83.
Jewett DM. A simple synthesis of [11C]methyl triflate. Int J Rad Appl Instrum A. 1992;43:1383–5.
Kikuchi T, Minegishi K, Hashimoto H, et al. The use of tetrabutylammonium fluoride to promote N- and O-11C-methylation reactions with iodo[11C]methane in dimethyl sulfoxide. J Labelled Comp Radiopharm. 2013;56:672–8.
Krohn M, Lange C, Hofrichter J, et al. Cerebral amyloid-beta proteostasis is regulated by the membrane transport protein ABCC1 in mice. J Clin Invest. 2011;121:3924–31.
Krohn M, Bracke A, Avchalumov Y, et al. Accumulation of murine amyloid-beta mimics early Alzheimer’s disease. Brain. 2015;138:2370–82.
Krohn M, Zoufal V, Mairinger S, et al. Generation and characterization of an Abcc1 humanized mouse model (hABCC1(flx/flx) ) with knockout capability. Mol Pharmacol. 2019;96:138–47.
Leslie EM, Deeley RG, Cole SP. Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol. 2005;204:216–37.
Löscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci. 2005;6:591–602.
Mairinger S, Sake JA, Hernandez Lozano I, et al. Assessing the activity of multidrug resistance-associated protein 1 at the lung epithelial barrier. J Nucl Med. 2020;61:1650–7.
Montgomery JA, Temple C Jr. Synthesis of potential anticancer agents. XXVI. The alkylation of 6-chloropurine. J Am Chem Soc. 1961;83:630–5.
Okamura T, Kikuchi T, Fukushi K, et al. Reactivity of 6-halopurine analogs with glutathione as a radiotracer for assessing function of multidrug resistance-associated protein 1. J Med Chem. 2009a;52:7284–8.
Okamura T, Kikuchi T, Okada M, et al. Noninvasive and quantitative assessment of the function of multidrug resistance-associated protein 1 in the living brain. J Cereb Blood Flow Metab. 2009b;29:504–11.
Okamura T, Kikuchi T, Okada M, et al. Imaging of activity of multidrug resistance-associated protein 1 in the lungs. Am J Respir Cell Mol Biol. 2013;49:335–40.
Okamura T, Okada M, Kikuchi T, et al. Mechanisms of glutathione-conjugate efflux from the brain into blood: involvement of multiple transporters in the course. J Cereb Blood Flow Metab. 2020;40:116–25.
Qosa H, Miller DS, Pasinelli P, et al. Regulation of ABC efflux transporters at blood-brain barrier in health and neurological disorders. Brain Res. 2015;1628:298–316.
Seckarova P, Marek R, Malinakova K, et al. Direct determination of tautomerism in purine derivatives by low-temperature NMR spectroscopy. Tetrahedron Lett. 2004;45:6259–63.
Tuncbilek M, Ates-Alagoz Z, Altanlar N, et al. Synthesis and antimicrobial evaluation of some new substituted purine derivatives. Bioorg Med Chem. 2009;17:1693–700.
van der Deen M, Marks H, Willemse BW, et al. Diminished expression of multidrug resistance-associated protein 1 (MRP1) in bronchial epithelium of COPD patients. Virchows Arch. 2006;449:682–8.
Zhang Q, Cheng G, Huang YZ, et al. Regioselective N9 alkylation of purine rings assisted by beta-cyclodextrin. Tetrahedron. 2012;68:7822–6.
Zhang W, Deng J, Sunkara M, et al. Loss of multidrug resistance-associated protein 1 potentiates chronic doxorubicin-induced cardiac dysfunction in mice. J Pharmacol Exp Ther. 2015;355:280–7.
Zoufal V, Mairinger S, Krohn M, et al. Influence of multidrug resistance-associated proteins on the excretion of the ABCC1 Imaging probe 6-bromo-7-[11C]methylpurine in mice. Mol Imaging Biol. 2019;21:306–16.
Zoufal V, Mairinger S, Krohn M, et al. Measurement of cerebral ABCC1 transport activity in wild-type and APP/PS1-21 mice with positron emission tomography. J Cereb Blood Flow Metab. 2020;40:954–65.
Acknowledgements
We are grateful to the technical team of the Cyclotron Section of the Institute for Quantum Medical Science for their support during cyclotron operation and for the production of radioisotopes. Victoria Muir, PhD, from Edanz (https://jp.edanz.com/ac) edited a draft of this manuscript.
Funding
This work was partially supported by JSPS KAKENHI Grant Number JP22K07785 to TO and AMED Moonshot Research and Development Program (Grant No. 21zf0127003h001 to MRZ).
Author information
Authors and Affiliations
Contributions
TO designed the study, performed the synthesis, analyzed the data, and wrote the manuscript. MO performed the automated synthesis of [11C]7m6BP using [11C]CH3OTf. TK, MO, and MRZ analyzed data and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Table S1: Fraction of [11C]7m6BP determined by TLC; Figure S1: Schematic diagram of the automated system used for [11C]7m6BP synthesis; Figure S2: Typical image of a developed TLC plate spotted with a reaction mixture.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Okamura, T., Kikuchi, T., Ogawa, M. et al. Improved synthesis of 6-bromo-7-[11C]methylpurine for clinical use. EJNMMI radiopharm. chem. 9, 10 (2024). https://doi.org/10.1186/s41181-024-00240-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41181-024-00240-8