
Abstract
Ribonucleotides, which are RNA precursors, are often incorporated into DNA during replication. Although embedded ribonucleotides in the genome are efficiently removed by canonical ribonucleotide excision repair (RER), inactivation of RER causes genomic ribonucleotide accumulation, leading to various abnormalities in cells. Mutation of genes encoding factors involved in RER is associated with the neuroinflammatory autoimmune disorder Aicardi–Goutières syndrome. Over the last decade, the biological impact of ribonucleotides in the genome has attracted much attention. In the present review, we particularly focus on recent studies that have elucidated possible mechanisms of ribonucleotide incorporation and repair and their significance in mammals.
Similar content being viewed by others
Background
In eukaryotic cells, the concentrations of ribonucleotide triphosphates (rNTPs), i.e., RNA precursors, are approximately two orders of magnitude higher than those of DNA precursors, deoxyribonucleotide triphosphates (dNTPs) [1, 2]. Although DNA polymerases (pols) can accurately discriminate the correct substrate dNTPs against rNTPs, the great abundance of rNTPs in cellular nucleotide pools enables them to be incorporated into genomic DNA. Indeed, numerous rNTPs are incorporated into the genome; approximately 13,000 and > 1000,000 ribonucleotides are embedded into the genomes of yeast and mouse embryonic fibroblast cells, respectively [3, 4]. In humans, hypomorphic mutations of the genes encoding subunits of RNase H2, the enzyme essential for initiation of canonical ribonucleotide excision repair (RER), are associated with the serious autoimmune disease Aicardi–Goutières syndrome (AGS) [5]. The AGS autoimmune phenotype is believed to be caused by the accumulation of endogenous nucleic acid species, which activate intracellular Toll-like receptors, and/or DNA damage responses induced by the embedded ribonucleotides, stimulating interferon production in RNase H2-compromised cells [6]. In mouse models, early embryonic lethality results from the complete disruption of RNase H2 [3, 7]. Additionally, tissue-specific inactivation of RNase H2 can progress to tumorigenesis [8, 9]. Mammalian cells deficient in RER accumulate ribonucleotides in the genome and display various abnormalities, such as DNA replication delay, enhanced DNA damage, chronic activation of DNA damage responses, and epigenetic dysfunction [3, 7, 10,11,12]. Thus, genomic ribonucleotide accumulation is a disastrous event in cells, and molecular mechanisms underlying ribonucleotide-induced genome instability have been of a great interest over the last decade. Essential studies in this field have been well summarized in several reviews [13,14,15,16,17,18,19]. In this article, we focused on mammals in particular and recent research that has investigated the possible mechanisms underlying ribonucleotide incorporation and their processing pathways has been described.
Review
Source of ribonucleotide incorporation into DNA
Eukaryotic DNA pols are classified into six families (A, B, X, Y, RT, and AEP) on the basis of amino acid sequence comparisons [20, 21]; family A (pols γ, θ, and ν), family B (pols α, δ, ε, and ζ), family X (pols β, λ, μ, and TdT), family Y (pols η, κ, ι, and Rev1), family RT including telomerase, and family AEP including PrimPol. Most pols possess a conserved “steric gate” amino acid residue, which prevents ribonucleotide incorporation into DNA [22]. Although pols β and λ lack an aromatic steric gate amino acid side chain, both pols utilize a protein backbone segment to discriminate among sugars [23,24,25].
Although pols have a discrimination system against rNTPs, they can incorporate rNTPs into DNA at a non-negligible rate. For the human replicative pol α from family B, rNTPs are inserted with a 500-fold lower frequency than dNTPs during DNA synthesis [26]. The other replicative pols, δ and ε, are prone to incorporate rNTPs at physiological nucleotide concentrations similar to those of yeast replicative pols that incorporate one ribonucleotide for every thousands of deoxyribonucleotides [27, 28]. Therefore, millions of ribonucleotides may be embedded into the human genome. Notably, 3′-exonuclease activities of these pols cannot efficiently remove the inserted ribonucleotides [27, 28], which suggests that the proofreading during replication does not protect the genome from the aberrant ribonucleotide incorporation.
The mitochondrial pol γ, a member of family A, discriminates rNTPs with 1000- to 77,000-fold preference for dNTPs depending on the identity of nucleotides [26, 29]. As observed in family B pols, the 3′-exonuclease activity of pol γ does not contribute to the protection from ribonucleotide incorporation [30]. Based on previous studies, for 16.5 kb of mitochondrial DNA (mtDNA), pol γ is predicted to incorporate roughly 10–20 ribonucleotides during replication. However, the number of ribonucleotides in mtDNA (54, 36, and 65 ribonucleotides in one mtDNA molecule of human fibroblasts, HeLa cells, and mouse liver, respectively) was shown to be much higher than the expected frequency [30, 31]. This difference is expected to result from the presence of the other pols participating in mtDNA replication and/or the influence of varying nucleotide concentrations inside mitochondria [30].
Family X pols, involved in DNA repair processes such as base excision repair (BER) and non-homologous end joining (NHEJ), have also been suggested to play roles in inserting ribonucleotides into DNA. Pols β and λ have substrate selectivity in the range of 3,000- to 50,000-fold preference for dNTPs in comparison with rNTPs [22]. Although they strongly discriminate against ribonucleotides, a recent study showed that pol β, rather than pol λ, has an impact on the activity of ribonucleotide insertion opposite 7,8-dihydro-8-oxo-2′-deoxyguanosine (8-oxo-dG), a base resulting from oxidative damage, in cellular extracts [32]. Additionally, oxidative ribonucleotide 8-oxo-rGTP can be utilized as a substrate for DNA synthesis by pol β [33]. Notably, pol μ and TdT, unlike other pols, favorably incorporate rNTPs into DNA (only 1- to 10-fold discrimination against rNTPs) [22, 34]. Importantly, ribonucleotides are primarily utilized by both pols during NHEJ in cells [35], leading to beneficial consequences for DNA strand break repair; the insertion of ribonucleotides increases the fidelity of pol μ and promotes the ligation step during NHEJ [35, 36]. Although DNA repair processes, as well as DNA replication, can be sources of ribonucleotide incorporation, the transient presence of ribonucleotides contributes to the efficient repair of DNA maintaining genome integrity.
Family Y pols can replicate across DNA lesions via a process known as translesion DNA synthesis (TLS). Despite the presence of the steric gate residue in the active site [37,38,39], TLS pols can insert rNTPs into DNA in the following specific situations [38, 40]: Pol ι can incorporate rNTPs opposite undamaged template DNA depending on the sequence context. During TLS, the insertion of rNTPs by Pol ι is also observed across damaged DNA such as an abasic site (AP-site) and 8-oxo-dG. Another TLS Pol η can insert rCTP opposite 8-oxo-dG and cisplatin intrastrand guanine crosslinks. In addition, the activity of RNase H2-mediated cleavage of the inserted ribonucleotide decreases in the presence of these types of DNA damage. Thus, the TLS pathway may contribute to genomic ribonucleotide accumulation.
Repair/tolerance mechanisms of embedded ribonucleotides
RNase H2-initiated ribonucleotide excision repair
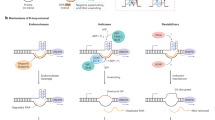
Embedded ribonucleotides are primarily repaired by RNase H2-mediated RER (Fig. 1 (1)) [41]. In vitro studies have revealed the detailed mechanism underlying the RER pathway: RNase H2 recognizes the ribonucleotide in DNA and cuts the DNA 5′-phosphodiester bond of the ribonucleotide [42, 43]. This incision reaction is followed by strand displacement synthesis by pols δ or ε, flap DNA cleavage by flap endonuclease FEN1 or the exonuclease Exo1, and nick sealing by DNA ligase I [41].

Overview of processing mechanisms of ribonucleotides embedded in DNA. (1) Embedded ribonucleotides are repaired by RNase H2-dependent RER. (2) In the absence of RNase H2, ribonucleotides in DNA are processed by topoisomerases, resulting in genomic instability. (3) The BER factor APE1 excises the damaged ribonucleotides in DNA. (4) The involvement of NER on ribonucleotide removal is under debate. (5) APTX resolves abortive ligation intermediates created at 5′-ribonucleotide termini. (6) Ribonucleotides on the template DNA strand impact on DNA synthesis
Eukaryotic RNase H2 is a heteromeric complex containing a catalytic subunit RNASEH2A and auxiliary subunits RNASEH2B and RNASEH2C [43]. RNASEH2B physically interacts with PCNA via the PCNA-interacting motif [44], indicating that RER is coupled with DNA replication. Indeed, mammalian cell studies suggest that RNase H2 is recruited and co-localized to replication and repair foci, not only via the interaction of RNASEH2B and PCNA but also via the catalytic site of RNASEH2A [45, 46]. Notably, RNase H2 is constitutively expressed throughout the cell cycle in HeLa cells [3], implying the possible role of RER in replication-independent repair.
Reportedly, RER is required for efficient mismatch repair (MMR). A single ribonucleotide in close proximity to a mismatch is processed by RNase H2 for generating a nick, which provides a strand discrimination signal for MMR of nascent strand replication errors [47, 48]. Hence, as also observed during NHEJ (see the section above) [35], ribonucleotide insertion is not merely an erroneous event occurring during replication, but it is an important biological process in maintaining genome stability.
Topoisomerase-mediated excision repair
In the absence of functional RNase H2, the embedded ribonucleotides are repaired by an alternate pathway involving DNA topoisomerase, the enzyme that relaxes negatively supercoiled DNA by transiently cleaving and re-ligating one or both strands of DNA (Fig. 1 (2)) [49,50,51]. Yeast and human topoisomerase 1 (TOP1) incise the DNA 3′-side of a ribonucleotide, generating a nick and a covalent protein-DNA cleavage complex (TOP1cc) between the TOP1 tyrosyl moiety and the 3′-phosphate of the ribonucleotide [52, 53]. Upon cleavage, the 2′-hydroxyl of the ribose sugar attacks the phosphotyrosyl linkages, generates a 2′,3′-cyclic phosphate, and releases TOP1 [52, 53].
Recent studies using purified human TOP1 suggest further distinct processing of the released DNA (Fig. 2): (1) re-ligation of the nick; (2) strand cleavage by TOP1 a few nucleotides upstream from the nick, leading to the formation of a second TOP1cc; and (3) sequential cleavage on the opposite strand of the nick [54, 55]. Specifically, the re-ligation of the nick by TOP1 allows a second attempt of the excision repair. Second, TOP1cc formation upstream from the nick leads to the release of a short DNA fragment containing 2′,3′-cyclic phosphate, which generates short deletions at repetitive sequences through TOP1-mediated false ligation. Lastly, cleavage of the opposite strand by TOP1 results in the formation of a severe DNA strand break with TOP1cc at the strand terminus. These models have been supported by studies with yeast TOP1, which induces 2–5-nt deletion mutations at the repetitive sequences, as well as DNA double strand breaks in the genome [54, 56, 57]. Furthermore, mouse and human cells lacking RNase H2 had elevated levels of 53BP1 or phosphorylated histone (γH2AX) foci, indicating the formation of DNA strand breaks in the mammalian genome [3, 7, 10, 12]. According to these studies, a question arises as to whether such deletion mutations can be caused by ribonucleotide accumulation in vivo. Findings of a recent study have revealed that deletions are induced by aberrant ribonucleotide incorporation into mouse mitochondrial DNA [58]. In contrast, base substitutions (T:A → G:C base substitutions at GTG trinucleotides), but not deletion mutations, have been detected through whole exome sequencing of tumor cells derived from Rnaseh2b knock-out mice [9]. Taken together, TOP1-dependent ribonucleotide excision repair can be highly mutagenic and possibly induces severe genomic instability in the absence of RER; however, its biological consequences in mammalian cells require further investigation.

Models depicting the processing of ribonucleotide by mammalian topoisomerase 1. (1) A nick containing 2′,3′-cyclic phosphate and 5′-OH ends is re-ligated by TOP1. (2) Strand cleavage by TOP1 upstream from the nick leads to the formation of a second TOP1cc. Re-ligation across the gap by TOP1 causes a short-deletion. (3) Cleavage of the opposite strand by TOP1 results in the formation of the DNA strand break with TOP1cc at the strand terminus
On the basis of a recent study, the depletion in TOP1 reduces the number of γH2AX foci in RER-deficient human cells [59], which provides evidence of the false processing of embedded ribonucleotides by TOP1 in mammals. Interestingly, the lack of RNase H2 desensitizes human cells to poly(ADP-ribose) polymerase (PARP) inhibitors that form PARP1-trapping DNA lesions [59]. Therefore, DNA damage created by TOP1-mediated ribonucleotide excision induces PARP1 activation. Because mono-allelic or bi-allelic loss of RNASEH2B is frequently observed in chronic lymphocytic leukemia and castration-resistant prostate cancers, genomic ribonucleotides may be a therapeutic target in tumors [59].
It has been reported that the presence of ribonucleotides in DNA stimulates the cleavage activity of type II topoisomerase (TOP2) and leads to the formation of a TOP2 cleavage complex (TOP2cc) at 5′-ribonucleotides [60, 61], possibly causing DNA strand breaks. For repairing this ribonucleotide-induced TOP2cc, TOP2 has to be proteolyzed. The consequent degradation of TOP2cc allows the processing of the TOP2-DNA crosslinks by tyrosyl-DNA phosphodiesterase 2 (TDP2) that hydrolyzes the 5′-tyrosine phosphodiester bonds between DNA 5′-phosphates and the active site tyrosine of TOP2 [61]. Therefore, TDP2 plays a protective role against the toxic effects of ribonucleotide-induced DNA damage in cells.
Base excision repair
BER is a primary repair pathway that is involved in correcting damage to endogenous bases such as oxidative and alkylated bases, e.g., 7,8-dihydro-8-oxoguanine and N3-methyladenine [62, 63]. BER is initiated by excision of the damaged or mismatched base by DNA glycosylases. The AP-site produced is further processed by apurinic/apyrimidinic endonuclease 1 (APE1), which catalyzes the cleavage of the sugar-phosphate backbone 5′ at the AP-site. For the mechanism of BER, the question that arises is whether the embedded ribonucleotides are recognized as the substrate of BER factors (Fig. 1 (3)). Reportedly, 8-oxoguanine DNA glycosylase (OGG1) can bind to an oxidized ribonucleotide, i.e., 8-oxoriboguanosine (8-oxo-rG), in DNA but showed no glycosylase/lyase activity in vitro [64]. Similarly, the human MutY homolog (MUTYH), which removes mispaired adenine opposite 8-oxoguanine, is fully inactive against riboadenosine (rA) paired with 8-oxoguanine [33]. Interestingly, APE1 cleaves an abasic ribonucleotide (rAP-site) in DNA and also has weak endonuclease and 3′-exonuclease activities on the embedded 8-oxo-rG, while mammalian RNase H2 has no activity against either rAP-site or 8-oxo-rG [65]. Therefore, among BER mechanisms, APE1 is a candidate for being the back-up repair mechanism for processing damaged ribonucleotides that cannot be removed by RNase H2.
Nucleotide excision repair
Nucleotide excision repair (NER) is involved in the removal of helix-distorting DNA lesions such as UV-induced cyclobutane pyrimidine dimers. Because NER factors can recognize a nearly infinite variety of DNA damages, ribonucleotides misincorporated into DNA may serve as the substrate for NER. The possibility of this alternative repair pathway has been debated among researchers (Fig. 1 (4)) [66]. Purified NER proteins derived from thermophilic eubacteria recognize and excise ribonucleotides in DNA [67]. In E. coli cells, the disruption of NER factors increases spontaneous mutagenesis in the absence of RNase HII [67]. However, a recent in vitro study revealed that ribonucleotide-containing DNA is a very poor substrate for purified E. coli and human NER systems [68], which indicates that NER is not a major repair pathway in mammals. The precise role of NER in the repair of embedded ribonucleotides is presently being debated.
Processing of ribonucleotide-induced abortive ligation
During RER, RNase H2 cleaves the 5′-side of a ribonucleotide and creates a nick, i.e., a RNA-DNA junction. In such conditions, the presence of a ribonucleotide on the 5′-terminus impairs the sealing of the nick by human DNA ligases I and III (Fig. 1 (5)). This abortive ligation results in the formation of a toxic 5′-adenylation (5′-AMP) at the ribonucleotide terminus [69]. Human aprataxin (APTX), the enzyme that removes 5′-AMP from abortive ligation intermediates, has been known to efficiently repair the 5′-AMP at RNA-DNA junctions generated during RER. The study indicated that the potential role of APTX is to protect genome integrity against the complex types of damage that can be generated during RER.
DNA synthesis across embedded ribonucleotides
In the absence of RER, the accumulation of ribonucleotides into the genome leads to replication stress in cells [3]. On the basis of in vitro experiments, human replicative pol δ pauses slightly during DNA synthesis across a single ribonucleotide on the template DNA (Fig. 1 (6)) [27]. Although human pol α and mitochondrial pol γ are also able to bypass a template ribonucleotide [30, 64], physiological concentrations of rNTPs have been shown to inhibit DNA synthesis by pol γ [30]. Furthermore, multiple consecutive ribonucleotides hinder the primer extension reaction catalyzed by pol δ [27].
The oxidation of ribonucleotides in DNA can be more problematic for replication; the oxidative ribonucleotide 8-oxo-rG strongly blocks primer extension catalyzed by pol α [64]. For TLS pols, pol κ inefficiently bypasses rG and 8-oxo-rG [64]. Interestingly, pol η rapidly bypasses both undamaged and damaged ribonucleotides [64]. Both TLS pols can bypass 8-oxo-rG in a more error-free manner than 8-oxo-dG. Therefore, the ribonucleotide sugar backbone influences fidelity during TLS. These studies suggest that the ribonucleotides in the genome impede replication by pols, possibly stalling replication forks. In this scenario, TLS pols are required as ribonucleotide-tolerance mechanisms.
Conclusions
There is increasing interest in the impact of ribonucleotide incorporation into DNA. The possible mechanisms underlying ribonucleotide-induced genomic instability and its consequences to the cell have been reported in numerous in vitro and in vivo studies. The recent noteworthy studies described in this review demonstrated that ribonucleotides that are transiently present in the genome are not only problematic lesions but may also be beneficial to the maintenance of genome integrity. However, the inactivation of canonical RER results in various deleterious effects in cells, which likely result from the unwanted processing of ribonucleotides, and may cause severe symptoms in humans. Further studies will be necessary for providing a better understanding of the biological action of the ribonucleotides, e.g., mutagenic potential, in the mammalian genome.
Abbreviations
- APE1:
-
apurinic/apyrimidinic endonuclease 1
- 8-oxo-dG:
-
7,8-dihydro-8-oxo-2'-deoxyguanosine
- 8-oxo-rG:
-
8-oxoriboguanosine
- AGS:
-
Aicardi–Goutières syndrome
- APTX:
-
aprataxin
- BER:
-
base excision repair
- dNTPs:
-
deoxyribonucleotide triphosphates
- MMR:
-
mismatch repair
- MUTYH:
-
MutY homologue
- NER:
-
Nucleotide excision repair
- NHEJ:
-
non-homologous end joining
- OGG1:
-
8-oxoguanine DNA glycosylase
- PARP:
-
poly(ADP-ribose) polymerase
- pol:
-
DNA polymerase
- RER:
-
ribonucleotide excision repair
- rNTPs:
-
ribonucleotide triphosphates
- TDP2:
-
tyrosyl-DNA phosphodiesterase 2
- TOP1:
-
topoisomerase 1
- TOP2:
-
type II topoisomerase
References
Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140:1–22.
Nick McElhinny SA, Watts BE, Kumar D, Watt DL, Lundstrom EB, Burgers PM, Johansson E, Chabes A, Kunkel TA. Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc Natl Acad Sci U S A. 2010;107:4949–54.
Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F, et al. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell. 2012;149:1008–22.
Williams JS, Smith DJ, Marjavaara L, Lujan SA, Chabes A, Kunkel TA. Topoisomerase 1-mediated removal of ribonucleotides from nascent leading-strand DNA. Mol Cell. 2013;49:1010–5.
Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet. 2006;38:910–6.
Feng S, Cao Z. Is the role of human RNase H2 restricted to its enzyme activity? Prog Biophys Mol Biol. 2016;121:66–73.
Hiller B, Achleitner M, Glage S, Naumann R, Behrendt R, Roers A. Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J Exp Med. 2012;209:1419–26.
Hiller B, Hoppe A, Haase C, Hiller C, Schubert N, Muller W, Reijns MAM, Jackson AP, Kunkel TA, Wenzel J, et al. Ribonucleotide excision repair is essential to prevent squamous cell carcinoma of the skin. Cancer Res. 2018;78:5917–26.
Aden K, Bartsch K, Dahl J, Reijns MAM, Esser D, Sheibani-Tezerji R, Sinha A, Wottawa F, Ito G, Mishra N, et al. Epithelial RNase H2 maintains genome integrity and prevents intestinal tumorigenesis in mice. Gastroenterology. 2019;156:145–159.
Lim YW, Sanz LA, Xu X, Hartono SR, Chedin F. Genome-wide DNA hypomethylation and RNA:DNA hybrid accumulation in Aicardi-Goutieres syndrome. Elife. 2015;4:e08007.
Gunther C, Kind B, Reijns MA, Berndt N, Martinez-Bueno M, Wolf C, Tungler V, Chara O, Lee YA, Hubner N, et al. Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. J Clin Invest. 2015;125:413–24.
Pizzi S, Sertic S, Orcesi S, Cereda C, Bianchi M, Jackson AP, Lazzaro F, Plevani P, Muzi-Falconi M. Reduction of hRNase H2 activity in Aicardi-Goutieres syndrome cells leads to replication stress and genome instability. Hum Mol Genet. 2015;24:649–58.
Cerritelli SM, Crouch RJ. The balancing act of ribonucleotides in DNA. Trends Biochem Sci. 2016;41:434–45.
Klein HL. Genome instabilities arising from ribonucleotides in DNA. DNA Repair (Amst). 2017;56:26–32.
Potenski CJ, Klein HL. How the misincorporation of ribonucleotides into genomic DNA can be both harmful and helpful to cells. Nucleic Acids Res. 2014;42:10226–34.
Wallace BD, Williams RS. Ribonucleotide triggered DNA damage and RNA-DNA damage responses. RNA Biol. 2014;11:1340–6.
Williams JS, Kunkel TA. Ribonucleotides in DNA: origins, repair and consequences. DNA Repair (Amst). 2014;19:27–37.
Williams JS, Lujan SA, Kunkel TA. Processing ribonucleotides incorporated during eukaryotic DNA replication. Nat Rev Mol Cell Biol. 2016;17:350–63.
Vaisman A, Woodgate R. Ribonucleotide discrimination by translesion synthesis DNA polymerases. Crit Rev Biochem Mol Biol. 2018;53:382–402.
Garcia-Diaz M, Bebenek K. Multiple functions of DNA polymerases. CRC Crit Rev Plant Sci. 2007;26:105–22.
Shanbhag V, Sachdev S, Flores JA, Modak MJ, Singh K. Family a and B DNA polymerases in Cancer: opportunities for therapeutic interventions. Biology (Basel). 2018;7.
Brown JA, Suo Z. Unlocking the sugar "steric gate" of DNA polymerases. Biochemistry. 2011;50:1135–42.
Brown JA, Fiala KA, Fowler JD, Sherrer SM, Newmister SA, Duym WW, Suo Z. A novel mechanism of sugar selection utilized by a human X-family DNA polymerase. J Mol Biol. 2010;395:282–90.
Bebenek K, Pedersen LC, Kunkel TA. Structure-function studies of DNA polymerase lambda. Biochemistry. 2014;53:2781–92.
Cavanaugh NA, Beard WA, Batra VK, Perera L, Pedersen LG, Wilson SH. Molecular insights into DNA polymerase deterrents for ribonucleotide insertion. J Biol Chem. 2011;286:31650–60.
Richardson FC, Kuchta RD, Mazurkiewicz A, Richardson KA. Polymerization of 2′-fluoro- and 2'-O-methyl-dNTPs by human DNA polymerase alpha, polymerase gamma, and primase. Biochem Pharmacol. 2000;59:1045–52.
Clausen AR, Zhang S, Burgers PM, Lee MY, Kunkel TA. Ribonucleotide incorporation, proofreading and bypass by human DNA polymerase delta. DNA Repair (Amst). 2013;12:121–7.
Goksenin AY, Zahurancik W, LeCompte KG, Taggart DJ, Suo Z, Pursell ZF. Human DNA polymerase epsilon is able to efficiently extend from multiple consecutive ribonucleotides. J Biol Chem. 2012;287:42675–84.
Kasiviswanathan R, Copeland WC. Ribonucleotide discrimination and reverse transcription by the human mitochondrial DNA polymerase. J Biol Chem. 2011;286:31490–500.
Forslund JME, Pfeiffer A, Stojkovic G, Wanrooij PH, Wanrooij S. The presence of rNTPs decreases the speed of mitochondrial DNA replication. PLoS Genet. 2018;14:e1007315.
Berglund AK, Navarrete C, Engqvist MK, Hoberg E, Szilagyi Z, Taylor RW, Gustafsson CM, Falkenberg M, Clausen AR. Nucleotide pools dictate the identity and frequency of ribonucleotide incorporation in mitochondrial DNA. PLoS Genet. 2017;13:e1006628.
Crespan E, Furrer A, Rosinger M, Bertoletti F, Mentegari E, Chiapparini G, Imhof R, Ziegler N, Sturla SJ, Hubscher U, et al. Impact of ribonucleotide incorporation by DNA polymerases beta and lambda on oxidative base excision repair. Nat Commun. 2016;7:10805.
Cilli P, Minoprio A, Bossa C, Bignami M, Mazzei F. Formation and repair of mismatches containing ribonucleotides and oxidized bases at repeated DNA sequences. J Biol Chem. 2015;290:26259–69.
Nick McElhinny SA, Ramsden DA. Polymerase mu is a DNA-directed DNA/RNA polymerase. Mol Cell Biol. 2003;23:2309–15.
Pryor JM, Conlin MP, Carvajal-Garcia J, Luedeman ME, Luthman AJ, Small GW, Ramsden DA. Ribonucleotide incorporation enables repair of chromosome breaks by nonhomologous end joining. Science. 2018;361:1126–9.
Martin MJ, Garcia-Ortiz MV, Esteban V, Blanco L. Ribonucleotides and manganese ions improve non-homologous end joining by human Polmu. Nucleic Acids Res. 2013;41:2428–36.
Su Y, Egli M, Guengerich FP. Mechanism of ribonucleotide incorporation by human DNA polymerase eta. J Biol Chem. 2016;291:3747–56.
Donigan KA, McLenigan MP, Yang W, Goodman MF, Woodgate R. The steric gate of DNA polymerase iota regulates ribonucleotide incorporation and deoxyribonucleotide fidelity. J Biol Chem. 2014;289:9136–45.
Brown JA, Fowler JD, Suo Z. Kinetic basis of nucleotide selection employed by a protein template-dependent DNA polymerase. Biochemistry. 2010;49:5504–10.
Mentegari E, Crespan E, Bavagnoli L, Kissova M, Bertoletti F, Sabbioneda S, Imhof R, Sturla SJ, Nilforoushan A, Hubscher U, et al. Ribonucleotide incorporation by human DNA polymerase eta impacts translesion synthesis and RNase H2 activity. Nucleic Acids Res. 2017;45:2600–14.
Sparks JL, Chon H, Cerritelli SM, Kunkel TA, Johansson E, Crouch RJ, Burgers PM. RNase H2-initiated ribonucleotide excision repair. Mol Cell. 2012;47:980–6.
Eder PS, Walder JA. Ribonuclease H from K562 human erythroleukemia cells. Purification, characterization, and substrate specificity. J Biol Chem. 1991;266:6472–9.
Jeong HS, Backlund PS, Chen HC, Karavanov AA, Crouch RJ. RNase H2 of Saccharomyces cerevisiae is a complex of three proteins. Nucleic Acids Res. 2004;32:407–14.
Chon H, Vassilev A, DePamphilis ML, Zhao Y, Zhang J, Burgers PM, Crouch RJ, Cerritelli SM. Contributions of the two accessory subunits, RNASEH2B and RNASEH2C, to the activity and properties of the human RNase H2 complex. Nucleic Acids Res. 2009;37:96–110.
Bubeck D, Reijns MA, Graham SC, Astell KR, Jones EY, Jackson AP. PCNA directs type 2 RNase H activity on DNA replication and repair substrates. Nucleic Acids Res. 2011;39:3652–66.
Kind B, Muster B, Staroske W, Herce HD, Sachse R, Rapp A, Schmidt F, Koss S, Cardoso MC, Lee-Kirsch MA. Altered spatio-temporal dynamics of RNase H2 complex assembly at replication and repair sites in Aicardi-Goutieres syndrome. Hum Mol Genet. 2014;23:5950–60.
Ghodgaonkar MM, Lazzaro F, Olivera-Pimentel M, Artola-Boran M, Cejka P, Reijns MA, Jackson AP, Plevani P, Muzi-Falconi M, Jiricny J. Ribonucleotides misincorporated into DNA act as strand-discrimination signals in eukaryotic mismatch repair. Mol Cell. 2013;50:323–32.
Lujan SA, Williams JS, Clausen AR, Clark AB, Kunkel TA. Ribonucleotides are signals for mismatch repair of leading-strand replication errors. Mol Cell. 2013;50:437–43.
Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. 2010;17:421–33.
Leppard JB, Champoux JJ. Human DNA topoisomerase I: relaxation, roles, and damage control. Chromosoma. 2005;114:75–85.
Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3:430–40.
Sekiguchi J, Shuman S. Site-specific ribonuclease activity of eukaryotic DNA topoisomerase I. Mol Cell. 1997;1:89–97.
Kim N, Huang SN, Williams JS, Li YC, Clark AB, Cho JE, Kunkel TA, Pommier Y, Jinks-Robertson S. Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science. 2011;332:1561–4.
Huang SN, Williams JS, Arana ME, Kunkel TA, Pommier Y. Topoisomerase I-mediated cleavage at unrepaired ribonucleotides generates DNA double-strand breaks. EMBO J. 2017;36:361–73.
Huang SY, Ghosh S, Pommier Y. Topoisomerase I alone is sufficient to produce short DNA deletions and can also reverse nicks at ribonucleotide sites. J Biol Chem. 2015;290:14068–76.
Nick McElhinny SA, Kumar D, Clark AB, Watt DL, Watts BE, Lundstrom EB, Johansson E, Chabes A, Kunkel TA. Genome instability due to ribonucleotide incorporation into DNA. Nat Chem Biol. 2010;6:774–81.
Sparks JL, Burgers PM. Error-free and mutagenic processing of topoisomerase 1-provoked damage at genomic ribonucleotides. EMBO J. 2015;34:1259–69.
Moss CF, Dalla Rosa I, Hunt LE, Yasukawa T, Young R, Jones AWE, Reddy K, Desai R, Virtue S, Elgar G, et al. Aberrant ribonucleotide incorporation and multiple deletions in mitochondrial DNA of the murine MPV17 disease model. Nucleic Acids Res. 2017;45:12808–15.
Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, Tarnauskaite Z, Muir M, Fluteau A, Aregger M, McEwan A, et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature. 2018;559:285–9.
Wang Y, Knudsen BR, Bjergbaek L, Westergaard O, Andersen AH. Stimulated activity of human topoisomerases IIalpha and IIbeta on RNA-containing substrates. J Biol Chem. 1999;274:22839–46.
Gao R, Schellenberg MJ, Huang SY, Abdelmalak M, Marchand C, Nitiss KC, Nitiss JL, Williams RS, Pommier Y. Proteolytic degradation of topoisomerase II (Top2) enables the processing of Top2.DNA and Top2.RNA covalent complexes by tyrosyl-DNA-phosphodiesterase 2 (TDP2). J Biol Chem. 2014;289:17960–9.
Iyama T, Wilson DM 3rd. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst). 2013;12:620–36.
Sassa A, Beard WA, Prasad R, Wilson SH. DNA sequence context effects on the glycosylase activity of human 8-oxoguanine DNA glycosylase. J Biol Chem. 2012;287:36702–10.
Sassa A, Caglayan M, Rodriguez Y, Beard WA, Wilson SH, Nohmi T, Honma M, Yasui M. Impact of ribonucleotide backbone on Translesion synthesis and repair of 7,8-Dihydro-8-oxoguanine. J Biol Chem. 2016;291:24314–23.
Malfatti MC, Balachander S, Antoniali G, Koh KD, Saint-Pierre C, Gasparutto D, Chon H, Crouch RJ, Storici F, Tell G. Abasic and oxidized ribonucleotides embedded in DNA are processed by human APE1 and not by RNase H2. Nucleic Acids Res. 2017;45:11193–212.
Cai Y, Geacintov NE, Broyde S. Ribonucleotides as nucleotide excision repair substrates. DNA Repair (Amst). 2014;13:55–60.
Vaisman A, McDonald JP, Huston D, Kuban W, Liu L, Van Houten B, Woodgate R. Removal of misincorporated ribonucleotides from prokaryotic genomes: an unexpected role for nucleotide excision repair. PLoS Genet. 2013;9:e1003878.
Lindsey-Boltz LA, Kemp MG, Hu J, Sancar A. Analysis of ribonucleotide removal from DNA by human nucleotide excision repair. J Biol Chem. 2015;290:29801–7.
Tumbale P, Williams JS, Schellenberg MJ, Kunkel TA, Williams RS. Aprataxin resolves adenylated RNA-DNA junctions to maintain genome integrity. Nature. 2014;506:111–5.
Acknowledgements
We are grateful to Dr. Kiyoe Ura (Chiba University) and Mr. Hiroaki Tanuma (Chiba University) for critically reading the manuscript. We thank Enago (www.enago.jp) for English-language review.
Funding
This work was partly supported by JSPS KAKENHI Grant Numbers 16 K16195 and 25281022.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
AS, MY, and MH wrote or contributed to the writing of the review. MY and MH were involved in the critical revision of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sassa, A., Yasui, M. & Honma, M. Current perspectives on mechanisms of ribonucleotide incorporation and processing in mammalian DNA. Genes and Environ 41, 3 (2019). https://doi.org/10.1186/s41021-019-0118-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41021-019-0118-7