
Abstract
Background
Tuberculosis pleural effusion (TPE) and malignant pleural effusion (MPE) are very common clinical complications. Considering the totally different prognosis and clinical treatment of TPE and MPE, the accurate and non-invasive diagnosis are very critical for patients with pleural effusion to initiate efficient management and treatment. However, effective clinical biomarkers were rarely explored to distinguish benign from MPE. The purpose of this study is to identify potential miRNAs which can probably be used to differentiate malignant pleural effusion from TPE.
Results
A total of 23 significantly differentially expressed miRNAs were identified in MPE, with 18 up-expressed and 5 down-expressed. And the target genes of the miRNAs mainly involved in the biology process of nervous system, cancer, immune system and metabolic process etc. Three high confident target genes, AGO4, FGF9 and LEF1 can be regulated by miR-195-5p, miR-182-5p and miR-34a-5p respectively. And these genes participate in the canonical pathway of regulation of the Epithelial-Mesenchymal and the biological functions of apoptosis, growth of tumor and cell proliferation of tumor cell lines. Further, RT-PCR validation results based on 64 collected individuals showed that the expression levels of the three miRNAs were 2–5 times higher in MPE samples, which were consistent with the microarray results. In addition, ROC curve analysis demonstrated that the combination of the three miRNAs can achieve higher AUC of 0.93 (p-value< 0.0001) to differentiate MPE from TPE.
Conclusions
The identified miR-195-5p, miR-182-5p and miR-34a-5p can become potential diagnostic biomarkers for MPE with further evidences.
Similar content being viewed by others


Background
Various causes can lead to pleural effusion, and one type of effusion caused by tuberculous pleurisy was complicate and time-consuming to differentiate from the other type caused by cancer [20]. Tuberculosis caused Tuberculosis pleural effusion (TPE) is endemic especially in the developing countries [35], whereas malignant disease like lung cancer is another major factor of pleural effusion, namely malignant pleural effusion (MPE) [17, 25].
The traditional methods to diagnose TPE are based on the microbiological analyses of M.tuberculosis in pleural fluid, sputum and pleural biopsy specimens [25]. MPE is characterized by the manifestation of neoplastic cells in the pleural region, and the golden standard diagnostic approach is cytology, which can be used to confirm 30–60% MPE in advanced disease stages [29]. In addition, various biomarkers including vascular endothelial growth factor (VEGF), tumor necrosis factor (TNF), endothelin, interleukin (IL)-27, IFN-γ and IL-6 have also been used in the distinction analysis of TPE and MPE [14, 33]. Given the low diagnostic yield of pleural fluid cytology and the invasive nature of pleural biopsy, it has limited the clinical application of biochemical tests [2, 22].
Our primary objective of this study was to identify potential miRNA biomarkers which can probably be used to differentiate MPE from TPE, considering the wide difference in prognosis and clinical treatment between TPE and MPE and the limitation of biochemical testing. MicroRNAs (miRNAs) are a type of small non-coding RNA with good stableness, which can mediate RNA silencing and regulate gene expression. And some specific miRNAs have been found to be critical in the development of MPE in recent years [1, 9, 26, 27]. However, most of the previous studies were still mainly based on low-throughput technologies.
Results
Identification of differentially expressed miRNAs
A total of 23 miRNAs were identified significantly differentially expressed in MPE comparing with TPE after background correction and normalization. Among the miRNAs, 18 were high-expressed, and 5 were down-expressed (Fig. 1a). The chromosome locations of the miRNAs were annotated and shown in Fig. 1b. Meanwhile, based on the expression values of the differentially expressed miRNAs the enrolled samples can also be clearly clustered into two groups, one belongs to MPE and the other one was TPE (Fig. 2).

The identified differentially expressed miRNAs. a Volcano plot of differentially expressed miRNAs in malignant pleural effusion. b Chromosome locations of differentially expressed miRNAs

Heatmap of differentially expressed miRNAs. The group of N represents tuberculosis pleural effusion and the group of T represents malignant pleural effusion
miRNA target gene predication and functional enrichment
miRWalk2.0 analysis showed that a total of 1356 genes can be regulated by the differentially expressed miRNAs, of which three genes, LEF1, FGF9 and AGO4, were predicted to be high confident target genes because they were predicted to be miRNA target by all 12 tools. Also, these three genes participate in the canonical pathway of regulation of the Epithelial-Mesenchymal and the biological functions of apoptosis, growth of tumor and cell proliferation of tumor cell lines (Fig. 3). Functional annotation of all target genes using DAVID showed that the genes mainly involved in the biology process of nervous system, cancer, immune system and metabolic process etc. (Fig. 4).

Regulation mechanism of three high confident target genes and miRNAs, and related biological functions

Function annotation results of differentially expressed miRNAs target genes. a top 30 significantly enriched diseases. b top 30 significantly enriched pathways. c top 30 significantly enriched Gene Ontology terms
RT-PCR validation and ROC curve analysis
Further, the expression levels of the three identified key miRNAs, miR-195-5p, miR-182-5p and miR-34a-5p, were validated using 65 collected individuals. As shown in Fig. 5, the expression level of miR-195-5p was higher in MPE group (1.10 ± 0.67) than TPE group (0.35 ± 0.30). The expression level of miR-182-5p was 1.24 ± 0.76 in MPE group and 0.45 ± 0.38 in TPE group. Similarly, higher expression level of miR-34a-5p was observed in MPE (0.79 ± 0.48) than TPE (0.35 ± 0.16). And, the differences for the three miRNAs were statistically significant between the two groups.

The expression levels of three key miRNAs in 65 tuberculosis pleural effusion and malignant pleural effusion individuals
ROC curve analysis was further carried out to assess diagnostic ability of the three miRNAs. As shown in Fig. 6, the AUC values for miRNA-195-5p, miRNA-182-5p and miRNA-34a-5p were 0.87 (95% CI, 0.79 to 0.97), 0.81 (95% CI, 0.71 to 0.92) and 0.78 (95% CI, 0.67 to 0.89), respectively. While, the AUC for the combination of the three miRNAs can achieve 0.93 with 95% CI range from 0.87 to 0.98 (Fig. 6d).

Comparison of the sensitivity and specificity of the three miRNAs and combination in differentiating tuberculosis pleural effusion from malignant pleural effusion
Discussion
In this study, a total of 23 significantly DE miRNAs were identified between MPE and TPE. Further functional annotation showed that the target genes of the differentially expressed miRNAs mainly involved in the process of cancer, immune system, regulation of metabolic process, and positive regulation of biological process, all of which are associated with benign or malignant lesions. In addition, three high confident target genes, AGO4, FGF9 and LEF1, can be regulated by differentially expressed miR-195-5p, miR-182-5p and miR-34a-5p respectively. Also, the genes participate in the canonical pathway of regulation of the Epithelial-Mesenchymal and the biological functions of apoptosis, growth of tumor and cell proliferation of tumor cell lines. RT-PCR validation and ROC curve analysis results further demonstrated that the combination of the three differentially expressed miRNAs could become good potential non-invasive biomarkers for the diagnosis of MPE, with AUC as high as 0.93.
The expression level of miR-195-5p was significantly up-expressed in MPE than TPE. It has been demonstrated that miR-195-5p could result in a 20% decrease in VEGF expression via VEGF 3′-UTR [34] and downregulate the protein level of HSP90B1 [5]. VEGF can be released by both malignant and inflammatory cells, and various studies indicated that VEGF is associated with survival and pleurodesis outcomes in MPE [6, 13]. Both VEGF and HSP90B1 are critical in the development of MPE by regulating angiogenesis [36]. VEGF has also been used as drug target of Bevacizumab and Cisplatin for the treatment of MPE in advanced stage non-small-cell lung cancer from one clinical trial by Chinese PLA General Hospital (NCT01661790). In addition, Noriko Hirayama’s study showed that the dysregulation of VEGF could be useful for the diagnosis of malignant pleural mesothelioma [12]. Another target gene of miR-195-5p AGO4, as an microRNA machinery gene, was demonstrated to be upregulated in ovarian carcinogenesis and apoptosis process [15]. In addition, miR-195-5p has been widely identified as tumor suppressor in osteosarcoma, prostate cancer, and bladder cancer [4, 7, 23].
miR-182-5p which has been reported to be an oncogene in several cancers was also highly up-regulated in MPE. The dysregulation of miR-182-5p in MPE probably participated in the development of different cancers by regulating the downstream genes. In-vitro studies showed that the knock-down of miR-182-5p in prostate cancer cell can promote cell proliferation, migration and invasion by regulating the potential targets of FOXF2, RECK and MTSS1 [10]. While in bladder cancer miR-182-5p can regulate the expression levels of Smad4 and RECK, and high-expression of miR-182-5p can significantly shorten the overall survival of bladder cancer patients [11]. By regulate the expression of FGF9, miR-182-5p can probably induce malignant lesions by inducing Epithelial-Mesenchymal, cell proliferation and growth of tumor cells, as various studies have identified FGF9 participate in the tumor development such as gastric cancer [28], hepatocellular carcinoma [32], NSCLC [21] and so on. All these results indicated that as an important upstream regulator, miR-182-5p participate in the biology functions related to malignant lesions development. It will significantly simplify the testing by detecting the expression of miRNA rather than tens of gene expression in the diagnosis of malignant lesions development.
Another important miRNA identified in this study was over-expressed miR-34a-5p in MPE group which showed a malignant tendency in lesions. This statement was supported by some previous publications [22–24]. In non-small cell lung cancer, high-expression of miR-34a-5p can induce cell apoptosis by targeting MDM4, and increase p53 and p21 protein expressions [16]. Also, p53 can indirectly regulates Fra-1 expression via miR-34a, and eventually inhibit cell migration, invasion in colon cancer [30]. In addition, low-expression of miR-34a-5p is correlated with high probability of relapse (p = 0.04) and can be used as a novel prognostic marker in NSCLC patients [8]. The oncogenic transcription factor LEF1 as the target gene of miR-34a-5p can also promote malignant lesions via Wnt signal pathway in various cancers [18, 19]. Via the canonical pathways such p53 and Wnt signal pathway, miR-34a-5p can regulate the development of malignant lesions. Each of the three miRNAs have a high accuracy, but with the miRNA panel can achieve higher AUC of 0.93 (p-value< 0.0001) to differentiate TPE from MPE.In summary, three potential miRNA biomarkers in differentiating TPE from MPE were identified based on miRNA microarray, RT-PCR validation and ROC curve analysis. The miRNA panel can provide an easy and non-invasive method for the diagnosis of MPE. But the results were still preliminary due to small sample size and lack of other experiments validation such as western blot.
Methods
Sample collection
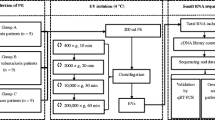
Six patients were enrolled for miRNA microarray analysis in this study between Sep. 30, 2017 and May. 3, 2018 from the department of Respiratory Medicine, Jiangsu University Hospital. Among these patients, three were diagnosed as TPE, and the rest were diagnosed as lung adenocarcinoma classified as MPE. Detailed clinical characterslike age, gender, cytology, pleural effusion protein, pleural LDH, pleural ADA and pleural glucose antigen were listed in Table 1. The present study was approved by the Ethics Committee of Jiangsu University Hospital, and informed consent was obtained from everyone involved in this study.
RNA extraction and miRNA microarray experiment
At least 50 mL of pleural fluid was collected from each patient during thoracentesis using a sterile syringe. Total RNA was extracted using the RNeasy Mini Kit (QIAGEN, Valencia, CA) according to the manufacturer’s instructions. Then RNA samples were quantified by Qubit (Thermo Fisher Scientific Inc., Wilmington, DE) and checked for integration by Agilent Bioanalyzer 2100 (Agilent Technologies Inc., Santa Clara, CA). The amplified miRNAs were hybrid to GeneChip® miRNA 4.0 array, which containing 30,424 total mature miRNA probe sets including 2578 from human and the rest from 202 other organisms. In brief, 245 ng of total RNA was labeled with Biotin using the 3DNA Array Detection FlashTag™ Biotin HSR kit (Genisphere, Hatfield, PA, U.S.) and subsequently hybridized overnight. After washing and staining processes, the GeneChip® miRNA 4.0 arrays were scanned with the Affymetrix GeneChip Scanner 3000 7G (Affymetrix, Santa Clara, CA, U.S.). Finally, the intensity was calculated and collected using AGCC software.
Identification of differentially expressed miRNAs
To identify the differentially expressed miRNAs, we adopted our internal optimized R script by integrating affy, Limma, Biobase etc. In general, the intensity of each probe and background probes for these samples were compared using boxplot. And the scale factors were retrieved to equalize the mean intensities of these arrays. Then, GCRMA (GeneChip Robust Multi-array Analysis) which is based on RMA were used to normalize the probe intensities by incorporating non-specific binding information [31]. Also, uninformative or duplicated probes were processed to better calculate expression levels. Finally, Limma (Linear Models for Microarray Analysis) package was used to identify differentially expressed miRNAs using t-test [24]. In this study, the cutoff for differentially expressed miRNAs were set to adjusted p-value ≤0.05 and |log2 fold change| ≥ 1.
miRNA target gene predication and function enrichment
To explore the potential biological functions of differentially expressed miRNAs, the target genes were predicted using widely adopted miRWalk2.0 software. The software integrates 12 different predication tools to improve the predication accuracy, including miRWalk, DIANA-microTv4.0, miRanda-rel2010, mirBridge, miRDB4.0, miRmap, miRNAMap, PicTar2, PITA, FNA22v2, RNAhybrid2.1 and TargetScan6.2. Here a plausible target gene was defined as at least six tools predicted to be target of differentially expressed miRNAs. Then the GO and function enrichment were carried out using online tool DAVID (Database for Annotation, Visualization and Integrated Discovery) and Ingenuity Pathway Analysis (QIAGEN, Redwood City, CA, USA). Significantly enriched diseases, pathways and GO functions were screened out with p-value less than 0.05.
RT-PCR validation and ROC curve analysis
To validate the results, the expression levels of three key miRNAs weremeasured in 65 collected individuals using RT-PCR (Additional file 1). Among these individuals, 24 were classified as TPE and 41 were classified as MPE. Similarly, total RNA was extracted, and reverse transcribed to cDNA using cDNA synthesis kit (QIAGEN, Valencia, CA). The experiment was carried out on Bio-Rad CFX384 Real-time PCR system with primers designed using miRprimer2 software [3]. And the expression levels were normalized using internal control miR-16 and processed using 2-Δct method.
To assess diagnostic ability of the three key miRNAs for differentiating between TPE and MPE, the ROC (Receiver-Operator Characteristic) and AUC (Area Under Curve) were calculated using GraphPad Prism 8 (GraphPad Software, La Jolla, CA, USA). The combinations of miRNAs as predictor was analyzed using logistic regression algorithm.
Availability of data and materials
All analyzed data related to this paper are included in this paper and additional file.
Abbreviations
- AUC:
-
Area under curve
- DAVID:
-
Database for annotation, visualization and integrated discovery
- GCRMA:
-
GeneChip robust multi-array analysis
- Limma:
-
Linear models for microarray analysis
- MPE:
-
Malignant pleural effusion
- ROC:
-
Receiver-operator characteristic
- TB:
-
Tuberculosis
- TNF:
-
Tumor necrosis factor
- TPE:
-
Tuberculosis pleural effusion
- VEGF:
-
Vascular endothelial growth factor
References
Ak G, Tomaszek SC, Kosari F, Metintas M, Jett JR, Metintas S, et al. MicroRNA and mRNA features of malignant pleural mesothelioma and benign asbestos-related pleural effusion. Biomed Res Int. 2015;2015:635748.
American Thoracic S. Management of malignant pleural effusions. Am J Respir Crit Care Med. 2000;162:1987–2001.
Busk PK. A tool for design of primers for microRNA-specific quantitative RT-qPCR. BMC Bioinformatics. 2014;15:29.
Cai C, Chen QB, Han ZD, Zhang YQ, He HC, Chen JH, et al. miR-195 inhibits tumor progression by targeting RPS6KB1 in human prostate cancer. Clin Cancer Res. 2015;21:4922–34.
Calin GA, Cimmino A, Fabbri M, Ferracin M, Wojcik SE, Shimizu M, et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci U S A. 2008;105:5166–71.
Cheng D, Kong H, Li Y. Prognostic values of VEGF and IL-8 in malignant pleural effusion in patients with lung cancer. Biomarkers. 2013;18:386–90.
Fei X, Qi M, Wu B, Song Y, Wang Y, Li T. MicroRNA-195-5p suppresses glucose uptake and proliferation of human bladder cancer T24 cells by regulating GLUT3 expression. FEBS Lett. 2012;586:392–7.
Gallardo E, Navarro A, Vinolas N, Marrades RM, Diaz T, Gel B, et al. miR-34a as a prognostic marker of relapse in surgically resected non-small-cell lung cancer. Carcinogenesis. 2009;30:1903–9.
Guan P, Yin Z, Li X, Wu W, Zhou B. Meta-analysis of human lung cancer microRNA expression profiling studies comparing cancer tissues with normal tissues. J Exp Clin Cancer Res. 2012;31:54.
Hirata H, Ueno K, Shahryari V, Deng G, Tanaka Y, Tabatabai ZL, et al. MicroRNA-182-5p promotes cell invasion and proliferation by down regulating FOXF2, RECK and MTSS1 genes in human prostate cancer. PLoS One. 2013;8:e55502.
Hirata H, Ueno K, Shahryari V, Tanaka Y, Tabatabai ZL, Hinoda Y, et al. Oncogenic miRNA-182-5p targets Smad4 and RECK in human bladder cancer. PLoS One. 2012;7:e51056.
Hirayama N, Tabata C, Tabata R, Maeda R, Yasumitsu A, Yamada S, et al. Pleural effusion VEGF levels as a prognostic factor of malignant pleural mesothelioma. Respir Med. 2011;105:137–42.
Hooper CE, Elvers KT, Welsh GI, Millar AB, Maskell NA. VEGF and sVEGFR-1 in malignant pleural effusions: association with survival and pleurodesis outcomes. Lung Cancer. 2012;77:443–9.
Hu Y, Hu MM, Shi GL, Han Y, Li BL. Imbalance between vascular endothelial growth factor and endostatin correlates with the prognosis of operable non-small cell lung cancer. Eur J Surg Oncol. 2014;40:1136–42.
Jeong W, Bae H, Lim W, Song G. Dicer1, AGO3, and AGO4 microRNA machinery genes are differentially expressed in developing female reproductive organs and overexpressed in cancerous ovaries of chickens. J Anim Sci. 2017;95:4857–68.
Jiang ZQ, Li MH, Qin YM, Jiang HY, Zhang X, Wu MH. Luteolin inhibits tumorigenesis and induces apoptosis of non-small cell lung cancer cells via regulation of MicroRNA-34a-5p. Int J Mol Sci. 2018;19:447–65.
Li M, Wang H, Wang X, Huang J, Wang J, Xi X. Diagnostic accuracy of tumor necrosis factor-alpha, interferon-gamma, interleukin-10 and adenosine deaminase 2 in differential diagnosis between tuberculous pleural effusion and malignant pleural effusion. J Cardiothorac Surg. 2014;9:118.
Li TW, Ting JH, Yokoyama NN, Bernstein A, van de Wetering M, Waterman ML. Wnt activation and alternative promoter repression of LEF1 in colon cancer. Mol Cell Biol. 2006;26:5284–99.
Liu Y, Yan W, Zhang W, Chen L, You G, Bao Z, et al. MiR-218 reverses high invasiveness of glioblastoma cells by targeting the oncogenic transcription factor LEF1. Oncol Rep. 2012;28:1013–21.
Liu YC, Shin-Jung Lee S, Chen YS, Tu HZ, Chen BC, Huang TS. Differential diagnosis of tuberculous and malignant pleurisy using pleural fluid adenosine deaminase and interferon gamma in Taiwan. J Microbiol Immunol Infect. 2011;44:88–94.
Marek L, Ware KE, Fritzsche A, Hercule P, Helton WR, Smith JE, et al. Fibroblast growth factor (FGF) and FGF receptor-mediated autocrine signaling in non-small-cell lung cancer cells. Mol Pharmacol. 2009;75:196–207.
Ong KC, Indumathi V, Poh WT, Ong YY. The diagnostic yield of pleural fluid cytology in malignant pleural effusions. Singap Med J. 2000;41:19–23.
Qu Q, Chu X, Wang P. MicroRNA-195-5p suppresses osteosarcoma cell proliferation and invasion by suppressing naked cuticle homolog 1. Cell Biol Int. 2017;41:287–95.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
Tang Y, Zhang J, Huang H, He X, Zhang J, Ou M, et al. Pleural IFN-gamma release assay combined with biomarkers distinguished effectively tuberculosis from malignant pleural effusion. BMC Infect Dis. 2019;19:55.
Ulivi P, Zoli W. miRNAs as non-invasive biomarkers for lung cancer diagnosis. Molecules. 2014;19:8220–37.
Vosa U, Vooder T, Kolde R, Vilo J, Metspalu A, Annilo T. Meta-analysis of microRNA expression in lung cancer. Int J Cancer. 2013;132:2884–93.
Wang R, Sun Y, Yu W, Yan Y, Qiao M, Jiang R, et al. Downregulation of miRNA-214 in cancer-associated fibroblasts contributes to migration and invasion of gastric cancer cells through targeting FGF9 and inducing EMT. J Exp Clin Cancer Res. 2019;38:20.
Wang Y, Xu YM, Zou YQ, Lin J, Huang B, Liu J, et al. Identification of differential expressed PE exosomal miRNA in lung adenocarcinoma, tuberculosis, and other benign lesions. Medicine (Baltimore). 2017;96:e8361.
Wu J, Wu G, Lv L, Ren YF, Zhang XJ, Xue YF, et al. MicroRNA-34a inhibits migration and invasion of colon cancer cells via targeting to Fra-1. Carcinogenesis. 2012;33:519–28.
Wu Z, Irizarry RA. Preprocessing of oligonucleotide array data. Nat Biotechnol. 2004;22:656–8 author reply 658.
Yang H, Fang F, Chang R, Yang L. MicroRNA-140-5p suppresses tumor growth and metastasis by targeting transforming growth factor beta receptor 1 and fibroblast growth factor 9 in hepatocellular carcinoma. Hepatology. 2013;58:205–17.
Yang WB, Liang QL, Ye ZJ, Niu CM, Ma WL, Xiong XZ, et al. Cell origins and diagnostic accuracy of interleukin 27 in pleural effusions. PLoS One. 2012;7:e40450.
Ye W, Lv Q, Wong CK, Hu S, Fu C, Hua Z, et al. The effect of central loops in miRNA:MRE duplexes on the efficiency of miRNA-mediated gene regulation. PLoS One. 2008;3:e1719.
Zhai K, Lu Y, Shi HZ. Tuberculous pleural effusion. J Thorac Dis. 2016;8:E486–94.
Zhang Y, Yu LK, Lu GJ, Xia N, Xie HY, Hu W, et al. Prognostic values of VEGF and endostatin with malignant pleural effusions in patients with lung cancer. Asian Pac J Cancer Prev. 2014;15:8435–40.
Acknowledgements
Thanks all for the contribution to the data analysis and manuscript preparation.
Funding
No.
Author information
Authors and Affiliations
Contributions
All authors participate in data collection, data analysis and manuscript preparation. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All the authors have consented for the publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
The detailed clinical characteritics of the validation group samples.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bao, Q., Xu, Y., Ding, M. et al. Identification of differentially expressed miRNAs in differentiating benign from malignant pleural effusion. Hereditas 157, 6 (2020). https://doi.org/10.1186/s41065-020-00119-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41065-020-00119-z