
Abstract
Chronic kidney diseases (CKD) are an economic burden and occur worldwide in all age groups, and the advancement of kidney disease at some point leads to deregulate or influence the function of other body organs and to find a specific target to halt the disease progression which is a tedious challenge. Regardless of the underlying mechanisms, it is essential to consider and evaluate the involvement and association of individual endogenous mediators and environmental factors in the progression of CKD to accumulate the required knowledge. More than a dozen pathways leading to relentless progression of CKD have been identified so far, but the association of serotonin 5-HT2A receptor with progressive renal injury is still under process.
Scientific reports demonstrated that the 5-HT2A receptor plays a significant role in renal metabolism, glomerular function, and renal vascular tone. So a better understanding of the evolving role of serotonin 5-HT2A-mediated pathophysiological mechanisms of CKD may be a helpful tool to identify new therapeutic targets. In this review, we will discuss recent interventions, pharmacological target, and the possible implication of serotonin 5-HT2A receptors with associated mechanistic trails leading to CKD.
Similar content being viewed by others


Introduction
Serotonin or 5-hydroxytryptamine (5-HT) is a well-reported endogenous autacoid that exerts a plethora of physiological and pathophysiological effects on different organs. 5-HT is released by epithelial enterochromaffin cells upon activation of mucosal processes by both intrinsic and extrinsic primary afferent neurons. 5-HT predominantly mediates signaling to the central nervous system, serotonergic transmission within the enteric nervous system, and the activation of myenteric intrinsic primary afferent neurons. It has seven families of receptors in the brain, numbering at least 14 distinct proteins (Table 1). Although most of the studies concerning its multiple functions in the CNS, high levels of receptor expression in other areas (intestine, platelets, and endothelial cells) suggest that it could play crucial roles in other aspects of physiology [1]. For instance, 5-HT2A has been implicated in the etiology of numerous disease states like neurodysfunction and hypertension, type 2 diabetes mellitus (T2DM), eating disorders, vomiting, and irritable bowel disease (IBD) [2,3,4]. It also regulates cell proliferation, migration, and maturation in a variety of cell types, including renal proximal tubular cells, endothelial cells, mast cells, neurons, and astrocytes. Recent in vitro studies examining 5-HT2 receptors in the kidney have shown the presence of serotonergic 5-HT2A mRNA expression in cultured renal cells, in addition to 5-HT2A protein in renal mesangial cells [5, 6]. Similarly, Banes et al. [7] have also demonstrated the presence of 5-HT2A receptors in vascular smooth muscle cells (VSMCs) [7]. Furthermore, scientific evidence revealed the presence of 5-HT2A receptor mRNA expression in rat renal artery and involvement of renal proximal tubules in the biosynthesis of 5-HT and its storage in the adrenal medulla [8,9,10] (Table 2). Previous reports demonstrated that 5-HT has significant effects on renal metabolism, glomerular function, and renal vascular tone [10, 22]. 5-HT stimulates mitogenesis in rat renal mesangial cells through the 5-HT2A receptor to induce the transcription of mitochondrial genes [23, 24]. Additionally, 5-HT stimulation upregulates PAI-1 expression in proximal tubular epithelial cells, ERK phosphorylation, and cellular proliferation in renal mesangial cells [19, 25]. Generally, the kidney remains protected from hypertensive injury as long as the blood pressure remains within the auto-regulatory range [26]. However, evidence has shown that activation of the 5-HT2A receptor, along with an increase in 5-HT2A receptor density results in an increase in blood pressure [14, 27].
Thus, targeting 5-HT2A receptors in nephrotoxic conditions may provide potential therapeutics for various pathological diseases associated with the kidney. However, 5-HT2A-mediated CKDs involve more than single pathological mechanisms. In this review, we have discussed the role and potential mechanisms involving serotonin 5-HT2A receptor-mediated CKDs.
Mechanisms involved in renal pathophysiology
There are several mechanisms proposed by different research groups correlating CKDs and 5-HT receptors.
Autoregulation (intrarenal mechanism)
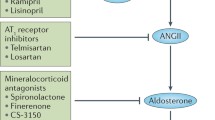
Renal autoregulation, necessary for the normal renal function and volume homeostasis, has long been a cornerstone of renal physiology. Damage of the vascular smooth muscle cells, acute tubular necrosis, and suppression of vasodilatory autoregulation of renal blood flow (RBF) results in renal dysfunction [8]. Stimulation of 5-HT2 receptors on smooth muscle cells elicits vasoconstriction and further amplifies the vasoconstrictive effects of other chemical mediators [27]. There are enough pieces of evidence that 5-HT is vital for the valid expression of autoregulation of RBF in normal rats [11, 28]. In a renal artery clamp model of ischemic acute renal failure (ARF), 5-HT2-antagonist (ketanserin) treatment showed a beneficial effect on the loss of RBF autoregulation, i.e., administration of ketanserin reduces renal perfusion pressure (RPP) which results in improved autoregulation [13] (Fig. 1). Similarly, Verbeke et al.’s study also states that in cyclosporin A-induced renal dysfunction, autoregulation is suppressed by 5-HT2A-mediated vasoconstriction [14].

Effect of serotonin 5HT2A receptors on renal autoregulation and nitric oxide pathway
Involvement of NO in 5-HT2A-mediated CKD (intracellular signaling)
Nitric oxide (NO) is one amongst the foremost necessary molecules created within the human body which regulates numerous essential cell functions, including the regulation of healthy blood flow and blood pressure levels. There are two primary pathways for the production of nitric oxide. Nitric oxide synthase (NOS) pathway: Nitric oxide synthase (NOS) is an enzyme found in the endothelial cells, which line blood vessels throughout the body and NOS converts l-arginine into nitric oxide through a series of reaction [29]. Nitrate-nitrite-pathway: Inorganic nitrate, when consumed through diet (i.e., green leafy vegetables such as beets, kale, or spinach), enabled food to be converted into nitrite and nitric oxide by oral bacteria [30].
Several shreds of evidence have reported impaired NOS pathway in animals suffering from CKD-induced [17]. Literature suggests that 5-HT2A receptor activates PLC through Gq and leads to an accumulation of IP3, di-acylglycerol (DAG), and activation of protein kinase C (PKC), and in normal physiological conditions, eNOS is responsible for NO production and further activation of various signaling molecules like increased intracellular cGMP, which inhibits calcium entry into the cell and reduces intracellular calcium concentrations. Followed by activation of K+ channels, which ends up in hyperpolarization and relaxation, stimulates a cGMP-dependent protein enzyme that activates myosin light-chain phosphates via de-phosphorylation of myosin light chains leading to smooth muscle relaxation, and all the abovementioned physiological mechanisms are restored by serotonin 5-HT2A antagonists (sarpogrelate) [19, 31, 32] (Fig. 1). The Umrani et al., in 2003, observed improved NO bioavailability as well as enhanced endothelial eNOS expression in diabetic rats after serotonin antagonist treatment [33].
Mitochondrial damage (intracellular mechanism)
In the renal cortex and renal proximal tubular cells (RPTCs), 5-HT2A receptors are responsible for mitochondrial biogenesis [23], which is a common consequence of ischemia/reperfusion (I/R) injury, drug-induced, toxicant-induced, and oxidant injury. Studies have shown that activation of serotonin 5-HT2A receptors induces transcription of mitochondrial genes and helps to promote recovery of cellular respiration in RPTCs after oxidant injury [34]. 5-HT2A is known to stimulate the release of calcium ions, which further activates cGMP, which is an essential factor for mitochondrial biogenesis [31]. 5-HT2A receptor also induces mitochondrial biogenesis via various signaling pathways, including peroxisome proliferator-activated receptor-γ-coactivator-1α (PPAR-γ-coactivator-1α) by activation of Src, P38 mitogen-activated protein kinase, and epidermal growth factor; cAMP-, calcineurin A-, and calcium/calmodulin-dependent protein kinase. So, these literature reports indicate that modulation of 5-HT2A signaling in the kidney may promote mitochondrial biogenesis [5, 32, 35]. On the contrary, some studies reported that DOI, a specific 5-HT2A agonist, improves the mitochondrial biogenesis after acute or chronic kidney injury [23, 36] (Fig. 2).

Schematic representation of serotonin 5HT2A receptor-mediated mitochondrial biogenesis
JAK/STAT (intracellular signaling) pathway
JAK/STAT pathway includes a cascade of protein interactions and is associated with immunity, cell death, cell division, and tumor formation. Previous studies have shown that 5-HT2A receptors in vascular smooth muscle cells are involved in the activation of the JAK/STAT pathway. 5-HT2A enhances phosphorylation of JAK2, JAK1, and STAT1 and activates Na+/H+ pump type-I [7]. Activation of JAK/STAT stimulates TGF-β1 and fibronectin synthesis in mesangial cells, which are responsible for increased cellular proliferation and increased DNA synthesis. Enhanced TGF-β1 is a primary pathological marker for proliferative glomerulonephritis, anti-glomerular basement membrane glomerulonephritis, and human renal cell carcinoma [37, 38] (Fig. 3). Furthermore, renal serotonin 5HT2A-mediated JAK/STAT pathway also plays a crucial role in the crosstalk between other signaling cascades like PI3k-Akt (involved in tubulointerstitial fibrosis, glomerulosclerosis results in a progressive decline in kidney functions lead to CKD) [39], EGF-mediated phosphorylation of STAT3, and activation of interleukin-6 (IL-6) [40, 41].

Activation of the JAK/STAT pathway, MCP-1, and adiponectin via stimulation of serotonin 5HT2A receptor
ERK (extracellular signal-regulated kinase) phosphorylation
Receptor tyrosine kinases are cell surface receptors, which include epidermal growth factor (EGF) receptor (EGFR) [25]. PKC transactivates EGFR in mesangial cells, which helps to regulate the extracellular matrix. Studies have shown that renal mesangial cell expresses mitogenic GPCRs, including angiotensin II, bradykinins, and 5-HT2A receptors, and they all participate in proliferative phases of chronic renal failure. Mesangial cells maintain structural integrity and help in ultrafiltration. Typically, mesangial cells are quiescent but can alter ECM by TGF-β1-mediated proliferation and fibrosis, which leads to irreversible renal injury [6]. 5-HT2A potently activates ERK and induces concentration and time-dependent expression of TGF-β1 mRNA and protein in mesangial cells. 5-HT activation also induces PKC which stimulates NADPH-oxidase and simultaneously increases the expression levels of MEK and ERK [21]. In contrast, serotonin 5-HT2A transactivates EFR and increases collagen IV via PKC and increases cell proliferation (Fig. 4).

Serotonin 5HT2Areceptor activation-mediated oxidative stress, ERK phosphorylation, collagen type IV activation, and alteration in inflammatory cytokines levels
Collagen type IV (extracellular mechanism)
Collagen type IV is present in basal lamina and reported to be involved in autoimmune disease- (Goodpasture syndrome), in which the immune system attacks the basement membrane of glomeruli and alveoli, and it is also associated with nephritic syndrome and hemoptysis. 5-HT is responsible for the increased induction and secretion of collagen type IV via activation of phospholipase C. Further, this well-known mechanism is linked with enhanced action of DAG and calcium, along with the activation of PKC [20]. Increased response of TGF-β1 is associated with collagen type IV-mediated PKC activation, which further leads to mesangial cell proliferation. Kasho et al., in 1998, reported that the 5-HT2A receptor is responsible for the activation of collagen type IV, and ketanserin (5-HT2A receptor antagonist) could be helpful to overcome the enhanced levels of collagen. Furthermore, several scientific studies suggest that collagen deposition-mediated mesangial cell proliferation and tubulointerstitial fibrosis are major pathogenic factors in the advancement of CKD [20, 42, 43] (Fig. 4). Kim et al. [44] also demonstrated that the activation of serotonin receptors in diabetic mice leads to cortex collagen deposition, and further, the levels of collagen IV and collagen I were consistently reduced by sarpogrelate (5-HT2A receptor antagonist) [45].
Adiponectin (metabolic pathway) and platelets (blood cell-mediated pathway)
Adiponectin is an adipocyte-derived multifunctional peptide which has an anti-inflammatory and anti-atherogenic effect and also involved in insulin sensitization. Studies suggest that higher levels of adiponectin are the indication of progressive CKD and ESRD. The underlying mechanism for adiponectin is found to be related to AMPK and NADPH oxidase-mediated pathway in the kidney. Clinical and preclinical studies suggest that adiponectin levels get reduced in T2DM and obesity [46,47,48,49,50,51,52]. Moreover, in mesenteric adipose tissue, the release of adiponectin is declined via activation of 5-HT2A receptors, and similarly, in genetically modified obese mice, increased 5-HT2A receptor expression levels result in markedly reduced adiponectin levels. Moreover, 5-HT2A receptor knockdown or inhibition of 5-HT2A receptor signaling increases adiponectin expression [53]. Furthermore, scientific studies also supported the fact that 5-HT2A receptor inhibition using sarpogrelate helps to improve early stages of diabetic nephropathy, glomerular endothelial function by increasing adiponectin levels in the blood and reducing albuminuria and glomerular platelet activation in a diabetic animal model [36, 54].
Chronic kidney disease (CKD) is known to associate with numerous hemostatic disorders including increased clotting tendency and defective fibrinolysis due to a decline in natural coagulation inhibitor levels [55,56,57]. Further, during hemodialysis treatment platelet disturbances are commonly aggravated in CKD patients and it is stated that platelets get activated immediately after hemodialysis treatment which is illustrated by upregulation of surface receptors followed by the release of degranulation product results in increased platelet turnover. The release of serotonin from granule stores potentiates activation of platelet further lead to platelet aggregation by recruiting more platelets. [19]. In a similar context, scientific evidences reveal that the release of serotonin from platelet significantly decreases in patients with diabetes mellitus (DM)-associated atherosclerosis and acute and chronic renal failure. Furthermore, the involvement of serotonin (released by platelets) on fibrotic and inflammatory processes in proximal tubular endothelial cells has been explained by Erikci et al. [10]. Proliferation initiates during the healing process of renal tissue platelet aggregates, and neutrophils move towards the injury site and basal epithelial cells. Moreover, serotonin-mediated stimulation of PTECs results in a dose-dependent increase in both TGF-β1 protein levels and relative gene expressions, suggesting the significance of platelet-derived serotonin in the regulation of TGF-β1 and inflammatory mediator levels. Thus, further investigation is required in the delineation of platelet-derived serotonin mechanisms and also in the evaluation of adiponectin impact in progressive CKD.
Inflammatory cytokines, chemokines, and 5-HT2A receptors in CKD
TGF-β1
Transforming growth factor-β (TGF-β) is critical for healthy kidney development and function. Thompson et al. identified TGF-β protein in distal tubular epithelial cells (within the cytoplasm of a subset) at the plasma membranes of cells in the corticomedullary junction in the embryonic murine kidney and the mammalian metanephros during tubular construction. TGF-β stimulation leads to extracellular matrix (ECM) accumulation, activation of many functions in the nearby cells, apoptosis, and cell dedifferentiation followed by loss of function which results in disruption of the orchestrated structural and functional balance of cells [44]. Yang et al., in 2017, reported that 5-HT2A receptors are responsible for the induction of TGF-β1 expression via extracellular signal-regulated kinases, and evidence proves that mitogenic signaling components have a direct relation with TGF-β1 regulation which acts as crucial mediators for proliferation and fibrosis in renal mesangial cells [36]. Schiffer et al. in 2001 presented that TGF-β1 can indirectly affect the permeability properties through apoptosis which is associated with mesangial cell expansion, increased proteinuria, and podocyte depletion [58]. These pathological events are the major hallmark for progressive renal diseases in humans [59, 60], such as glomerulosclerosis [59]and increased expression of cellular ROS.
IL-6 and TNF-α
TNF-α is a cytokine involved in the systemic inflammation, while IL-6 is both a cytokine and a myokine in nature, and their levels get altered during renal complications [61]. Inhibition of TNF-α is responsible for IL-6-mediated anti-inflammatory effect, which also plays a primary role in immune cells, acute-phase reaction, and apoptotic cell death. Kubera et al. [18] have explored the involvement of serotonin receptors in the production of TNF-α and IL-6 by observing the effects of both 5-HT serotonin agonists and antagonists in humans [18]. Their study disclosed that at physiological concentrations, serotonin might partly enhance the production of TNF-α and IL-6 via activation of 5-HT2 receptors, but above the baseline levels, serotonin may suppress the synthesis of these cytokines. Some previous studies have reported a decrease in levels of pro-inflammatory markers as a result of serotonin 5-HT2A receptor antagonism via sarpogrelate hydrochloride [62, 63]. In addition to the determination of the relationship between serotonin receptors and cytokines, IL-6 and TNF-α, dissimilar scientific evidence is also available, suggesting that activation of serotonin by DOI (5-HT2A receptor agonist) leads to inhibition of TNF-α-mediated inflammation and it also inhibits IL-6 [64, 65] (Fig. 4).
MCP-1
MCP-1 is one of the potent chemokines which plays an important role in the kidney, and there are enough pieces of preclinical and clinical evidences describing the role of MCP-1 of renal disease. Inflammation mediated by MCP-1 initiated from the release of monocytes from the bone marrow, and then monocytes migrate towards the site of inflammation via endothelial glycocalyx generated gradient, thereby reducing the migration of blood leukocytes into the inflamed tissue. In addition, MCP-1 has a direct role in migration, proliferation, signaling pathway of monocytes, and differentiation of leukocytes. MCP-1 inhibition is a promising and valid strategy in renal inflammatory disease. Furthermore, several shreds of evidence also reveal the positive correlation between the upregulation of MCP-1 and obesity. Glomerular proteinuria is the postulated link between the increased lysosome and MCP-1 release by PTECs and eventually starts MCP-1-mediated tubulointerstitial fibrosis by stimulating the release of TGF-β via activation of macrophages [66, 67]. Moreover, serotonin-mediated activation of JAK/STAT pathway followed by the activation of monocyte chemoattractant protein-1 (MCP-1), which plays a significant role in macrophage accumulation. However, treatment with 5-HT2A antagonist sarpogrelate was found to be helpful in the recovery of plasma adiponectin levels, maintenance of albumin to Cr ratio, and also MCP-1 levels in the plasma and urine [68].
Oxidative stress
Reactive oxygen species (ROS) are reactive molecules and free radicals derived from molecular oxygen. Recent studies disclosed that renal ROS production in kidney dysfunction is predominantly mediated by various NADPH oxidases (NOXs), mitochondrial dysfunction, NF-κB pathway, and compromised antioxidant system [69,70,71,72]. Kirkman et al., in 2017, observed that mitochondrial dysfunction appeared as a potential source of oxidative stress responsible for the impaired vascular function and mitochondria-derived reactive oxygen species contribute to microvascular dysfunction in stage 3–5 CKD [73]. As previous studies suggest, 5-HT2A receptors are involved in the activation and phosphorylation of ERK; during this pathway, reactive oxygen species is generated as intermediate via NADPH [6]. Various clinical and preclinical evidences support the fact that inhibition of 5-HT2A receptor declines the ROS production, enhances the availability of antioxidant enzymes, and as well as helps to improve mitochondrial dysfunction [15, 36]. Kobayashi and his coworkers reported the involvement of 5-HT2A receptors in decreased bioavailability of NO in glomeruli and ROS-mediated endothelial dysfunction in diabetic rats [16]. Upregulation of ROS also results in overexpression of PAI-1 via enhanced levels of TGF-β, which is associated with tubulointerstitial fibrosis, a major pathological event of progressive kidney injury [19] (Fig. 4). 5-HT2A can mediate ROS via mitochondrial oxidative phosphorylation or NADPH oxidases or both.
Clinical relevance of 5-HT receptor modulators in CKD
There are several serotonin modulators available and used for therapeutic purposes, out of which the most popular one is selective serotonin reuptake inhibitors (SSRIs) and are clinically used as anti-depressants. There are clinical pieces of evidence in which sertraline and escitalopram are mentioned to be used cautiously in renal disease patients [74]. On the contrary, some scientific studies suggest that 5-HT2A receptor antagonists help to reduce albuminuria and improve GFR and also long-term outcomes in diabetic patients with early-stage diabetic nephropathy [75, 76]. Bennet et al. in 2015 clinically investigated different gene expressions and enzymes and also conducted transcriptional mapping of 5-HT receptors in diabetes. In results, the author suggested that increased expression of 5-HT1D and 5-HT2A is either cause or consequence of islet dysfunction in type 2 diabetes and alter overall islet hormone secretion in non-diabetic individuals [77]. Additionally, SSRIs have also been studied in depression associated with ESRD and CKD patients [74, 78, 79].
Future perspective for 5-HT2A receptor modulators
A long list of 5-HT2A agonists and antagonists with their respective derivatives has been developed and extensively reviewed. However, most of the drugs are used to evaluate the different mechanisms associated with kidney diseases using cell cultures. Long-term in vivo studies are still needed to do to determine the effect and specific doses of these modulators in chronic therapies. Specific agonists and antagonists, targeting individual PKC mediator for EGFR transactivation will also be helpful. Modern reconstructed lifestyle, patient compliance, MDT (multidrug therapy), and fixed-dose combination therapy will be a positive approach to optimize control of the several risk factors for CKDs. The centerpiece renoprotective treatment for nephropathy-associated proteinuria often includes ACE inhibitors and ARBs. Intriguingly, recent preclinical and clinical findings shown that serotonin antagonism will be useful to avert structural changes and remodeling of the glomerular architecture which offers completely novel perspectives for CKD treatments. However, all the above-listed mechanisms or pathways are interrelated directly or indirectly. Pre- and pro-inflammatory mediators can work as an essential target in every mechanistic pathway associated with CKDs. Furthermore, the number of clinical complications is known to influence different pathological processes of CKDs in various manners. In that case, we have to make a distinct approach to nullify the causes and consequences of CKDs or related complication by using emerging advanced or detailed knowledge of serotonin-mediated pathways.
Conclusion
There is a growing repertoire of proofs supporting the fact that serotonin 5-HT2A receptors are strongly related to the physiological and pathophysiological process of the kidney. Pharmacological inhibition or activation of 5-HT2A-mediated renal autoregulation, mitochondrial biogenesis, inflammatory response via altered pre- or pro-inflammatory cytokines, and accelerated ROS generation and modulation in ERK, MAPK, and JAK/STAT pathways present an appealing therapeutic strategy to attenuate the development of CKDs and renal complications. In conclusion, in the future, with the increasing knowledge of structural systems, all the above-listed pathways, including serotonin 5-HT2A receptor allied with kidney impairment, will provide vital information to explore newer targets, risk factors, and treatment strategies to combat or halt development kidney diseases.
Availability of data and materials
Not applicable
Abbreviations
- ARF:
-
Acute renal failure
- CKD:
-
Chronic kidney disease
- DAG:
-
Di-acylglycerol
- EGF:
-
Epidermal growth factor
- ERK:
-
Extracellular receptor kinase
- I/R:
-
Ischemia/reperfusion
- IBD:
-
Irritable bowel disease
- IL:
-
Interleukin
- NO:
-
Nitric oxide
- NOS:
-
Nitric oxide synthase
- NOXs:
-
NADPH oxidases
- PKC:
-
Protein kinase C
- PPAR-γ-coactivator-1α:
-
Peroxisome proliferator-activated receptor-Γ-coactivator-1α
- RBF:
-
Renal blood flow
- ROS:
-
Reactive oxygen species
- RPP:
-
Renal perfusion pressure
- RPTCs:
-
Renal proximal tubular cells
- SSRIs:
-
Selective serotonin reuptake inhibitors
- STAT:
-
Signal transducer and activator of transcription 3
- T2DM :
-
Type 2 diabetes mellitus
- TGF-β:
-
Transforming growth factor-Β
- TNF-α:
-
Tumor necrosis factor-Α
- VSMCs:
-
Vascular smooth muscle cells
References
Hoyer D, Clarke DE, Fozard JR, Hartig PR, Martin GR, Mylecharane EJ, Saxena PR, Humphrey PP. International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (Serotonin). Pharmacol rev. 1994;46(2):157–203.
Schreiber R, Melon C, De Vry J. The role of 5-HT receptor subtypes in the anxiolytic effects of selective serotonin reuptake inhibitors in the rat ultrasonic vocalization test. Psychopharmacology. 1998 Feb 1;135(4):383–91.
Popa D, Léna C, Fabre V, Prenat C, Gingrich J, Escourrou P, Hamon M, Adrien J. Contribution of 5-HT2 receptor subtypes to sleep–wakefulness and respiratory control, and functional adaptations in knock-out mice lacking 5-HT2A receptors. J Neurosci. 2005;25(49):11231–8.
Bubar MJ, Cunningham KA. Serotonin 5-HT2A and 5-HT2C receptors as potential targets for modulation of psychostimulant use and dependence. Current topics in medicinal chemistry. 2006;6(18):1971–85.
Harmon JL, Wills LP, McOmish CE, Demireva EY, Gingrich JA, Beeson CC, Schnellmann RG. 5-HT2 receptor regulation of mitochondrial genes: unexpected pharmacological effects of agonists and antagonists. J Pharmacol Exp Ther. 2016;357(1):1–9.
Greene EL, Houghton O, Collinsworth G, Garnovskaya MN, Nagai T, Sajjad T, Bheemanathini V, Grewal JS, Paul RV, Raymond JR. 5-HT2A receptors stimulate mitogen-activated protein kinase via H2O2 generation in rat renal mesangial cells. Am J Physiol-Ren Physiol. 2000;278(4):F650–8.
Banes AK, Shaw SM, Tawfik A, Patel BP, Ogbi S, Fulton D, Marrero MB. Activation of the JAK/STAT pathway in vascular smooth muscle by serotonin. Am J Physiol-Cell Physiol. 2005;288(4):C805–12.
Morán A, de Urbina AV, Martín ML, Rodríguez-Barbero A, San RL. Characterization of the contractile 5-hydroxytryptamine receptor in the autoperfused kidney of L-NAME hypertensive rats. Eur J Pharmacol. 2009;620(1-3):90–6.
Hannon J, Hoyer D. Molecular biology of 5-HT receptors. Behav Brain Res. 2008;195(1):198–213.
Erikci A, Ucar G, Yabanoglu-Ciftci S. Role of serotonin in the regulation of renal proximal tubular epithelial cells. Ren fail. 2016;38(7):1141–50.
Lameire NH, Matthys E, Kesteloot D, Waterloos MA. Effect of a serotonin blocking agent on renal hemodynamics in the normal rat. Kidney international. 1990;38(5):823–9.
Endlich K, Kühn R, Steinhausen M, Dussel R. Visualization of serotonin effects on renal vessels of rats. Kidney Int. 1993;43(2):314–23.
Verbeke M, Smöllich B, Van de Voorde J, De Ridder L, Lameire N. Beneficial influence of ketanserin on autoregulation of blood flow in post-ischemic kidneys. J Am Soc Nephrol. 1996;7(4):621–7.
VERBEKE M, Van de Voorde J, De Ridder L, Lameire N. Beneficial effect of serotonin 5-HT2-receptor antagonism on renal blood flow autoregulation in cyclosporin-treated rats. J Am Soc Nephrol. 1999;10(1):28–34.
Ali T, Shaheen F, Mahmud M, Waheed H, Ishtiaq M, Javed Q, Murtaza I. Serotonin-promoted elevation of ROS levels may lead to cardiac pathologies in diabetic rat. Archives of Biological Sciences. 2015;67(2):655–61.
Kobayashi S, Satoh M, Namikoshi T, Haruna Y, Fujimoto S, Arakawa S, Komai N, Tomita N, Sasaki T, Kashihara N. Blockade of serotonin 2A receptor improves glomerular endothelial function in rats with streptozotocin-induced diabetic nephropathy. Clin Exp Nephrol. 2008;12(2):119–25.
Lee ES, Lee MY, Kwon MH, Kim HM, Kang JS, Kim YM, Lee EY, Chung CH. Sarpogrelate hydrochloride ameliorates diabetic nephropathy associated with inhibition of macrophage activity and inflammatory reaction in db/db mice. PloS one. 2017;12(6):e0179221.
Kubera M, Maes M, Kenis G, Kim YK, Lasoń W. Effects of serotonin and serotonergic agonists and antagonists on the production of tumor necrosis factor α and interleukin-6. Psychiatry res. 2005;134(3):251–8.
Hamasaki Y, Doi K, Maeda-Mamiya R, Ogasawara E, Katagiri D, Tanaka T, Yamamoto T, Sugaya T, Nangaku M, Noiri E. A 5-hydroxytryptamine receptor antagonist, sarpogrelate, reduces renal tubulointerstitial fibrosis by suppressing PAI-1. Am J Physiol-Ren Physiol. 2013;305(12):F1796–803.
Kasho M, Sakai M, Sasahara T, Anami Y, Matsumura T, Takemura T, Matsuda H, Kobori S, Shichiri M. Serotonin enhances the production of type IV collagen by human mesangial cells. Kidney int. 1998;54(4):1083–92.
Grewal JS, Mukhin YV, Garnovskaya MN, Raymond JR, Greene EL. Serotonin 5-HT2A receptor induces TGF-β1 expression in mesangial cells via ERK: proliferative and fibrotic signals. Am J Physiol-Ren Physiol. 1999;276(6):F922–30.
Watts SW, Thompson JM. Characterization of the contractile 5-hydroxytryptamine receptor in the renal artery of the normotensive rat. J pharmacol exp ther. 2004;309(1):165–72.
Rasbach KA, Funk JA, Jayavelu T, Green PT, Schnellmann RG. 5-hydroxytryptamine receptor stimulation of mitochondrial biogenesis. J pharmacol exp ther. 2010;332(2):632–9.
Cameron RB, Peterson YK, Beeson CC, Schnellmann RG. Structural and pharmacological basis for the induction of mitochondrial biogenesis by formoterol but not clenbuterol. Scientific reports. 2017;7(1):1–1.
Göőz M, Göőz P, Luttrell LM, Raymond JR. 5-HT2A receptor induces ERK phosphorylation and proliferation through ADAM-17 tumor necrosis factor-α-converting enzyme (TACE) activation and heparin-bound epidermal growth factor-like growth factor (HB-EGF) shedding in mesangial cells. J Biol Chem. 2006;281(30):21004–12.
Loutzenhiser R, Griffin K, Williamson G, Bidani A. Renal autoregulation: new perspectives regarding the protective and regulatory roles of the underlying mechanisms. Am J Physiol Regul Integr Comp Physiol. 2006;290(5):R1153–67.
Morán A, Restrepo B, de Urbina AV, García M, Martín ML, San RL. Pharmacological profile of 5-hydroxytryptamine-induced inhibition on the pressor effect elicited by sympathetic stimulation in long-term diabetic pithed rats. Eur J Pharmacol. 2010;643(1):70–7.
Van JN. Serotonin and the blood vessel wall. Journal of cardiovascular pharmacology. 1985;7:S49–51.
Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33(7):829–37.
Vanderpool R, Gladwin MT. Harnessing the nitrate–nitrite–nitric oxide pathway for therapy of heart failure with preserved ejection fraction. 2015;334–6.
Raote I, Bhattacharya A, Panicker MM. Serotonin 2A (5-HT2A) receptor function: ligand-dependent mechanisms and pathways. Serotonin receptors in neurobiology. 2007;17:105–32.
Scarpulla RC. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim Biophys Acta. 2002;(1-2):1576, 1–4.
Umrani DN, Bodiwala DN, Goyal RK. Effect of sarpogrelate on altered STZ-diabetes induced cardiovascular responses to 5-hydroxytryptamine in rats. InBiochemistry of Diabetes and Atherosclerosis 2003 (pp. 53-57). Springer, Boston, MA.
Rasbach KA, Schnellmann RG. Signaling of mitochondrial biogenesis following oxidant injury. J Biol Chem. 2007;282(4):2355–62.
Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92(6):829–39.
Yang Y, Huang H, Xu Z, Duan JK. Serotonin and its receptor as a new antioxidant therapeutic target for diabetic kidney disease. J diabetes res. 2017;2017.
Garnovskaya MN, Mukhin YV, Turner JH, Vlasova TM, Ullian ME, Raymond JR. Mitogen-induced activation of Na+/H+ exchange in vascular smooth muscle cells involves Janus kinase 2 and Ca2+/calmodulin. Biochemistry. 2003;42(23):7178–87.
Shaw S, Wang X, Redd H, Alexander GD, Isales CM, Marrero MB. High glucose augments the angiotensin II-induced activation of JAK2 in vascular smooth muscle cells via the polyol.
Bousoik E, Aliabadi HM. “Do we know jack” about JAK? A closer look at JAK/STAT signaling pathway. Front Oncol. 2018;8:287.
Huang JS, Chuang LY, Guh JY, Huang YJ, Hsu MS. Antioxidants attenuate high glucose-induced hypertrophic growth in renal tubular epithelial cells. Am J Physiol-Ren Physiol. 2007;293(4):F1072–82.
Lan A, Du J. Potential role of Akt signaling in chronic kidney disease. Nephrology Dialysis Transplantation. 2014;30(3):385–94.
Scindia YM, Deshmukh US, Bagavant H. Mesangial pathology in glomerular disease: targets for therapeutic intervention. Adv. Drug Deliv. Rev. 2010;62(14):1337–43.
Shimizu F, Kawachi H, Orikasa M. Role of mesangial cell damage in progressive renal disease. Kidney and Blood Pressure Research. 1999;22(1-2):5–12.
Kim DH, Choi BH, Ku SK, Park JH, Oh E, Kwak MK. Beneficial effects of sarpogrelate and rosuvastatin in high fat diet/streptozotocin-induced nephropathy in mice. PLoS One. 2016;11(4):e0153965.
Wilmer WA, Tan LC, Dickerson JA, Danne M, Rovin BH. Interleukin-1β induction of mitogen-activated protein kinases in human mesangial cells. Role of oxidation. J Biol Chem. 1997;272(16):10877–81.
Yoon YS, Ryu D, Lee MW, Hong S, Koo SH. Adiponectin and thiazolidinedione targets CRTC2 to regulate hepatic gluconeogenesis. Experimental & molecular medicine. 2009;41(8):577–83.
Nomura S, Shouzu A, Omoto S, Nishikawa M, Iwasaka T. 5-HT2A receptor antagonist increases circulating adiponectin in patients with type 2 diabetes. Blood coagulation & fibrinolysis. 2005;16(6):423–8.
Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, Tataranni PA. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab. 2001;86(5):1930–5.
Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, Iwahashi H, Kuriyama H, Ouchi N, Maeda K, Nishida M, Kihara S, Sakai N, Nakajima T, Hasegawa K, Muraguchi M, Ohmoto Y, Nakamura T, Yamashita S, Hanafusa T, Matsuzawa Y. Plasma concentrations of a novel, adiposespecific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol. 2000;20(6):1595–9.
Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P, Kadowaki T. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7(8):941–6.
Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraguchi M, Ohmoto Y, Funahashi T, Matsuzawa Y. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257(1):79–83.
Milan G, Granzotto M, Scarda A, Calcagno A, Pagano C, Federspil G, Vettor R. Resistin and adiponectin expression in visceral fat of obese rats: effect of weight loss. Obes Res. 2002;10(11):1095–103.
Uchida-Kitajima S, Yamauchi T, Takashina Y, Okada-Iwabu M, Iwabu M, Ueki K, Kadowaki T. 5-Hydroxytryptamine 2A receptor signaling cascade modulates adiponectin and plasminogen activator inhibitor 1 expression in adipose tissue. FEBS Lett. 2008;582(20):3037–44.
Adams MJ, Irish AB, Watts GF, et al. Hypercoagulability in chronic kidney disease is associated with coagulation activation but not endothelial function. Thromb Res. 2008;123:374–80.
Kushiya F, Wada H, Sakakura M, et al. Atherosclerotic and hemostatic abnormalities in patients undergoing hemodialysis. Clin Appl Thromb Hemost. 2003;9:53–60.
Undas A, Kolarz M, Kopec G, et al. Altered fibrin clot properties in patients on long-term haemodialysis: relation to cardiovascular mortality. Nephrol Dial Transplant. 2008;23:2010–5.
Schoorl M, Schoorl M, Bartels PC. Changes in platelet volume, morphology and RNA content in subjects treated with haemodialysis. Scand J Clin Lab Invest. 2008;68:335–42.
Schiffer M, Bitzer M, Roberts IS, Kopp JB, ten Dijke P, Mundel P, Böttinger EP. Apoptosis in podocytes induced by TGF-β and Smad7. J clin investig. 2001;108(6):807–16.
Yamamoto T, Nakamura T, Noble NA, Ruoslahti E, Border WA. Expression of transforming growth factor beta is elevated in human and experimental diabetic nephropathy. Proceedings of the National Academy of Sciences. 1993;90(5):1814–8.
Kopp JB, Factor VM, Mozes M, Nagy P, Sanderson N, Böttinger EP, Klotman PE, Thorgeirsson SS. Transgenic mice with increased plasma levels of TGF-beta 1 develop progressive renal disease. Lab invest. 1996;74(6):991–1003.
Chu C, Li D, Zhang S, Ikejima T, Jia Y, Wang D, Xu F. Role of silibinin in the management of diabetes mellitus and its complications. Archives of pharmacal research. 2018;41(8):785–96.
Marconi A, Darquenne S, Boulmerka A, Mosnier M, d’Alessio P. Naftidrofuryl-driven regulation of endothelial ICAM-1 involves nitric oxide. Free Radical Biology and Medicine. 2003;34(5):616–25.
Akiyoshi T, Zhang Q, Inoue F, Aramaki O, Hatano M, Shimazu M, Kitajima M, Shirasugi N, Niimi M. Induction of indefinite survival of fully mismatched cardiac allografts and generation of regulatory cells by sarpogrelate hydrochloride. Transplantation. 2006;82(8):1051–9.
Nau F Jr, Yu B, Martin D, Nichols CD. Serotonin 5-HT2A receptor activation blocks TNF-α mediated inflammation in vivo. PloS one. 2013;8(10):e75426.
Yu B, Becnel J, Zerfaoui M, Rohatgi R, Boulares AH, Nichols CD. Serotonin 5-hydroxytryptamine2A receptor activation suppresses tumor necrosis factor-α-induced inflammation with extraordinary potency. J Pharmacol Exp Ther. 2008;327(2):316–23.
Cockwell P, Howie AJ, Adu D, Savage CO. In situ analysis of CC chemokine mRNA in human glomerulonephritis. Kidney int. 1998;54(3):827–36.
Rovin B, Rumancik M, Tan L, Dickerson J. Glomerular expression of monocyte chemoattractant protein-1 in experimental and human glomerulonephritis. Lab Invest. 1994;71:536–42.
Ogawa S, Mori T, Nako K, Ishizuka T, Ito S. Reduced albuminuria with sarpogrelate is accompanied by a decrease in monocyte chemoattractant protein-1 levels in type 2 diabetes. Clin J Am Soc Nephrol. 2008;3(2):362–8.
Hong J, Wang X, Zhang N, Fu H, Li W. D-ribose induces nephropathy through RAGE-dependent NF-κB inflammation. Archives of pharmacal research. 2018;41(8):838–47.
Ozbek E. Induction of oxidative stress in kidney. Int j nephrol. 2012;2012:465897.
You YH, Sharma K. Reactive oxygen species and chronic kidney disease. InSystems Biology of Free Radicals and Antioxidants. Berlin Heidelberg: Springer-Verlag; 2012. p. 2645–58.
Jha JC, Banal C, Chow BS, Cooper ME, Jandeleit-Dahm K. Diabetes and kidney disease: role of oxidative stress. Antioxidants & redox signaling. 2016;25(12):657–84.
Kirkman DL, Muth BJ, Ramick MG, Townsend RR, Edwards DG. Role of mitochondria-derived reactive oxygen species in microvascular dysfunction in chronic kidney disease. Am J Physiol-Ren Physiol. 2018;314(3):F423–9.
Hedayati SS, Yalamanchili V, Finkelstein FO. A practical approach to the treatment of depression in patients with chronic kidney disease and end-stage renal disease. Kidney international. 2012;81(3):247–55.
Watanabe H, Nakagawa K, Kakihana M. Effects of sarpogrelate, a selective serotonin receptor antagonist, on vascular function and renal function in diabetic patients with stable angina and chronic kidney disease. 2009;S973.
Takahashi T, Yano M, Minami J, Haraguchi T, Koga N, Higashi K, Kobori S. Sarpogrelate hydrochloride, a serotonin2A receptor antagonist, reduces albuminuria in diabetic patients with early-stage diabetic nephropathy. Diabetes research and clinical practice. 2002;58(2):123–9.
Bennet H, Balhuizen A, Medina A, Nitert MD, Laakso EO, Essén S, Spégel P, Storm P, Krus U, Wierup N, Fex M. Altered serotonin (5-HT) 1D and 2A receptor expression may contribute to defective insulin and glucagon secretion in human type 2 diabetes. Peptides. 2015;71:113–20.
Pena-Polanco JE, Mor MK, Tohme FA, Fine MJ, Palevsky PM, Weisbord SD. Acceptance of antidepressant treatment by patients on hemodialysis and their renal providers. Clin J Am Soc Nephrol. 2017;12(2):298–303.
Cukor D, Kimmel PL. Treatment of depression in CKD patients with an SSRI: why things don’t always turn out as you expect. Clin J Am Soc Nephrol. 2018;13(6):943–5.
Acknowledgements
The authors are grateful to the Indian Council of medical research (ICMR) and Department of Pharmaceutical Sciences and Drug Research, Punjabi University, Patiala, India, for supporting this study.
Funding
The authors are grateful to the Indian Council of Medical Research (ICMR) for funding assistance (Application no: 45/23/2018-PA/BMS/OL)
Author information
Authors and Affiliations
Contributions
Both authors contributed to read, edit, and approve this review article. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kaur, G., Krishan, P. Understanding Serotonin 5-HT2A Receptors-regulated cellular and molecular Mechanisms of Chronic Kidney Diseases. Ren Replace Ther 6, 25 (2020). https://doi.org/10.1186/s41100-020-00268-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41100-020-00268-x