
Abstract
Background
Chromatin accessibility is crucial for gene expression regulation in specific cells and in multiple biological processes. Assay for Transposase Accessible Chromatin with high-throughput sequencing (ATAC-seq) is an effective way to reveal chromatin accessibility at a genome-wide level. Through ATAC-seq, produced reads from a small number of cells reflect accessible regions that correspond to nucleosome positioning and transcription factor binding sites, due to probing hyperactive Tn5 transposase to DNA sequence.
Conclusion
In this review, we summarize both principle and features of ATAC-seq, highlight its applications in basic and clinical research. ATAC-seq has generated comprehensive chromatin accessible maps, and is becoming a powerful tool to understand dynamic gene expression regulation in stem cells, early embryos and tumors.
Similar content being viewed by others


Background
In eukaryotic cells, chromatin is a basic hereditary unit, which consists of DNA, histone proteins and other genetic materials, and regulates cell type-specific gene expression [1, 2]. Chromatin, as a dynamic nuclear structure, is transcriptionally active in the interphase, and is relatively inactive in the metaphase in a cell cycle [3]. Regulation of transcription is a dynamic interaction between chromatin structure and recruitment of numerous transcription factors to the enhancers, upstream activator sequences, and proximal promoter elements. These transcription factors recruit RNA polymerase to the core promoter for productive transcription [4].
In general, the regulatory elements selectively localize in the accessible chromatin, which is crucial to transcriptional regulation [5]. Although transcription factor occupancy is not necessarily positively correlated with chromatin accessibility [6], the maintenance of accessible chromatin configurations requires binding of transcription factors to activate target genes [7] (Fig. 1a). On the other hand, condensed chromatin, known as closed chromatin, restricts binding of transcription factors and transcriptional regulators to the promoter and/or enhancer, which results in gene silencing [8,9,10] (Fig. 1a). Moreover, chromatin accessibility is a substantial part of epigenetic regulation, which is marked by DNA methylation and histone modification [11]. Environmental pollution factors, such as polycyclic aromatic hydrocarbons (PAHs), can affect DNA methylation [12, 13]. Therefore, chromatin accessibility, which can be modified by some environmental and pathogenic factors, indicates positions of nucleosome and regulatory regions such as enhancers, reflects precise regulations of cell behaviors, and implies dynamic physiological processes or disease conditions [14,15,16,17,18,19].

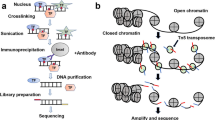
The mechanism of identifying chromatin accessibility using the Tn5 transposase. a Open and closed status of chromatin. b When the chromatin accessibility is increased, the Tn5 transposase transpose in the open chromatin more often than in the inaccessible chromatin. Then the Tn5 transposase cuts the open chromatin and tags the adaptors to it to generate DNA fragments. The green symbol represents “adapter 1” of the Tn5 transposase, while the red symbol represents “adapter 2” of theTn5 transposase
Changes of the chromatin structure occur at specific ribozyme accessibility sites that are associated with transcriptional initiation or some specific DNA structures such as specific hypersensitive sites [20]. These sites in DNA double strands can be digested by DNA enzyme I (DNase I), which reflects the accessibility of chromatin [21]. The hypersensitive sites, mostly in the promoter region, are related to gene expression [22]. To reveal accessible chromatin regions in real time and at a genome-wide level, a method named Assay for Transposase Accessible Chromatin with high-throughput sequencing (ATAC-seq) was developed and quickly applied in various studies of gene expression. ATAC-seq utilizes the Tn5 transposase and the transposable DNA as adapters, which allows the adapter introduced into the accessible chromatin [19].
Here we summarize the principle of the ATAC-seq method, highlight its usage in understanding basic transcription programs in specific cell types of humans and mice, and in revealing genetic reasoning of human diseases.
Summary of the ATAC-seq method
Principle and procedures of ATAC-seq
ATAC-seq is an innovative epigenetic technology, which is a method for mapping chromatin accessibility by probing hyperactive Tn5 transposase to DNA sequence at a genome-wide level [23] (Fig. 1b). DNA transposon is a phenomenon that transfers DNA sequence from one region of chromosome to another, which is assisted by DNA transposase [24]. DNA transposon requires that the chromatin at the insertion site is open, and the transposase carrying known DNA sequence tags needs to be artificially added to the nucleus, and then the open chromatin can be identified by using labels of known sequences to construct a library for sequencing [23]. At present, the most commonly used transposase is the Tn5 transposase, which can transpose in the accessible chromatin more often than in the inaccessible chromatin. The Tn5 transposase acts as a probe for measuring chromatin accessibility at the genome-wide level through the “cut and paste” mechanism, and the transposon can simultaneously fragment and tag the unprotected regions of DNA with sequencing adapters [25, 26] (Fig. 1b and Fig. 2).

The major procedures of ATAC-seq. a Nuclei preparation: target cells are lysed in the lysis buffer to collect nuclei. b Transposase reaction: adding the Tn5 transposase to tag the genomic DNA. The green symbol represents “adapter 1” of the Tn5 transposase, while the red symbol represents “adapter 2” of theTn5 transposase. c PCR amplification: PCR primer-1 and -2 are used to generate library for sequencing. Primer-1 and -2 are two universal PCR primers, which capture fragments with special length and add barcodes appropriate for the next generation sequencing
The construction of ATAC-seq library consists of three steps: nuclei preparation, transposition and amplification [19] (Fig. 2). Firstly, tissues or cells for examination are suspended into intact, homogenous single cells, which are subsequently incubated in the lysis buffer to generate crude nuclei (Fig. 2a). Secondly, the re-suspended nuclei are incubated in the transposition reaction mix to yield DNA fragment (Fig. 2b). Finally, transposed DNA is amplified to generate libraries for sequencing (Fig. 2c). The reaction of transposable enzyme to the chromatin of the sample is the key step of the ATAC experiment [27].
Quality control of the ATAC-seq library should be performed prior to sequencing to guarantee that the library concentration reaches the sequencing criteria. After library sequencing, raw reads are collected through sequencing the qualified library. After filtering data through sequencing data quality assessment, clean reads are further obtained by evaluating sequencing quality and summarizing data production [18, 28, 29]. After removing adapter sequences and low quality reads, high-quality reads about 150 nucleotides (nts) in length are processed for further analysis [30]. The peak calling reads are mapped to the reference genome and accessible chromatin regions, such as promoters, enhancers and insulators [31,32,33]. A series of detailed analysis can be further conducted, such as ascertaining distribution of reads across the whole genome, determining distribution of the peak length, functional analysis of genes with identified peaks, distribution of peaks on functional elements of genes, and analysis of differential peaks among samples [34, 35].
Advantages of ATAC-seq
The ATAC-seq method was first developed as an alternative or supplement to sequencing of Micrococcal Nuclease sensitive sites (MNase-seq), Formaldehyde-Assisted Isolation of Regulatory Elements (FAIRE-seq), and Deoxyribonuclease I hypersensitive sites sequencing (DNase-seq) [36,37,38] (Table 1). For the MNase-seq and DNase-seq, when the aggregation of DNA and histone is decreased, the unprotected DNA is exposed and cut by DNA enzymes such as MNase and DNase [39, 40]. The accessible chromatin regions are recognized by sequencing the cleaved DNA fragments and comparing the sequence reads to the reference genome [39, 41, 42]. Drawbacks of these two methods are time-consuming and poor repeatability [38, 43, 44]. The FAIRE-seq uses formaldehyde immobilization, phenol chloroform extraction and separation to obtain exposed DNA. However, its background is high, and sequencing signal-to-noise ratio is low [45,46,47].
Noteworthy, ATAC-seq has several advantages: first, the transposase method can reduce experimental time to 2–3 h to achieve DNA fragmentation using a simple enzymatic reaction, which avoids the tedious conventional DNA fragmentation, terminal repairing and adapter connection reaction [38]. On the other hand, it usually takes 2–3 days to prepare the DNase-seq and MNase-seq experiments, and 3–4 days for the FAIRE-seq experiment. Second, the simplified experimental procedure reduces the duration of sample preparation and decreases the probability of errors, which significantly improves the successful rate and repeatability of an experiment. Third, the sample size is reduced by at least 1000 times, by decreasing from 1 to 50 million cells (FAIRE-seq) and 50 million cells (DNase-seq) to as low as about 500 cells [38, 48,49,50]. When sample collection is challenging, this advantage is particularly prominent. Fourth, ATAC-seq can use paired-end sequencing technology to map nucleosome positioning and occupancy [51]. Paired-end sequencing can sequence both ends of the DNA fragment, making the alignment of reads mapping over repetitive regions of the genome more accurate [52].
There are also some limitations of the ATAC-seq technology. First, the Tn5 transposase simultaneously fragments and tags unprotected regions of DNA with sequencing adapters through the “cut and paste” mechanism. The adapter joints at both ends of each DNA fragment are random, which leads to a 50% probability of that the adapters at both ends of one fragment are the same, generating half unusable fragments for enrichment, amplification and sequencing [53]. Second, studies have shown that “naked” DNA without nucleosomes and transcription factors is easier to be cleaved by the Tn5 transposase [27]. Moreover, the Tn5 transposase tends to bind and cleave at transcription factor binding regions, which results in a loss of part of the transcription factor information [54, 55]. All these drawbacks make ATAC-seq difficult to detect the footprint of transcription factors, which can be used to identify potential binding motifs of transcription factors. Third, due to presence of mitochondrial DNA, data obtained by ATAC-seq inevitably contains some mitochondrial reads. Depending on the cell type, ATAC-seq data may contain 20–80% of mitochondrial sequencing reads [56].
To obtain pure nuclear genome reads and to reduce mitochondrial contamination, two methods can be used: using the cell lysis buffer without detergent [19], and using the clustered regularly interspaced short palindromic repeats (CRISPR) technology [57,58,59]. The CRISPR/Cas9 technology uses guide RNA (gRNA), which can target the mitochondrial chromosome [60, 61]. By adding gRNA/Cas9 mix to prepared sequencing library, gRNA can target mitochondrial ribosomal DNA and Cas9 enzyme will cleavage the fragments [58]. Compared to the original protocol, CRISPR technology results in lower mitochondrial reads, and more reads in the nuclear genome [58, 60, 62].
Improvement of ATAC-seq
Since the ATAC-seq method was first developed, it has been improved in order to adapt broader usage in research. Single-cell ATAC-seq (scATAC-seq) provides the first insightful examination of cell-to-cell variability in chromatin organization, which can be achieved by a programmable microfluidic device or combinatorial cellular indexing scheme. The scATAC-seq can be used as a genome-wide vehicle to map chromatin accessibility in all specific cell types of an organism [50, 63,64,65]. Because it is still unclear exactly how many open chromatin regions exist in a single cell, and how chromatin accessibility differs between the two alleles in an individual cell, whether the scATAC-seq does capture a limited subset of open chromatin sites in single cells remains unclear [66].
Moreover, Omni-ATAC-seq is another improved ATAC-seq protocol to detect chromatin accessibility [56]. Based on the standard ATAC-seq protocol, the Omni-ATAC-seq adds a washing step using detergents after cell lysis to remove mitochondria from the transposition reaction. The Omni-ATAC-seq also uses phosphate-buffered saline (PBS) in the transposition reaction to increase the signal-to-background ratio and to reduce the background. Thus, the Omni-ATAC-seq eliminates mitochondrial interference and reduces background noise to obtain high quality data of chromatin accessibility [56]. Moreover, the standard ATAC-seq requires the transposition reaction to be performed on fresh cells, and slowly cooled cryopreserved cells, but poorly on snap-frozen cells [67]. The Omni-ATAC protocol can generate high-quality chromatin accessibility profiles from clinically relevant frozen tissues, such as brains [56].
ATAC-seq obtains the information of accessible chromatin by breaking up cells, so it cannot describe the three-dimensional structure of these accessible genomic regions. Assay for Transposase Accessible Chromatin with Visualization (ATAC-see) uses the same enzymatic methods as ATAC-seq, and adds fluorescent clusters together with DNA markers, which allows visualization of three-dimensional immobilized nuclei [68].
Applications
Applying ATAC-seq has advanced our understanding of the machinery of gene expression regulation, such as chromatin accessibility between different samples, nucleosome positions, and genome-wide binding sites of transcription factors [23, 69, 70]. It has provided meaningful insight into revealing the landscape of chromosome accessibility, epigenetic modification of embryonic development, epigenetic mechanism of tumorigenesis, and potential disease biomarkers [61, 71,72,73,74]. Here, we focus on applications of ATAC-seq in basic research and clinical usage.
ATAC-seq in mapping the accessible chromatin landscape
Mapping the accessible chromatin landscape can obtain information of spatial changes in chromatin structures and transcription factors associated with gene expression [59]. This information can reveal the network of relevant transcription factors, and mechanisms of chromatin structural regulation that governs gene expression programs [59]. For instance, in the human immune system, the accessible chromatin map of primary immune cells—T lymphocytes has been identified by using ATAC-seq [75]. A significant change of chromatin accessibility has been identified in regions near genes that are associated with B cell activation, especially in Systemic Lupus Erythematosus (SLE) patients [76].
In developmental biology, the lineage-specific open chromatin regions and changes have been mapped using ATAC-seq in epidermal differentiation, and in trophoblast stem cell differentiation in placenta [77, 78]. In the developing heart, transcription factor TBX20 has been identified to bind to the conserved long-range enhancer Vcan, and to co-regulate gene expression [79]. In the nervous system, induced activation of neurons leads to instantaneous changes in the chromatin structure, especially in the enhancer region [80]. Mapping the accessible chromatin landscape of the developing cerebral cortex has identified enhancers for FGFR2 and EOMES as important regulatory players in cortical neurogenesis [81]. Moreover, ATAC-seq has been used to obtain landscapes of accessible chromatin of endocrine cells and germ cells [32, 82,83,84,85,86].
In summary, applying ATAC-seq has generated comprehensive accessible chromatin landscapes of various cell types in different tissues and organs, which has provided valuable insights into the complexity of gene transcription.
ATAC-seq in embryonic development
Chromatin reprogramming actively occurs during early embryonic development [61]. Studies have shown that when chromatin reprogramming happens, regulatory factors that are associated with gene transcription and DNA recombination are recruited by chromatin [87, 88], and simultaneously the stability of nucleosome is altered [89]. During zygote gene activation, the activity of open chromatin is increased, in parallel with activities of cis-regulatory factors, which confirms that cis-regulatory elements play a significant role in early development [90, 91]. ATAC-seq has been used, together with the CRISP/Cas-9 technology, to detect mouse preimplantation embryos [61]. Chromatin atlas of mouse early embryos at different developmental stages have been drawn by ATAC-seq, and motifs of essential transcription factors for early development such as CTCF, NR5A2 and TEAD4 have been identified [61].
Moreover, to study embryonic genome activation, ATAC-seq has been used to detect transcriptome sequences [92]. These studies have shown that multiple copies of DUX4 are activated by endogenous genes KDM4E and ZSCAN4 that are expressed only in cleavage-stage of human embryos, which subsequently initiates transition of embryonic stem cells to 2-cells stage with totipotency [92,93,94,95,96,97].
ATAC-seq in cancer research
ATAC-seq is highly applicable to capture the tissue-specific chromatin activity of regulatory regions in tumors [18, 75, 98,99,100]. In Ras-dependent oncogenesis, 3778 over-activated regulatory regions are detected by using ATAC-seq [18, 101]. Recurrent mutations in RAD21 and STAG2 genes, which encode the chromosome cohesion complex, have been shown to be key elements in malignancy formation in acute myeloid leukemia (AML) [98, 102,103,104]. Studies have shown that mutant cohesin can increase chromatin accessibility of binding sites for transcription factors such as ERG, GATA2 and RUNX1, as detected by using ATAC-seq [105,106,107,108].
Moreover, ARID1A mutations usually occur in many kinds of tumors, such as melanoma, glioblastoma and other human malignancies [109,110,111,112,113], and ARID1B mutations are usually found in neuroblastoma, hepatocellular carcinoma and breast invasive ductal carcinoma [114,115,116,117]. Studies have shown that mutations of the ARID1A and ARID1B complex are frequently associated with tumorigenesis via altering promoter and enhancer activities to modulate downstream gene expression [118]. During the cell neoplastic transformation, down-regulation of ARID1A leads to H3K27ac reduction at enhancer regions of downstream genes for ARID1A [119, 120]. ARID1A plays an important role in maintaining chromatin accessibility at enhancers. In particular, the expression of MET gene has been changed in ARID1A mutant ovarian cancer cells, while ARID1B deletion displays the same effect only in the context of ARID1A mutation, indicating an important role of ARID1A in ovarian cancer cells [120].
Furthermore, p53 is a well-studied cancer suppressor gene. The protein encoded by p53 has a role of suppressing cancer under normal circumstances, and promoting cancer development when mutations occur [121, 122]. When DNA damage occurs, p53 initiates cell apoptosis by regulating gene expression [123]. Studies have shown that p53 can bind to the promoter and enhancer of a gene to excel function [124, 125]. It has been found that p53 has a prior binding to the enhancer in healthy fibroblasts detected using ATAC-seq [126]. When DNA damage occurs, chromatin is converted from inaccessible to accessible status, and simultaneously, p53 gene is activated to maintain genome stability [127].
Conclusion
ATAC-seq uses high-throughput sequencing approach to identify all active regulatory sequences in the genome using a small amount of cells. ATAC-seq has been widely used in the acquisition of open chromatin regions and transcription factor binding sites to reveal a real time profile of chromatin accessibility. It has been rapidly applied and accepted to investigate gene expression dynamics in stem cells, early embryos, and various tumors, and even to detect potential biomarkers. Taking advantage of optimization of ATAC-seq methodology to simplify the experimental procedure and to reduce the cost, ATAC-seq should soon have a broader usage in basic research and clinical diagnostics.
Availability of data and materials
Not applicable.
Abbreviations
- AML:
-
Acute myeloid leukemia
- ATAC-see:
-
Assay for Transposase Accessible Chromatin with Visualization
- ATAC-seq:
-
Assay for Transposase Accessible Chromatin with high-throughput sequencing
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- DNase-seq:
-
Deoxyribonuclease I hypersensitive sites sequencing
- FAIRE-seq:
-
Formaldehyde-Assisted Isolation of Regulatory Elements
- MNase-seq:
-
sequencing of Micrococcal Nuclease sensitive sites
- PAHs:
-
Polycyclic aromatic hydrocarbons
- PBS:
-
Phosphate-buffered saline
- SLE:
-
Systemic Lupus Erythematosus
References
Jackson DA. Nuclear organization: uniting replication foci, chromatin domains and chromosome structure. Bioessays. 1995;17(7):587–91.
Bartova E, Kozubek S. Nuclear architecture in the light of gene expression and cell differentiation studies. Biol Cell. 2006;98(6):323–36.
Pederson T. The spatial organization of the genome in mammalian cells. Curr Opin Genet Dev. 2004;14(2):203–9.
Gottesfeld JM, Carey MF. Introduction to the thematic Minireview series: chromatin and transcription. J Biol Chem. 2018;293(36):13775–7.
Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489(7414):75–82.
Hsiung CC, Morrissey CS, Udugama M, Frank CL, Keller CA, Baek S, et al. Genome accessibility is widely preserved and locally modulated during mitosis. Genome Res. 2015;25(2):213–25.
Morris SA, Baek S, Sung MH, John S, Wiench M, Johnson TA, Schiltz RL, Hager GL. Overlapping chromatin-remodeling systems collaborate genome wide at dynamic chromatin transitions. Nat Struct Mol Biol. 2014;21(1):73–81.
Stergachis AB, Neph S, Reynolds A, Humbert R, Miller B, Paige SL, et al. Developmental fate and cellular maturity encoded in human regulatory DNA landscapes. Cell. 2013;154(4):888–903.
Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–66.
Chen T, Dent SY. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat Rev Genet. 2014;15(2):93–106.
Pacis A, Tailleux L, Morin AM, Lambourne J, MacIsaac JL, Yotova V, et al. Bacterial infection remodels the DNA methylation landscape of human dendritic cells. Genome Res. 2015;25(12):1801–11.
Yauk CL, Polyzos A, Rowan-Carroll A, Kortubash I, Williams A, Kovalchuk O. Tandem repeat mutation, global DNA methylation, and regulation of DNA methyltransferases in cultured mouse embryonic fibroblast cells chronically exposed to chemicals with different modes of action. Environ Mol Mutagen. 2008;49(1):26–35.
Kondo K, Takahashi Y, Hirose Y, Nagao T, Tsuyuguchi M, Hashimoto M, Ochiai A, Monden Y, Tangoku A. The reduced expression and aberrant methylation of p16(INK4a) in chromate workers with lung cancer. Lung Cancer. 2006;53(3):295–302.
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705.
Song L, Zhang Z, Grasfeder LL, Boyle AP, Giresi PG, Lee BK, et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011;21(10):1757–67.
Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods. 2009;6(4):283–9.
Leung A, Parks BW, Du J, Trac C, Setten R, Chen Y, et al. Open chromatin profiling in mice livers reveals unique chromatin variations induced by high fat diet. J Biol Chem. 2014;289(34):23557–67.
Davie K, Jacobs J, Atkins M, Potier D, Christiaens V, Halder G, Aerts S. Discovery of transcription factors and regulatory regions driving in vivo tumor development by ATAC-seq and FAIRE-seq open chromatin profiling. PLoS Genet. 2015;11(2):e1004994.
Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol. 2015;109(21.9):1–9.
Tekel SJ, Haynes KA. Molecular structures guide the engineering of chromatin. Nucleic Acids Res. 2017;45(13):7555–70.
Chen A, Chen D, Chen Y. Advances of DNase-seq for mapping active gene regulatory elements across the genome in animals. Gene. 2018;667:83–94.
Pascoe CD, Obeidat M, Arsenault BA, Nie Y, Warner S, Stefanowicz D, et al. Gene expression analysis in asthma using a targeted multiplex array. BMC pulmonary medicine. 2017;17(1):189.
Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10(12):1213–8.
Chuong EB, Elde NC, Feschotte C. Regulatory activities of transposable elements: from conflicts to benefits. Nat Rev Genet. 2017;18(2):71–86.
Reznikoff WS. Transposon Tn5. Annu Rev Genet. 2008;42:269–86.
Haniford DB, Ellis MJ. Transposons Tn10 and Tn5. Microbiol Spectr. 2015;3(1):MDNA3–0002-2014.
Picelli S, Bjorklund AK, Reinius B, Sagasser S, Winberg G, Sandberg R. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 2014;24(12):2033–40.
Bell O, Tiwari VK, Thoma NH, Schubeler D. Determinants and dynamics of genome accessibility. Nat Rev Genet. 2011;12(8):554–64.
Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402.
Miskimen KL, Chan ER, Haines JL. Assay for Transposase-Accessible Chromatin Using Sequencing (ATAC-seq) Data Analysis. Curr Protoc Hum Genet. 2017;92:20 4 1–4 13.
Kumasaka N, Knights AJ, Gaffney DJ. Fine-mapping cellular QTLs with RASQUAL and ATAC-seq. Nat Genet. 2016;48(2):206–13.
Ackermann AM, Wang Z, Schug J, Naji A, Kaestner KH. Integration of ATAC-seq and RNA-seq identifies human alpha cell and beta cell signature genes. Molecular metabolism. 2016;5(3):233–44.
Quillien A, Abdalla M, Yu J, Ou J, Zhu LJ, Lawson ND. Robust identification of developmentally active endothelial enhancers in zebrafish using FANS-assisted ATAC-Seq. Cell Rep. 2017;20(3):709–20.
Pranzatelli TJF, Michael DG, Chiorini JA. ATAC2GRN: optimized ATAC-seq and DNase1-seq pipelines for rapid and accurate genome regulatory network inference. BMC Genomics. 2018;19(1):563.
Mu JC, Jiang H, Kiani A, Mohiyuddin M, Bani Asadi N, Wong WH. Fast and accurate read alignment for resequencing. Bioinformatics. 2012;28(18):2366–73.
Zaret K. Micrococcal nuclease analysis of chromatin structure. Curr Protoc Mol Biol. 2005; Chapter 21:Unit 21 1.
Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22(9):1813–31.
Song L, Crawford GE. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb Protoc. 2010; 2010(2):pdb prot5384.
Rizzo JM, Sinha S. Analyzing the global chromatin structure of keratinocytes by MNase-seq. Methods Mol Biol. 2014;1195:49–59.
Cumbie JS, Filichkin SA, Megraw M. Improved DNase-seq protocol facilitates high resolution mapping of DNase I hypersensitive sites in roots in Arabidopsis thaliana. Plant Methods. 2015;11:42.
Cui K, Zhao K. Genome-wide approaches to determining nucleosome occupancy in metazoans using MNase-Seq. Methods Mol Biol. 2012;833:413–9.
Koohy H, Down TA, Spivakov M, Hubbard T. A comparison of peak callers used for DNase-Seq data. PLoS One. 2014;9(5):e96303.
Hoeijmakers WAM, Bartfai R. Characterization of the nucleosome landscape by Micrococcal nuclease-sequencing (MNase-seq). Methods Mol Biol. 2018;1689:83–101.
Barozzi I, Bora P, Morelli MJ. Comparative evaluation of DNase-seq footprint identification strategies. Front Genet. 2014;5:278.
Murtha M, Strino F, Tokcaer-Keskin Z, Sumru Bayin N, Shalabi D, Xi X, Kluger Y, Dailey L. Comparative FAIRE-seq analysis reveals distinguishing features of the chromatin structure of ground state- and primed-pluripotent cells. Stem Cells. 2015;33(2):378–91.
Bianco S, Rodrigue S, Murphy BD, Gevry N. Global mapping of open chromatin regulatory elements by formaldehyde-assisted isolation of regulatory elements followed by sequencing (FAIRE-seq). Methods Mol Biol. 2015;1334:261–72.
Yang CC, Buck MJ, Chen MH, Chen YF, Lan HC, Chen JJ, Cheng C, Liu CC. Discovering chromatin motifs using FAIRE sequencing and the human diploid genome. BMC Genomics. 2013;14:310.
Nammo T, Rodriguez-Segui SA, Ferrer J. Mapping open chromatin with formaldehyde-assisted isolation of regulatory elements. Methods Mol Biol. 2011;791:287–96.
Simon JM, Giresi PG, Davis IJ, Lieb JD. Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat Protoc. 2012;7(2):256–67.
Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523(7561):486–90.
Brunelle M, Rodrigue S, Jacques PE, Gevry N. High-resolution genome-wide mapping of nucleosome positioning and occupancy level using paired-end sequencing technology. Methods Mol Biol. 2017;1528:229–43.
Li YL, Weng JC, Hsiao CC, Chou MT, Tseng CW, Hung JH. PEAT: an intelligent and efficient paired-end sequencing adapter trimming algorithm. BMC bioinformatics. 2015;16(Suppl 1):S2.
Lu Z, Hofmeister BT, Vollmers C, DuBois RM, Schmitz RJ. Combining ATAC-seq with nuclei sorting for discovery of cis-regulatory regions in plant genomes. Nucleic Acids Res. 2017;45(6):e41.
Meyer CA, Liu XS. Identifying and mitigating bias in next-generation sequencing methods for chromatin biology. Nat Rev Genet. 2014;15(11):709–21.
Ishii H, Kadonaga JT, Ren B. MPE-seq, a new method for the genome-wide analysis of chromatin structure. Proc Natl Acad Sci U S A. 2015;112(27):E3457–65.
Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. 2017;14(10):959–62.
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–23.
Montefiori L, Hernandez L, Zhang Z, Gilad Y, Ober C, Crawford G, Nobrega M, Jo SN. Reducing mitochondrial reads in ATAC-seq using CRISPR/Cas9. Sci Rep. 2017;7(1):2451.
Grbesa I, Tannenbaum M, Sarusi-Portuguez A, Schwartz M, Hakim O. Mapping genome-wide accessible chromatin in primary human T lymphocytes by ATAC-Seq. Journal of visualized experiments : JoVE. 2017. https://doi.org/10.3791/56313(129.
Gu W, Crawford ED, O'Donovan BD, Wilson MR, Chow ED, Retallack H, DeRisi JL. Depletion of abundant sequences by hybridization (DASH): using Cas9 to remove unwanted high-abundance species in sequencing libraries and molecular counting applications. Genome Biol. 2016;17:41.
Wu J, Huang B, Chen H, Yin Q, Liu Y, Xiang Y, et al. The landscape of accessible chromatin in mammalian preimplantation embryos. Nature. 2016;534(7609):652–7.
Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet. 2016;48(10):1193–203.
Pott S, Lieb JD. Single-cell ATAC-seq: strength in numbers. Genome Biol. 2015;16:172.
Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, Steemers FJ, Trapnell C, Shendure J. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science. 2015;348(6237):910–4.
Fiers M, Minnoye L, Aibar S, Bravo Gonzalez-Blas C, Kalender Atak Z, Aerts S. Mapping gene regulatory networks from single-cell omics data. Brief Funct Genomics. 2018. https://doi.org/10.1093/bfgp/elx046.
Yuan GC, Cai L, Elowitz M, Enver T, Fan G, Guo G, et al. Challenges and emerging directions in single-cell analysis. Genome Biol. 2017;18(1):84.
Milani P, Escalante-Chong R, Shelley BC, Patel-Murray NL, Xin X, Adam M, et al. Cell freezing protocol suitable for ATAC-Seq on motor neurons derived from human induced pluripotent stem cells. Sci Rep. 2016;6:25474.
Chen X, Shen Y, Draper W, Buenrostro JD, Litzenburger U, Cho SW, et al. ATAC-see reveals the accessible genome by transposase-mediated imaging and sequencing. Nat Methods. 2016;13(12):1013–20.
Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The epigenetic landscape of T cell exhaustion. Science. 2016;354(6316):1165–9.
Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell. 2016;164(1–2):57–68.
Criscione SW, Teo YV, Neretti N. The chromatin landscape of cellular senescence. Trends in genetics: TIG. 2016;32(11):751–61.
Wu J, Xu J, Liu B, Yao G, Wang P, Lin Z, et al. Chromatin analysis in human early development reveals epigenetic transition during ZGA. Nature. 2018;557(7704):256–60.
Rendeiro AF, Schmidl C, Strefford JC, Walewska R, Davis Z, Farlik M, Oscier D, Bock C. Chromatin accessibility maps of chronic lymphocytic leukaemia identify subtype-specific epigenome signatures and transcription regulatory networks. Nat Commun. 2016;7:11938.
Dirks RA, Stunnenberg HG, Marks H. Genome-wide epigenomic profiling for biomarker discovery. Clin Epigenetics. 2016;8:122.
Qu K, Zaba LC, Satpathy AT, Giresi PG, Li R, Jin Y, et al. Chromatin accessibility landscape of cutaneous T cell lymphoma and dynamic response to HDAC inhibitors. Cancer Cell. 2017;32(1):27–41 e4.
Scharer CD, Blalock EL, Barwick BG, Haines RR, Wei C, Sanz I, Boss JM. ATAC-seq on biobanked specimens defines a unique chromatin accessibility structure in naive SLE B cells. Sci Rep. 2016;6:27030.
Bao X, Rubin AJ, Qu K, Zhang J, Giresi PG, Chang HY, Khavari PA. A novel ATAC-seq approach reveals lineage-specific reinforcement of the open chromatin landscape via cooperation between BAF and p63. Genome Biol. 2015;16:284.
Nelson AC, Mould AW, Bikoff EK, Robertson EJ. Mapping the chromatin landscape and Blimp1 transcriptional targets that regulate trophoblast differentiation. Sci Rep. 2017;7(1):6793.
Boogerd CJ, Aneas I, Sakabe N, Dirschinger RJ, Cheng QJ, Zhou B, Chen J, Nobrega MA, Evans SM. Probing chromatin landscape reveals roles of endocardial TBX20 in septation. J Clin Invest. 2016;126(8):3023–35.
Su Y, Shin J, Zhong C, Wang S, Roychowdhury P, Lim J, Kim D, Ming GL, Song H. Neuronal activity modifies the chromatin accessibility landscape in the adult brain. Nat Neurosci. 2017;20(3):476–83.
de la Torre-Ubieta L, Stein JL, Won H, Opland CK, Liang D, Lu D, Geschwind DH. The dynamic landscape of open chromatin during human cortical neurogenesis. Cell. 2018;172(1–2):289–304 e18.
Raurell-Vila H, Ramos-Rodriguez M, Pasquali L. Assay for transposase accessible chromatin (ATAC-Seq) to chart the open chromatin landscape of human pancreatic islets. Methods Mol Biol. 2018;1766:197–208.
Varshney A, Scott LJ, Welch RP, Erdos MR, Chines PS, Narisu N, et al. Genetic regulatory signatures underlying islet gene expression and type 2 diabetes. Proc Natl Acad Sci U S A. 2017;114(9):2301–6.
Ruf-Zamojski F, Fribourg M, Ge Y, Nair V, Pincas H, Zaslavsky E, et al. Regulatory architecture of the LbetaT2 Gonadotrope cell underlying the response to gonadotropin-releasing hormone. Front Endocrinol. 2018;9:34.
Guo J, Grow EJ, Yi C, Mlcochova H, Maher GJ, Lindskog C, et al. Chromatin and single-cell RNA-Seq profiling reveal dynamic signaling and metabolic transitions during human Spermatogonial stem cell development. Cell Stem Cell. 2017;21(4):533–46 e6.
Maezawa S, Yukawa M, Alavattam KG, Barski A, Namekawa SH. Dynamic reorganization of open chromatin underlies diverse transcriptomes during spermatogenesis. Nucleic Acids Res. 2018;46(2):593–608.
Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142(6):967–80.
Musselman CA, Lalonde ME, Cote J, Kutateladze TG. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol. 2012;19(12):1218–27.
Tropberger P, Pott S, Keller C, Kamieniarz-Gdula K, Caron M, Richter F, et al. Regulation of transcription through acetylation of H3K122 on the lateral surface of the histone octamer. Cell. 2013;152(4):859–72.
Funaya S, Aoki F. Regulation of zygotic gene activation by chromatin structure and epigenetic factors. The Journal of reproduction and development. 2017;63(4):359–63.
Hug CB, Grimaldi AG, Kruse K, Vaquerizas JM. Chromatin architecture emerges during zygotic genome activation independent of transcription. Cell. 2017;169(2):216–28 e19.
Hendrickson PG, Dorais JA, Grow EJ, Whiddon JL, Lim JW, Wike CL, et al. Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat Genet. 2017;49(6):925–34.
Matoba S, Liu Y, Lu F, Iwabuchi KA, Shen L, Inoue A, Zhang Y. Embryonic development following somatic cell nuclear transfer impeded by persisting histone methylation. Cell. 2014;159(4):884–95.
Chung YG, Matoba S, Liu Y, Eum JH, Lu F, Jiang W, et al. Histone demethylase expression enhances human somatic cell nuclear transfer efficiency and promotes derivation of pluripotent stem cells. Cell Stem Cell. 2015;17(6):758–66.
Zalzman M, Falco G, Sharova LV, Nishiyama A, Thomas M, Lee SL, et al. Zscan4 regulates telomere elongation and genomic stability in ES cells. Nature. 2010;464(7290):858–63.
Kalmbach K, Robinson LG Jr, Wang F, Liu L, Keefe D. Telomere length reprogramming in embryos and stem cells. Biomed Res Int. 2014;2014:925121.
Macfarlan TS, Gifford WD, Driscoll S, Lettieri K, Rowe HM, Bonanomi D, et al. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature. 2012;487(7405):57–63.
Ye M, Zhang H, Yang H, Koche R, Staber PB, Cusan M, et al. Hematopoietic differentiation is required for initiation of acute myeloid leukemia. Cell Stem Cell. 2015;17(5):611–23.
Garcia E, Hayden A, Birts C, Britton E, Cowie A, Pickard K, et al. Authentication and characterisation of a new oesophageal adenocarcinoma cell line: MFD-1. Sci Rep. 2016;6:32417.
Bhattacharyya S, Pradhan K, Campbell N, Mazdo J, Vasantkumar A, Maqbool S, et al. Altered hydroxymethylation is seen at regulatory regions in pancreatic cancer and regulates oncogenic pathways. Genome Res. 2017;27(11):1830–42.
Britton E, Rogerson C, Mehta S, Li Y, Li X, Consortium O, Fitzgerald RC, Ang YS, Sharrocks AD. Open chromatin profiling identifies AP1 as a transcriptional regulator in oesophageal adenocarcinoma. PLoS genetics. 2017;13(8):e1006879.
Leeke B, Marsman J, O'Sullivan JM, Horsfield JA. Cohesin mutations in myeloid malignancies: underlying mechanisms. Exp Hematol Oncol. 2014;3:13.
Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet. 2013;45(10):1232–7.
Panigrahi AK, Pati D. Higher-order orchestration of hematopoiesis: is cohesin a new player? Exp Hematol. 2012;40(12):967–73.
Mazumdar C, Shen Y, Xavy S, Zhao F, Reinisch A, Li R, et al. Leukemia-associated Cohesin mutants dominantly enforce stem cell programs and impair human hematopoietic progenitor differentiation. Cell Stem Cell. 2015;17(6):675–88.
Wang Q, Li W, Liu XS, Carroll JS, Janne OA, Keeton EK, Chinnaiyan AM, Pienta KJ, Brown M. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell. 2007;27(3):380–92.
Wunsche P, Eckert ESP, Holland-Letz T, Paruzynski A, Hotz-Wagenblatt A, Fronza R, et al. Mapping active gene-regulatory regions in human repopulating long-term HSCs. Cell Stem Cell. 2018;23(1):132–46 e9.
Lichtinger M, Ingram R, Hannah R, Muller D, Clarke D, Assi SA, et al. RUNX1 reshapes the epigenetic landscape at the onset of haematopoiesis. EMBO J. 2012;31(22):4318–33.
Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45(6):592–601.
Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov. 2015;5(7):752–67.
Wu RC, Wang TL, Shih IM. The emerging roles of ARID1A in tumor suppression. Cancer Biol Ther. 2014;15(6):655–64.
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–12.
Wei X, Walia V, Lin JC, Teer JK, Prickett TD, Gartner J, et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat Genet. 2011;43(5):442–6.
Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44(7):760–4.
Sausen M, Leary RJ, Jones S, Wu J, Reynolds CP, Liu X, et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet. 2013;45(1):12–7.
Shao F, Guo T, Chua PJ, Tang L, Thike AA, Tan PH, Bay BH, Baeg GH. Clinicopathological significance of ARID1B in breast invasive ductal carcinoma. Histopathology. 2015;67(5):709–18.
Ho L, Jothi R, Ronan JL, Cui K, Zhao K, Crabtree GR. An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proc Natl Acad Sci U S A. 2009;106(13):5187–91.
Wang X, Nagl NG, Wilsker D, Van Scoy M, Pacchione S, Yaciuk P, Dallas PB, Moran E. Two related ARID family proteins are alternative subunits of human SWI/SNF complexes. Biochem J. 2004;383(Pt 2):319–25.
Lakshminarasimhan R, Andreu-Vieyra C, Lawrenson K, Duymich CE, Gayther SA, Liang G, Jones PA. Down-regulation of ARID1A is sufficient to initiate neoplastic transformation along with epigenetic reprogramming in non-tumorigenic endometriotic cells. Cancer Lett. 2017;401:11–9.
Kelso TWR, Porter DK, Amaral ML, Shokhirev MN, Benner C, Hargreaves DC. Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. Elife. 2017;6.
Pieri M, Castiglia L, Basilicata P, Sannolo N, Acampora A, Miraglia N. Biological monitoring of nurses exposed to doxorubicin and epirubicin by a validated liquid chromatography/fluorescence detection method. Ann Occup Hyg. 2010;54(4):368–76.
Staib F, Robles AI, Varticovski L, Wang XW, Zeeberg BR, Sirotin M, et al. The p53 tumor suppressor network is a key responder to microenvironmental components of chronic inflammatory stress. Cancer Res. 2005;65(22):10255–64.
Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9(5):402–12.
Zeron-Medina J, Wang X, Repapi E, Campbell MR, Su D, Castro-Giner F, et al. A polymorphic p53 response element in KIT ligand influences cancer risk and has undergone natural selection. Cell. 2013;155(2):410–22.
Melo CA, Drost J, Wijchers PJ, van de Werken H, de Wit E, Oude Vrielink JA, et al. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol Cell. 2013;49(3):524–35.
Younger ST, Rinn JL. p53 regulates enhancer accessibility and activity in response to DNA damage. Nucleic Acids Res. 2017;45(17):9889–900.
Attardi LD, de Vries A, Jacks T. Activation of the p53-dependent G1 checkpoint response in mouse embryo fibroblasts depends on the specific DNA damage inducer. Oncogene. 2004;23(4):973–80.
Acknowledgments
We thank members of the Sun laboratory for their valuable discussions and advice.
Funding
This work was supported by China Postdoctoral Science Foundation (2017 M622053) and the National Natural Science Foundation of China (81471152 and 31771141).
Author information
Authors and Affiliations
Contributions
Wrote the paper: YS and TS, Edited paper: NM and TS. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sun, Y., Miao, N. & Sun, T. Detect accessible chromatin using ATAC-sequencing, from principle to applications. Hereditas 156, 29 (2019). https://doi.org/10.1186/s41065-019-0105-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41065-019-0105-9