
Abstract
Chronic kidney disease (CKD) has been known to be associated with an increased risk of cardiovascular (CV) mortality and morbidity in Japan. Although traditional risk factors contribute to the development of CV disease in CKD patients, they cannot fully explain the unacceptably high incidence of CV mortality. Recently, non-traditional risk factors, including abnormal mineral metabolism, have been suggested to be involved in the increased risk of CV events. The medical treatment of CKD-mineral bone disorders (CKD-MBD) has been associated with encouraging, but inconsistent, improvement in CV disease complications and patient survival. A better understanding of the biomarkers and mechanisms involved in left ventricular hypertrophy (LVH) and vascular calcification might improve the diagnosis and treatment of the CV disease secondary to CKD-MBD, thus improving patient survival. Recent insights into fibroblast growth factor 23 (FGF23) and its co-receptor, Klotho, have led to marked advancements in the interpretation of data about CKD-MBD and CV damage.
Similar content being viewed by others

Background
Japan is one of the countries with the highest incidence of end-stage renal disease (ESRD) globally, and the number of Japanese patients with ESRD is continuously increasing [1]. Chronic kidney disease (CKD) is known to be associated with an increased risk for cardiovascular (CV) mortality and morbidity in Japan [2]. CV diseases account for 30–50% of all-cause mortality in CKD patients, especially dialysis patients worldwide [1, 3]. Although traditional risk factors contribute to the development of CV disease in CKD patients, they cannot fully explain the unacceptably high incidence of CV mortality in these patients. Hence, non-traditional risk factors, including abnormal mineral metabolism, are thought to be involved in the increased risk of CV events [4, 5].
There is growing interest in the issues surrounding CKD-mineral and bone disorder (CKD-MBD) [6]. CKD has been shown to be associated with bone and mineral disorder characterized by (i) laboratory abnormalities of calcium, phosphate, parathyroid hormone (PTH), and vitamin D; (ii) evidence of bone disease; and (iii) vascular calcification. Vascular calcification is associated with adverse clinical outcomes, including ischemic CV events and subsequent vascular mortality [7]. The pathogenesis of vascular calcification in CKD is complex, and rather than being attributable to a simple process of calcium and phosphate precipitation, involves an active process where vascular smooth muscle cells (VSMCs) undergo apoptosis with release of matrix vesicles and are transformed into osteoblast-like cells [7].
The complex mechanisms by which aging leads to increased vascular calcification remain uncertain. However, there is no doubt that patients with ESRD are at high risk of and have a high prevalence of vascular calcification because of multiple risk factors that induce the phenotypic transformation of VSMCs into osteoblast-like cells capable of carrying out tissue mineralization [8]. Vascular calcification has been associated with numerous traditional CV risk factors, including advanced age, hypertension, diabetes, and dyslipidemia, as well as with non-traditional CV risk factors, including hyperphosphatemia, hyperparathyroidism, and excessive calcium intake [9]. There are two patterns of vascular calcification. One occurs in the intimal layer and the other occurs in the medial layer of the vessel wall as in Mönckeberg’s sclerosis, which is very common in ESRD patients. The hemodynamic consequences of vascular calcification include loss of arterial elasticity, an increase in pulse wave velocity, left ventricular hypertrophy, a decrease in coronary artery perfusion, and myocardial ischemia (Fig. 1).

Schematic representation of the clinical effects of arterial intimal and medial calcification. Cited from [7]
Recent studies have shown that fibroblast growth factor 23 (FGF23), a bone-derived phosphatonin, and its co-receptor Klotho play an important role in the regulation of calcium and phosphate metabolism in CKD-MBD [10]. This review summarizes the pathophysiology involving the FGF23-Klotho axis, with particular focus on the recent advances elucidating its role in CV disease, which are characterized by left ventricular hypertrophy (LVH) and vascular calcification.
Mineral metabolism and vascular calcification
As earlier mentioned, numerous risk factors for vascular calcification have been reported. Traditional risk factors include advanced age, hypertension, diabetes, and dyslipidemia. Non-traditional risk factors in CKD include disorders of mineral metabolism, elevated serum PTH levels, excessive intake of calcium supplements, inflammation, malnutrition, and oxidative stress [11]. Patients with advanced CKD develop hyperphosphatemia secondary to impaired renal phosphate excretion. There is strong evidence that vascular calcification is closely associated with high serum calcium and phosphate levels. High serum phosphate levels can be considered a vascular toxin [12], and clinical studies have shown that patients with the poorest phosphate control experience the most rapid progression of vascular calcification [13].
Two different mechanisms of vascular calcification have been proposed to explain the relationship between calcium and phosphate disorders and vascular calcification. Experimental studies have demonstrated that calcium plays a role in the development of vascular calcification by stimulating mineralization of VSMCs under normal phosphate conditions [14]. When phosphate levels are elevated, this calcium-driven mineralization is accelerated synergistically and hyperphosphatemia may directly induce vascular injury, and it indirectly stimulates osteoblastic differentiation through a type III sodium-dependent phosphate co-transporter (PiT-1) [15]. New discoveries related to extracellular vesicles, microRNAs, and calciprotein particles (CPPs) further reveal the mechanisms involved in the initiation and progression of vascular calcification in CKD [16]. Thus, elevated intracellular phosphate concentration may directly stimulate VSMCs to transform into calcifying cells by activating genes associated with osteoblastic functions. These findings provide strong evidence that excess phosphate and calcium load is probably the most important pathogenic factor in vascular calcification.
Interaction between FGF23 and Klotho
FGF23 is a 32 kDa protein synthesized primarily in osteocytes, and the corresponding gene encodes a 251-amino-acid protein, including a 24-amino-acid signal peptide [17]. FGF23 circulates in two distinct forms, a single, intact, full-length active protein and a shorter, inactive protein. Intact-FGF23 activates FGF receptors (FGFRs) 1, 3, and 4 to elicit tissue-specific responses through the subsequent induction of two main pathways: (1) Klotho-dependent activation of the mitogen-activated protein kinase (MAPK) cascade, leading to extracellular signal-regulated kinase (ERK) activation and Egr-1 expression, and (2) a Klotho-independent cascade, characterized by phosphoinositide-specific phospholipase C (PLC) γ- dependent activation of the calcineurin-nuclear factor of activated T-cells (NFAT) [18]. FGF23 appears to impair synthesis and accelerate degradation of 1, 25 (OH)2D. Recombinant FGF23 also has a phosphaturic effect, which is attributable to reduced renal phosphate reabsorption. FGF23 downregulates the expression of both type IIa and type IIc sodium-phosphate cotransporters on the apical surface of renal proximal tubular epithelial cells in vivo [19].
The effects of FGF23 are influenced by Klotho, which is itself a 130-kDa transmembrane β-glucuronidase that catalyzes the hydrolysis of steroid β-glucuronides. Klotho was discovered by Kuro-o et al. in 1997 [20]. The Klotho gene is expressed in a limited number of tissues, mainly the kidneys, and mutations cause multiple aging-related disorders in nearly all organs and tissues. FGF23 exerts its biological effects through activation of FGFRs in a Klotho-dependent manner (Fig. 2), because a Klotho/FGFR complex binds to FGF23 with higher affinity than FGF-R or Klotho alone [21]. Thus, activation of FGF23 receptors requires not only circulating FGF23, but also the presence of Klotho as a specific promoter whose affinity dictates the selectivity towards its targets.

Interaction between FGF23 and Klotho. Cited from [10]
Because of the reduction in functioning nephrons during CKD progression, each nephron is required to excrete an increasing amount of phosphate to maintain normal serum phosphate levels. This can be attained by increasing serum FGF23 levels, which is an early sign of changes in phosphate metabolism during CKD progression. However, because FGF23 is a counter-regulatory hormone for vitamin D, an increase in its levels is at the expense of vitamin D synthesis. Decreasing vitamin D in turn stimulates PTH secretion, leading to secondary hyperparathyroidism. In fact, decreases in serum 1, 25 (OH)2D levels and increases in serum PTH levels are observed in patients with early-stage CKD with normophosphatemia. The fact that Klotho expression in the parathyroid gland is decreased in CKD patients may explain why elevated serum FGF23 levels fail to suppress PTH. The increase of FGF23 was associated with the decrease of 1, 25 (OH)2D in CKD patients [22]. Moreover, the inhibition of FGF23 activity in the animal model of CKD increased serum 1, 25 (OH)2D [23]. While the reduction of 1, 25 (OH)2D by increased FGF23 is likely to reduce intestinal phosphate absorption and contribute to maintaining serum phosphate level, it may induce or aggravate secondary hyperparathyroidism. However, the role of FGF23 in the development of secondary hyperparathyroidism is complex because FGF23 was shown to directly inhibit production and secretion of PTH [24]. With the progression of CKD, the expression of Klotho in the kidneys and parathyroid glands were shown to decrease that likely leads to impaired actions of FGF23 [25].
Elevated serum FGF23 levels and low vitamin D may further reduce Klotho expression in the kidney and parathyroid tissue in CKD patients, because FGF23 and vitamin D can downregulate and upregulate Klotho expression, respectively. Decline in Klotho expression exacerbates FGF23 resistance in the kidney and parathyroid gland, further increasing FGF23, decreasing vitamin D, and increasing PTH. This vicious cycle (Fig. 3) may contribute to the pathophysiology of deranged phosphate metabolism in CKD patients [26]. In the advanced stages of CKD, increased FGF23 can no longer compensate for dietary phosphate overload, resulting in overt hyperphosphatemia, vitamin D deficiency, and parathyroid hyperplasia. The fact that patients with advanced stage CKD have extremely high serum levels of biologically active FGF23 has raised the possibility that the elevated serum FGF23 levels may activate FGF signaling in tissues that do not innately express Klotho, which may then contribute to systemic complications in ESRD.

Changes in phosphate-regulating factors during CKD progression. Cited from [26]
It has been reported that calcimimetics decrease plasma FGF23 concentration in hemodialysis patients, and the decrease in plasma FGF23 concentration seems to be related to the decrease in serum phosphate concentration [27]. Chertow et al. reported the results of the Evaluation of Cinacalcet Therapy to Lower Cardiovascular Events (EVOLVE) trial in 3883 patients receiving long-term hemodialysis treatment [28]. In an unadjusted intention-to-treat (ITT) analysis, cinacalcet had no significant benefit on the primary composite outcome (death or first non-fatal myocardial infarction, hospitalization for unstable angina, heart failure, or peripheral vascular events) but significantly reduced the incidence of heart failure. In an adjusted ITT analysis and lag-censoring analysis, the effect of cinacalcet on the primary outcome was significant. ITT analyses adjusted for baseline characteristics suggest that treating secondary hyperparathyroidism with cinacalcet results in significant reduction in the risk of death or cardiovascular events.
Role of FGF23 in CV disease
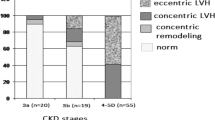
Serum FGF23 levels were independently associated with left ventricular mass index and the risk of new-onset LVH in the Chronic Renal Insufficiency Cohort (CRIC) study [29]. Negishi et al. reported that FGF23 was significantly associated with LVH, and they suggested that FGF23 could be a novel biomarker of left ventricular overload, which is closely associated with an increased risk of death in hemodialysis patients [30]. We demonstrated that hemodialysis patients with LVH were more likely to have higher systolic blood pressure and LVH was significantly associated with female gender and higher serum phosphate levels in addition to serum FGF23 levels (Fig. 4) [31]. An experimental study by Faul et al. demonstrated that FGF23 induced rat cardiomyocyte hypertrophy through FGFR-dependent activation of the calcineurin-NFAT signaling pathway in a Klotho-independent manner [32]. FGFR-4 is now considered the specific FGFR directly involved in the Klotho-independent activity of FGF23 in the myocardium [33]. FGF23 activates only FGFR-4 in cardiomyocytes, thereafter stimulating PLC-γ-calineurin-NFAT cell signaling [34]. Knock-out mice lacking FGFR-4 did not develop LVH in response to high FGF23 levels, while gain-of-function transgenic mutants of FGFR-4 spontaneously led to LVH [35]. Leifhelt-Nestler et al. recently reported that FGF23/FGFR4-mediated LVH is reversible [36]. Moreover, data from 24 deceased patients with childhood-onset ESRD provides evidence that expression of FGFR-4 and activation of the PLC-γ-calcineurin-NFAT pathway were associated with cardiac expression of FGF23 in situ [36]. Reduced expression of Klotho in the same cardiac tissues supports the notion that high FGF23 levels concomitant with Klotho downregulation may favor activation of Klotho-independent pathways that lead to LVH. Thus, high serum FGF23 levels exert toxic effects via Klotho-independent pathways in myocardium. However, a potential effect of LVH on FGF23 remains unclear. Matsui et al. recently evaluated the effect of LVH on FGF23 using cardiomyocyte-specific calcineurin A transgenic mice and showed severe LVH with elevated serum FGF23 levels [37]. The FGF23 levels were elevated in cardio myocytes, but not osteocytes, suggesting that FGF23 and LVH might be linked in a vicious cycle, particularly in the setting kidney disease: LVH itself could increase FGF23, which, in turn, cause LVH progression.

Serum fibroblast growth factor 23 (FGF23) level in hemodialysis patients with and without left ventricular hypertrophy (LVH). Cited from [26]
FGF23 has been associated with abdominal aortic calcification in healthy older men [38]. In a study of 545 African Americans with diabetes, Freedman et al. found an independent association of FGF23 with coronary artery-calcified atherosclerotic plaque, but not with carotid and aortoiliac-calcified plaque [39]. Among 142 patients in CKD stages 2 through 5D, intact FGF23 was elevated and independently associated with aortic calcification score, as assessed by multislice, spiral-computed tomography (CT) [40]. Morena et al. described a significant correlation between FGF23 expression and coronary artery calcification among 195 non-dialysis CKD patients [41]. Due to the considerable interaction between FGF23 and osteoprotegerin (OPG), these authors suggested that FGF23 expression represented a biomarker for severe vascular calcification, whereas OPG expression was associated with moderate coronary calcification [41]. More recently, Di Lullo et al. demonstrated an independent association between FGF23 expression and the extent of aortic valve calcification among 100 consecutive non-hospitalized CKD stage 3–4 patients [42].
Aortic calcification index, which is assessed on non-contrast CT, was independently associated with serum levels of intact FGF23 in 65 hemodialysis patients [43]. Although coronary artery and thoracic aortic calcium concentrations were not associated with serum FGF23 levels among 1501 patients from the CRIC study, secondary analysis revealed an association between serum FGF23 levels and thoracic aorta calcium concentrations among CKD patients with non-zero calcification scores [44]. This result was statistically significant, despite the significant association detected between serum phosphate levels and coronary calcium concentrations. The authors concluded that these types of studies are highly sensitive to the specific definition of vascular calcification, the site of interest, the sample size, and the proportion of subjects with a calcification score of zero [44].
FGF23 may even be associated with progression of vascular calcification, although there are discrepancies in these results. Khan et al. assessed coronary arterial calcification by electrocardiography-triggered multislice CT in 99 incident hemodialysis patients [45]. FGF23 was associated with progression of coronary calcification independently of serum phosphate levels and known risk factors [45]. Among 74 hemodialysis patients, progression of coronary artery calcification, assessed twice by scoring with a 1-year interval, was associated with serum levels of FGF23 and phosphate, as well as baseline coronary calcium scores [46]. Conversely, we reported higher FGF23 levels in hemodialysis patients with regression, rather than progression, of aortic arch calcification, at a 5-year follow-up, as assessed by simple chest radiographs [47].
Prognostic value of FGF23 in CKD patients
Previous studies suggest that elevated serum FGF23 level is strongly associated with increased mortality risk across the spectrum of CKD [48,49,50]. We have recently reported that serum FGF23 level was not associated with increased mortality risk in our cohort of prevalent hemodialysis patients [51], suggesting that the impact of FGF23 on mortality may be modified by gender and previous CV disease and is blunted by the grade of hyperphosphatemia in ESRD patients. However, the overall prognostic utility of FGF23 remains unclear. Previous observational studies have shown conflicting results. To resolve this uncertainty regarding FGF23 as a prognostic tool in patients with ESRD, Yang et al. conducted a systematic review to quantify the association between elevated FGF23 and long-term mortality among ESRD patients. They showed that elevated serum FGF23 levels were significantly associated with higher risk of all-cause mortality in ESRD patients, indicating FGF23 predicts poor prognosis in these patients [52]. The mechanism of the association between elevated serum FGF23 levels and mortality, however, remained unclear. Possible mechanisms may include that FGF23 suppresses vitamin D metabolism, stimulates the renin–angiotensin– aldosterone system, and increases production of inflammatory mediators. In addition, elevated circulating FGF23 concentration contributes directly to the high prevalence of LVH in CKD as mentioned above. These factors have shown a strong association with adverse outcomes in patients with CKD and ESRD.
FGF23 concentrations had been found to be associated with serum phosphate level, and FGF23 also leads to decreased synthesis of vitamin D as mentioned above. The effects of serum calcium, vitamin D, and PTH on serum FGF-23 concentration have not been studied. However, some medical administration including vitamin D supplementation, cinacalcet, and phosphate binders may influence serum calcium and phosphate level, further affect FGF23 concentration directly or indirectly [53]. Most of the included studies had adjusted for serum phosphate when calculating hazard ratios. However, four studies did not report medical treatment. Few studies adjusted medication. The medical treatment with phosphate binders and vitamin D may potentially be a confounding factor.
Conclusion
CV disease remains frequently complicated in CKD patients, representing the main cause of death in dialysis patients, because conventional therapies against atherosclerosis, vascular calcification, and CKD-MBD seem to provide no survival benefit to these patients. Thus, additional research is warranted to improve the understanding of vascular aging in CKD. Signaling through FGF23 and Klotho represents a critical element in the vascular consequences of CKD-MBD.
Results from clinical and experimental studies support a consistent, direct, protective effect of Klotho on LVH, atherosclerosis, and vascular calcification. However, additional research is needed to elucidate the precise localization of transmembrane Klotho expression within the heart and arterial walls, as well as the paracrine and endocrine effects of secreted Klotho on the vascular system. Concomitant reduction of Klotho and FGFR expression may be responsible for the onset and progression of (1) CKD-MBD via FGF23 resistance and (2) LVH, atherosclerosis, and vascular calcification resulting from impaired endogenous protection against vascular injury.
The classic Klotho-dependent activity of FGF23 on the kidney and parathyroid glands is accepted as a fundamental pathway for the maintenance of phosphate homeostasis and protection against the onset of CKD-MBD. However, many of the findings are conflicting. For example, experimental data and analysis of families with FGF23 deficiency confirm that defects in FGF23 signaling induce vascular damage with eventual critical impact. These data conflict with observational studies that report a direct association between high serum FGF23 levels and CV disease, suggesting a role for FGF23 as a biomarker of advanced CKD-MBD rather than a direct causative agent of vascular aging. As a second example, experimental data seem to suggest that LVH results from a direct Klotho-independent response to high serum FGF23 levels in cardiomyocytes. Although the direct activity exerted by FGF23 on the arteries is supported by several studies, data regarding whether this effect is protective or detrimental remain inconsistent.
Future perspectives
Future research in this field will shed light on several topics, such as Klotho-independent pathways activated by FGF23, and the precise and dynamic localization of Klotho-FGFR-FGF23 expression within the heart and arterial tissue. These studies should be designed to overcome several weaknesses in the literature, including (i) lack of information on gender, ethnicity, and genetic characteristics of human VSMC donors, which may interact with FGF23 on vascular calcification [54], (ii) frequent barriers encountered while dealing with translation from animal to human models and from in vitro to dynamic in vivo models, (iii) the specific characteristics and limitations of laboratory methods adopted for assessing Klotho and FGF23 levels, and (iv) the challenging reproduction and control of systemic conditions, such as inflammation, which may influence Klotho and FGF23 activity significantly in vivo.
Abbreviations
- 1, 25(OH)2D:
-
1, 25 Dihydroxyvitamin D
- CKD:
-
Chronic kidney disease
- CKD-MBD:
-
Chronic kidney disease-mineral and bone disorder
- CRIC:
-
Chronic Renal Insufficiency Cohort
- CV:
-
Cardiovascular
- ERK:
-
Extracellular signal-regulated kinase
- ESRD:
-
End-stage renal disease
- FGF23:
-
Fibroblast growth factor 23
- FGFR:
-
Fibroblast growth factor receptor
- LVH:
-
Left ventricular hypertrophy
- MAPK:
-
Mitogen-activated protein kinase
- NFAT:
-
Calcineurin-nuclear factor of activated T cells
- OPG:
-
Osteoprotegerin
- PiT:
-
Sodium-dependent phosphate co-transporter
- PLC:
-
Phospholipase C
- PTH:
-
Parathyroid hormone
- VSMC:
-
Vascular smooth muscle cell
References
Masakane I, Taniguchi M, Nakai S, Tsuchida K, Goto S, Wada A, Ogata S, Hasegawa T, Hamano T, Hanafusa N, Hoshino J, Minakuchi J, Nakamoto H. Annual Dialysis data report 2015, JSDT renal data registry (JRDR). Ren Replace Ther. 2018;4:19.
Tanaka K, Watanabe T, Takeuchi A, Ohashi Y, Nitta K, Akizawa T, Matsuo S, Imai E, Makino H, Hishida A, CKD-JAC Investigators. Cardiovascular events and death in Japanese patients with chronic kidney disease. Kidney Int. 2017;91:227–34.
Collins AJ, Foley RN, Gilbertson DT, Chen SC. United States Renal Data System public health surveillance of chronic kidney disease and end-stage renal disease. Kidney Int Suppl. 2011;5:2–7.
Briet M, Burns KD. Chronic kidney disease and vascular remodelling: molecular mechanisms and clinical implications. Clin Sci. 2012;123:399–416.
Rhee CM, Kovesdy CP. Epidemiology: spotlight on CKD deaths increasing ortality worldwide. Nat Rev Nephrol. 2015;11:199–200.
Ketteler M, Elder GJ, Evenepoel P, Ix JH, Jamal SA, Lafage-Proust MH, Shroff R, Thadhani RI, Tonelli MA, Kasiske BL, Wheeler DC, Leonard MB. Revising KDIGO clinical practice guideline on chronic kidney disease-mineral and bone disorder: a commentary from a Kidney Disease: improving Global Outcomes controversies conference. Kidney Int. 2015;87:502–28.
Nitta K. Vascular calcification in patients with chronic kidney disease. Ther Apher Dial. 2011;15:513–21.
Hruska KA, Sugatani T, Agapova O, Fang Y. The chronic kidney disease – mineral bone disorder (CKD-MBD): advances in pathophysiology. Bone. 2017;100:80–6.
Bover J, Evenepoel P, Urena-Torres P, Vervloet MG, Brandenburg V, Mazzaferro S, Covic A, Goldsmith D, Massy ZA, Cozzolino M, CKD-MBD Working Group of ERA-EDTA. Pro: cardiovascular calcifications are clinically relevant. Nephrol Dial Transplant. 2015;30:345–51.
Nitta K, Nagano N, Tsuchiya K. Fibroblast growth factor 23/klotho axis in chronic kidney disease. Nephron Clin Pract. 2014;128:1–10.
Raggi P. Cardiovascular disease: coronary artery calcification predicts risk of CVD in patients with CKD. Nat Rev Nephrol. 2017;13:324–6.
Shroff R. Phosphate is a vascular toxin. Pediatr Nephrol. 2013;28:583–93.
Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res. 2011;109:697–711.
Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ, Proudfoot D, Jahnen-Dechent W, Weissberg PL, Shanahan CM. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. 2004;15:2857–67.
Li X, Yang HY, Giachelli CM. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ Res. 2006;98:905–12.
Paloian NJ, Giachelli CM. A current understanding of vascular calcification in CKD. Am J Physiol Renal Physiol. 2014;307:F8915–F900.
Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113:561–8.
Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev. 2012;92:131–55.
Mace ML, Gravesen E, Hofman-Bang J, Olgaard K, Lewin E. Key role of the kidney in the regulation of fibroblast growth factor 23. Kidney Int. 2015;88:1304–13.
Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51.
Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro-o M. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281:6120–3.
Gutierrez O, Isakova T, Rhee E, Shah A, Holmes J, Collerone G, Juppner H, Wolf M. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol. 2005;16:2205–15.
Hasegawa H, Nagano N, Urakawa I, Yamazaki Y, Iijima K, Fujita T, Yamashita T, Fukumoto S, Shimada T. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int. 2010;78:975–80.
Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, Sirkis R, Naveh-Many T, Silver J. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;117:4003–8.
Komaba H, Goto S, Fujii H, Hamada Y, Kobayashi A, Shibuya K, Tominaga Y, Otsuki N, Nibu K, Nakagawa K, Tsugawa N, Okano T, Kitazawa R, Fukagawa M, Kita T. Depressed expression of Klotho and FGF receptor 1 in hyperplastic parathyroid glands from uremic patients. Kidney Int. 2010;77:232–8.
John GB, Cheng CY, Kuro-o M. Role of Klotho in aging, phosphate metabolism, and CKD. Am J Kidney Dis. 2011;58:127–34.
Kuczera P, Adamczak M, Wiecek A. Cinacalcet treatment decreases plasma fibroblast growth factor 23 concentration in haemodialysis patients with chronic kidney disease and secondary hyperparathyroidism. Clin Endocrinol. 2014;80:607–12.
Chertow GM, Block GA, Correa-Rotter R, Drueke TB, Floege J, Goodman WG, Herzog CA, Kubo Y, London GM, Mahaffey KW, Mix TC, Moe SM, Trotman ML, Wheeler DC, Parfrey PS. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N Engl J Med. 2012;367:2482–94.
Scialla JJ, Xie H, Rahman M, Anderson AH, Isakova T, Ojo A, Zhang X, Nessel L, Hamano T, Grunwald JE, Raj DS, Yang W, He J, Lash JP, Go AS, Kusek JW, Feldman H, Wolf M, Chronic Renal Insufficiency Cohort (CRIC) Study Investigators. Fibroblast growth factor-23 and cardiovascular events in CKD. J Am Soc Nephrol. 2014;25:349–60.
Negishi K, Kobayashi M, Ochiai I, Yamazaki Y, Hasegawa H, Yamashita T, Shimizu T, Kasama S, Kurabayashi M. Association between fibroblast growth factor 23 and left ventricular hypertrophy in maintenance hemodialysis patients. Comparison with B-type natriuretic peptide and cardiac troponin T. Circ J. 2010;74:2734–40.
Saito A, Onuki T, Echida Y, Otsubo S, Nitta K. Fibroblast growth factor 23 and left ventricular hypertrophy in hemodialysis patients. Int J Clin Med. 2014;5:1102–10.
Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121(11):4393–408.
Juppner H, Wolf M. Klotho: FGF23 coreceptor and FGF23-regulating hormone. J Clin Invest. 2012;122:4336–9.
Grabner A, Amaral AP, Schramm K, Singh S, Sloan A, et al. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab. 2015;22(6):1020–32.
Grabner A, Schramm K, Silswal N, Hendrix M, Yanucil C, Czaya B, Singh S, Wolf M, Hermann S, Stypmann J, Di Marco GS, Brand M, Wacker MJ, Faul C. FGF23/FGFR4-mediated left ventricular hypertrophy is reversible. Sci Rep. 2017;7:1993.
Leifheit-Nestler M, Grose Siemer R, Flasbart K, Richter B, Kirchhoff F, et al. Induction of cardiac FGF23/FGFR4 expression is associated with left ventricular hypertrophy in patients with chronic kidney disease. Nephrol Dial Transplant. 2016;31:1088–99.
Matsui I, Oka T, Kusunoki Y, Mori D, Hashimoto N, Matsumoto A, Shimada K, Yamaguchi S, Kubota K, Yonemoto S, Higo T, Sakaguchi Y, Takabatake Y, Hamano T, Isaka Y. Cardiac hypertrophy elevates serum levels of fibroblast growth factor 23. Kidney Int. 2018;94(1):60–71.
Schoppet M, Hofbauer LC, Brinskelle-Schmal N, Varennes A, Goudable J, Richard M, Hawa G, Chapurlat R, Szuic P. Serum level of the phosphaturic factor FGF23 is associated with abdominal aortic calcification in men: the STRAMBO study. J Clin Endocrinol Metab. 2012;97:575–983.
Freedman BI, Divers J, Russell GB, Palmer ND, Bowden DW, Carr JJ, Wagenknecht LE, Hightower RC, Xu J, Smith SC, Langefeld CD, Hruska KA, Register TC. Plasma FGF23 and calcified atherosclerotic plaque in African Americans with type 2 diabetes mellitus. Am J Nephrol. 2015;42:391–401.
Desjardins L, Liabeuf S, Renard C, Lenglet A, Lemke HD, Choukroun G, Drueke TB, Massy ZA, European Uremic Toxin (EUTox) Work Group. FGF23 is independently associated with vascular calcification but not bone mineral density in patients at various CKD stages. Osteoporos Int. 2012;23:2017–25.
Morena M, Jaussent I, Halkovich A, Dupuy AM, Bargnoux AS, Chenine L, Leray-Moragues H, Klouche K, Vernhet H, Canaud B, Cristol JP. Bone biomarkers help grading severity of coronary calcifications in non dialysis chronic kidney disease patients. PLoS One. 2012;7:e36175.
Di Lullo L, Gorini A, Bellasi A, Morrone LF, Rivera R, Russo L, Santoboni A, Russo D. Fibroblast growth factor 23 and parathyroid hormone predict extent of aortic valve calcifications in patients with mild to moderate chronic kidney disease. Clin Kidney J. 2015;8:732–6.
Nasrallah MM, El-Shehaby AR, Salem MM, Osman NA, El Sheikh E, Sharaf El Din UA. Fibroblast growth factor-23 (FGF-23) is independently correlated to aortic calcification in haemodialysis patients. Nephrol Dial Transplant. 2010;25:2679–85.
Scialla JJ, Lau WL, Reilly MP, Isakova T, Yang HY, et al. Fibroblast growth factor 23 is not associated with and does not induce arterial calcification. Kidney Int. 2013;83:1159–68.
Khan AM, Chirinos JA, Litt H, Yang W, Rosas SE. FGF-23 and the progression of coronary arterial calcification in patients new to dialysis. Clin J Am Soc Nephrol. 2012;7(12):2017–22.
Ozkok A, Kekik C, Karahan GE, Sakaci T, Ozel A, Unsal A, Yildiz A. FGF-23 associated with the progression of coronary artery calcification in hemodialysis patients. BMC Nephrol. 2013;14:241.
Tamei N, Ogawa T, Ishida H, Ando Y, Nitta K. Serum fibroblast growth factor-23 levels and progression of aortic arch calcification in non-diabetic patients on chronic hemodialysis. J Atheroscler Thromb. 2011;18:217–23.
Gutiérrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, Smith K, Lee H, Thadhani R, Jüppner H, Wolf M. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359:584–92.
Isakova T, Xie H, Yang W, Xie D, Anderson AH, Scialla J, Wahl P, Gutiérrez OM, Steigerwalt S, He J, Schwartz S, Lo J, Ojo A, Sondheimer J, Hsu CY, Lash J, Leonard M, Kusek JW, Feldman HI, Wolf M, Chronic Renal Insufficiency Cohort (CRIC) Study Group. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA. 2011;305:2432–9.
Kendrick J, Cheung AK, Kaufman JS, Greene T, Roberts WL, Smits G, Chonchol M; HOST investigators: FGF-23 associates with death, cardiovascular events, and initiation of chronic dialysis. J Am Soc Nephrol 2011; 22: 1913–1922.
Sugimoto H, Ogawa T, Iwabuchi Y, Otsuka K, Nitta K. Relationship between serum fibroblast growth factor-23 level and mortality in chronic hemodialysis patients. Int Urol Nephrol. 2014;46:99–106.
Yang H, Luo H, Tang X, Zeng X, Yu Y, Ma L, Fu P. Prognostic value of FGF23 among patients with end-stage renal disease: a systematic review and meta-analysis. Biomarker Med. 2016;10:547–56.
Moe SM, Chertow GM, Parfrey PS, Kubo Y, Block GA, Correa-Rotter R, Drueke TB, Herzog CA, London GM, Mahaffey KW, Wheeler DC, Stolina M, Dehmel B, Goodman WG, Floege J. Cinacalcet, fibroblast growth factor-23, and cardiovascular disease in hemodialysis: the Evaluation of Cinacalcet HCl Therapy to Lower Cardiovascular Events (EVOLVE) trial. Circulation. 2015;132:27–39.
Morita H, Takeda Y, Fujita S, Okamoto Y, Sakane K, Teramoto K, Ozeki M, Tasaki R, Kizawa S, Sohmiya K, Hoshiga M, Ishizaka N. Gender specific association between serum fibroblast growth factor 23/alpha-klotho and coronary artery and aortic valve calcification. J Atheroscler Thromb. 2015;22:1338–46.
Acknowledgements
The authors acknowledge the medical staffs who contributed to this work in the Department of Medicines, Kidney Center, Tokyo Women’s Medical University.
Author’s contributions
KN searched the literature and prepared this article. The author read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The author declares no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Nitta, K. Fibroblast growth factor 23 and cardiovascular disease in patients with chronic kidney disease. Ren Replace Ther 4, 31 (2018). https://doi.org/10.1186/s41100-018-0172-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41100-018-0172-9