
Abstract
Metformin, an inexpensive and well-tolerated oral agent commonly used in the first-line treatment of type 2 diabetes, has become the focus of intense research as a candidate anticancer agent. Here, we discuss the potential of metformin in cancer therapeutics, particularly its functions in multiple signaling pathways, including AMP-activated protein kinase, mammalian target of rapamycin, insulin-like growth factor, c-Jun N-terminal kinase/mitogen-activated protein kinase (p38 MAPK), human epidermal growth factor receptor-2, and nuclear factor kappaB pathways. In addition, cutting-edge targeting of cancer stem cells by metformin is summarized.
Similar content being viewed by others

Background
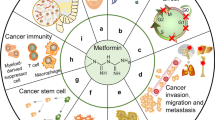
Metformin (1,1-dimethylbiguanide hydrochloride), a USA Food and Drug Administration (FDA)-approved biguanide derivative and the most widely prescribed antihyperglycemic drug, is used as first-line therapy for diabetes mellitus type 2. Metformin reduces blood glucose levels by inhibiting hepatic glucose production, increasing glucose uptake and utilization by the skeletal muscle, reducing insulin resistance in peripheral tissue, and suppressing gluconeogenesis in the liver [1–4]. Interestingly, metformin attracted increasing interests in recent years due to its anticancer effects [5–10]. The drug has been demonstrated to reduce the development of prostate cancer [11], lung cancer [12], breast cancer [13], esophageal cancer [14], colon cancer [15], and melanoma [16]. Several preclinical studies have reported that metformin reduced cell proliferation, induced apoptosis, and caused cell cycle arrest in vitro and also reduced occurrence and growth of experimental tumors in vivo [17–19]. Metformin can also be used as a sensitizer or be combined with conventional chemotherapeutic agents and radiotherapy to combat cancer [20–24]. Moreover, metformin plays an important role in targeting cancer stem cells (CSCs) [25] and reversing the epithelial-mesenchymal transition (EMT), a critical process in cancer metastasis [26]. The possible signaling pathways involved in the anticancer effects of metformin are outlined below and demonstrated in Fig. 1 and Table 1.

Signaling pathways through which metformin acts in cancer. IGF-1 insulin-like growth factor-1, MAPK mitogen-activated protein kinase, REDD1 regulated in development and DNA damage 1, AMPK adenosine monophosphate-activated protein kinase, mTOR mammalian target of rapamycin, NF-κB nuclear factor kappaB, MDR1 multidrug resistance 1, HER2 human epidermal growth factor receptor-2, IGF-1R IGF-1 receptor, IR insulin receptor
Activation of adenosine monophosphate-activated protein kinase (AMPK)
AMPK-dependent effects of metformin
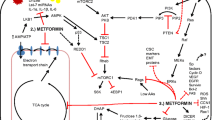
Activation of adenosine monophosphate-activated protein kinase, an intracellular energy sensor, is activated by elevating the ratio of adenosine monophosphate (AMP)/adenosine triphosphate (ATP). Once activated, AMPK restores cellular energy levels by inhibiting anabolic processes and promoting catabolic processes, e.g., glycolysis and fatty acid oxidation, to increase the AMP/ATP ratio [27, 28]. Vazquez-Martin et al. [29] reported that activation of AMPK inhibited cell mitosis and proliferation by directly influencing the dynamics of cell division during mitosis. Metformin has also been reported to exert its antineoplastic effects by stimulating AMPK [30–32] through up-regulation of the p53–p21 axis and down-regulation of cyclin D1 levels. Metformin inhibits the corresponding cyclin-dependent kinases and then induces G1-phase arrest of the cell cycle [33]. Moreover, Kisfalvi et al. [34] reported that metformin caused sustained and significant increases in AMPK activity through Thr172 phosphorylation and that the specific AMPK inhibitor compound C attenuated the effect of metformin on DNA synthesis, revealing an AMPK-dependent pathway for metformin treatment of pancreatic cancer. However, Klubo-Gwiezdzinska et al. [35] observed different results that AMPKα knockdown by small interfering RNA (siRNA) and compound C did not prevent the growth-inhibitory effects of metformin on medullary thyroid cancer cells. Shi et al. [36] observed that both molecular and pharmacologic knockdown of AMPK counteracted the metformin-induced growth inhibition and G0/G1 cell cycle arrest of lymphoma cells. Shi et al. [36] reported that AMPKα siRNA caused not only a striking attenuation of the lymphoma cell response to metformin but also a further growth inhibition when it was combined with doxorubicin. Moreover, in acute lymphoblastic leukemia (ALL), knockdown of AMPKα by short hairpin RNA (shRNA) rescued cells from metformin-induced apoptosis, which was associated with restoration of the unfolded protein response (UPR)/glucose-regulated protein 78 kDa (GRP78) function, down-regulation of UPR apoptotic markers inositol-requiring enzyme 1α (IRE1α) and C/EBP homologous protein (CHOP), and interruption of protein synthesis. Studies on breast cancer therapy have demonstrated that inhibition of AMPK with siRNA decreased the suppression of multidrug resistance 1 (MDR1) gene activation after exposure to metformin [37]. Furthermore, overexpression of a dominant-negative mutant of AMPK attenuated the inhibitory effects of metformin on the phosphorylation of cAMP-responsive element-binding protein (CREB) and the expression of MDR1 [37]. Taken together, the antidiabetic drug metformin exhibits anticancer effects that are associated with activation of the AMPK signaling pathway.
AMPK-independent effects of metformin
In contrast to the above findings, Sahra et al. [38] reported that the anti-proliferation effect of metformin was independent of the AMPK pathway. They used AMPK siRNA to inhibit the two catalytic subunits of AMPK, but AMPK inhibition did not block the G0/G1 cell cycle arrest induced by metformin. Their subsequent study showed that a negative regulator of mammalian target of rapamycin (mTOR), regulated in development and DNA damage 1 (REDD1), mediated the effects of metformin on the cell cycle arrest and cyclin D1 alteration [39]. Similarly, Yasmeen et al. [40] found that metformin-induced apoptosis of human ovarian cancer cells was independent of AMPK. In addition, AMPK deficiency sensitized cancer cells to the growth-inhibitory effects of metformin [41]. Arai et al. [42] demonstrated that metformin-mediated repression of chronic inflammatory responses was associated with inhibition of tumor necrosis factor alpha (TNFα) production in human monocytes, an event that was most likely independent of AMPK activation. Chronic inflammation may provide a basis for cancer progression, but there was no obvious change in phosphor-AMPKα observed after metformin treatment [43]. Collectively, these studies provide compelling evidence that certain antitumor effects of metformin are independent of the AMPK signaling pathway [38–43].
Inhibition of the mTOR pathway
mTOR plays a critical role in regulating cellular energy homeostasis by modulating cellular processes such as protein synthesis and autophagy [44–47]. mTOR signaling exerts significant positive regulation of cell proliferation and tumorigenesis in diverse cancers, and it is frequently aberrantly activated in cancers. Activation of mTOR is associated with malignant tumor progression, resistance to chemotherapy and molecularly targeted therapies, and dismal prognosis [48–52]. mTOR is involved in the formation of two functionally and biochemically discrete signaling complexes: rapamycin with either nutrient-sensitive mTOR1 or nutrient-insensitive mTOR2 [53]. Components upstream of mTOR1 include tuberous sclerosis complex 1 (TSC1) and 2 (TSC2) [54, 55]. The combination of TSC1 and TSC2 functions as a tumor inhibitory complex that suppresses mTOR activity. Such mTOR signaling suppression reduces the phosphorylation of major downstream substrates, such as the eukaryotic initiation factor 4E-binding protein 1 (4E-BP1), ribosomal protein S6 kinase (S6K), and initiation factor eIF4G, and net inhibition of global protein synthesis and proliferation in a large number of cancers [56–58].
Metformin-induced inhibition of the mTOR pathway has been demonstrated in different types of cancer, such as leukemia [30, 32, 59, 60], lung cancer [61, 62], breast cancer [63, 64], oral squamous cell carcinoma [65], lymphoma [36], and thyroid cancer [35, 66] in human, as well as in both papilloma and squamous cell carcinoma in mice [67]. Metformin induces the liver kinase B1 (LKB1)-mediated activation of AMPK, which in turn blocks mTOR signaling and protein synthesis in many cancer cell lines [58, 68, 69]. AMPK impacts mTOR through phosphorylation and activation of the tumor suppressor TSC2, which results in inhibition of a downstream small GTPase (RHEB), negatively regulating mTOR activity [70, 71]. In contrast, metformin can also inhibit mTOR in an AMPK-independent pathway by reducing the levels of insulin-like growth factor-1 (IGF-1) [72, 73]. Kalender et al. [74] have shown that the inhibitory effects of metformin on mTOR signaling were mediated by Rag GTPases in the absence of AMPK and TSC1/2. Of note, one study indicated that metformin directly influenced mTOR in a p53-dependent manner through an AMPK-independent mechanism to boost the level of REDD1, a negative regulator of mTOR [39]. In that report, REDD1 inactivation, using siRNA or REDD1−/− cells, abrogated cell cycle arrest independently of AMPK.
Suppression of the IGF signaling pathway
Insulin and IGFs are key regulators of metabolism and growth. A rapidly growing body of researches has revealed that insulin and IGFs are associated with cancer progression by activating signaling pathways that are associated with cell growth and proliferation [75]. There are two subtypes of IGF, IGF-1 and IGF-2, which are both mitogenic and antiapoptotic. IGF-1 receptor (IGF-1R) binds to the ligand IGF-1, IGF-2, or insulin to promote autophosphorylation of tyrosine at its kinase domain. This triggers tyrosine and serine phosphorylation to form binding sites for insulin receptor substrates (IRSs) and Src and concomitant activation of signaling through the phosphatidylinositol-3-kinase (PI3K)/Akt/mTOR and RAS/RAF/mitogen-activated protein kinase (MAPK) pathways [76–78]. Moreover, overexpression of IGF-1R can induce tumor formation and metastasis [79, 80]. Likewise, in endometrial cancer cells, overexpression of IGF-1R triggers endometrial hyperplasia and contributes to type I epithelial cell growth by activating PI3K/Akt/mTOR signaling [3, 81, 82]. In addition, activation of IGF accelerated YYH1 tumor progression by promoting vascular smooth muscle cell proliferation, migration, and angiogenesis [83].
Emerging evidence suggests that metformin can exert its anticancer functions by reducing the levels of IGF-1. Metformin, which acts as an insulin-sensitizing agent, decreases IGF-1 by indirectly down-regulating insulin and insulin-binding proteins to reverse hyperinsulinemia, which may be a mechanism for metformin’s anticancer effects [75]. In fact, Memmott et al. [62] observed that metformin acted by reducing the circulating levels of insulin and IGF-1 to block tumor growth and proliferation in a tobacco carcinogen-induced lung cancer model in A/J mice. Similarly, Malaguarnera et al. [84] recently confirmed that metformin reduced cell proliferation, invasion, and clonogenic capacity by preventing the androgen-mediated up-regulation of IGF-1R. Moreover, recent studies have shown that metformin-mediated activation of AMPK increased the phosphorylation of IRS-1, diminishing the IGF-1-induced activation of Akt/TSC1/mTOR [85–87]. Another mechanism relevant to IGF-1 could be disruption of the crosstalk between insulin receptor/IGF-1R and G protein-coupled receptor (GPCR) signaling via metformin-induced activation of AMPK [86, 88].
Inhibition of other signaling pathways
Metformin and the JNK/p38 MAPK pathway
Other possible mechanisms for the beneficial effects of metformin on cancer development have also been described. The MAPK-involved pathways are significant intracellular signaling pathways that regulate cell growth, differentiation, proliferation, apoptosis, and migration [89–92]. Four major MAPK pathways have been described: extracellular signal-regulated kinase (ERK, also known as p42/44 MAPK), big MAP kinase (BMK, also known as ERK5), p38 MAPK (also known as SAPK2/RK), and c-jun N-terminal kinase (JNK, also known as stress-activated protein kinase 1 [SAPK1]) pathways. Although not universally observed in all cells, metformin has been found to be relevant to MAPK signaling in certain malignancies such as lung cancer [61, 93], human fibrosarcoma [94], breast cancer [95–98], prostate cancer [99], endometrial cancer [100], and cutaneous squamous cell carcinoma [101]. Metformin has been shown to exert an apoptosis-mediated effect through activating the JNK/p38 MAPK pathway and enhancing expression of growth inhibition and DNA damage-inducible gene 153 (GADD153) [102]. Monteagudo et al. [99] used a dendrimer-vehiculized siRNA to block the MAPK signaling pathway and found that the blockade enhanced the anticancer effect of metformin. Other data from Tseng et al. [93] suggested that metformin could reduce paclitaxel-induced, p38 MAPK-mediated expression of excision repair cross complementary 1.
Metformin and the HER2 pathway
Human epidermal growth factor receptor-2 (HER2) belongs to the epidermal growth factor receptor family, the members of which possess tyrosine kinase activity. HER2 is overexpressed in approximately 20%–30% of breast cancers. As a significant biomarker of breast cancer, HER2 is a crucial therapeutic target in breast cancers that overexpress HER2. Vazquez-Martin et al. [103] studied the effects of metformin on cultured human breast cancer cells with HER2 amplification and observed that ectopic overexpression of the HER2 oncogene significantly enhanced metformin-induced growth inhibition. They also reported that metformin suppressed HER2 oncoprotein overexpression via AMPK-independent inhibition of mTOR in human breast cancer cells [103]. Interestingly, metformin notably blocked HER2 tyrosine kinase activity at low therapeutic concentrations [96]. In addition, it was found that metformin combination therapy with the anti-HER2 monoclonal antibody trastuzumab could eliminate stem/progenitor cell populations in HER2-amplified breast carcinoma cells [104].
Metformin and the NF-κB pathway
Nuclear factor kappaB (NF-κB) is a protein complex that functions as a signal-induced transcription factor to regulate proliferation and apoptosis [105]. It is an important potential target in cancer therapy [106–108]. Inhibition of NF-κB can induce cancer cells to halt proliferation and die or can sensitize cells to chemotherapeutic reagents [109]. Kim et al. [37] reported that metformin activated AMPK and inhibited mTOR by suppressing NF-κB and CREB. Later, Chaudhary et al. [101] observed that the growth-inhibitory effect of metformin repressed the NF-κB and mTOR signaling pathways. Additionally, Zheng et al. [110] showed that metformin dampened NF-κB signaling by boosting NF-κB inhibitor alpha (IκBa) in hepatocellular carcinoma cell lines. Moreover, forced expression of p65 or overexpression of an undegradable mutant form of IκBa was found to activate NF-κB signaling, thereby attenuating the antitumor effects of metformin.
Metformin targets cancer stem cells
Cancer stem cells, also called tumor-initiating cells, are a subset of cancer cells that are believed to have indefinite potential capacity to self-renew and result in tumorigenesis [111]. Compared with non-cancer stem cells, CSCs are both chemoresistant [112–116] and radioresistant [116–119]. CSCs are compelling candidates for tumor origination and may contribute to cancer metastasis and relapse, which are the main impediments to prolonging overall survival. Of note, self-renewal and inherent chemoresistance are responsible for tumor recurrence [120]. Therefore, development of non-toxic treatment strategies targeting CSCs will be of significant therapeutic benefit.
Metformin inhibition of CSCs was first demonstrated in 2009 in preclinical breast cancer models [121]. Subsequent reports indicated that metformin improved the response of human cancer xenografts to conventional chemotherapy by eradicating CSCs in multiple cancer types [104, 122]. In parallel, metformin down-regulates CSC marker genes in pancreatic cancer [123, 124], esophageal cancer [125], and breast cancer [126]. In pancreatic cancer, metformin inhibits cell proliferation, migration, and invasion by weakening CSC function mediated by deregulating miRNAs [123]. In esophageal cancer, metformin inhibits esophageal cancer cell growth and sensitizes cells to the cytotoxic effects of 5-fluorouracil (5-FU) by targeting CSCs and mTOR [125]. Regarding the mechanisms by which metformin targets CSCs, Song et al. [127] reported that metformin increased the sensitivity of cancer cells to radiotherapy and exhibited cytotoxicity toward CSCs, overcoming their radioresistance via activation of AMPK and suppression of mTOR. In contrast, Hirsch et al. [128] reported that metformin selectively suppressed NF-κB nuclear localization and Stat3 activity in CSCs.
Conclusions
In conclusion, in vitro and in vivo studies strongly indicate that metformin, a widely prescribed oral medication used as front-line therapy for type 2 diabetes, could be a valuable adjuvant therapy for cancer. Metformin may become a useful adjuvant drug in association with established anticancer therapies, and there are multiple clinical trials examining the effects of metformin on cancer outcomes. In general, most data support the hypothesis that metformin is protective against cancer. However, based on the current preliminary findings, it appears that metformin is not an effective treatment alone for unselected patient populations or larger number of patients. Therefore, we recommend that combination therapies with metformin as well as potential novel biomarkers that could identify patient populations sensitive to metformin treatment should be pursued. Further studies are needed to improve our understanding of the pathways linking high metformin efficacy and cancer development.
Overall, the biological effect of metformin on cancer cells is based on its ability to activate AMPK or inhibit downstream growth factor signaling through inhibition of mTOR. Metformin also has indirect effects on the IGF and JNK/p38 MAPK pathways; other possible mechanisms include inhibition of the HER2 and NF-κB signaling pathways. Further support for these observations is that metformin kills cancer stem cells and changes the properties of CSCs. Nonetheless, a large number of further translational studies are required to evaluate the potential of metformin as an additive antitumoral agent.
References
Dowling RJ, Goodwin PJ, Stambolic V. Understanding the benefit of metformin use in cancer treatment. BMC Med. 2011;9:33.
Hundal HS, Ramlal T, Reyes R, et al. Cellular mechanism of metformin action involves glucose transporter translocation from an intracellular pool to the plasma membrane in l6 muscle cells. Endocrinology. 1992;131(3):1165–73.
Li X, Zhang N, Li Y, et al. Effects of metformin and rosiglitazone on peripheral insulin resistance and beta-cell function in obesity: a double-blind, randomized, controlled study. J Int Med Res. 2011;39(2):358–65.
Witters LA. The blooming of the french lilac. J Clin Invest. 2001;108(8):1105–7.
Sahra IB, Le Marchand-Brustel Y, Tanti JF, et al. Metformin in cancer therapy: a new perspective for an old antidiabetic drug? Mol Cancer Ther. 2010;9(5):1092–9.
Gonzalez-Angulo AM, Meric-Bernstam F. Metformin: a therapeutic opportunity in breast cancer. Clin Cancer Res. 2010;16(6):1695–700.
Tomic T, Botton T, Cerezo M, et al. Metformin inhibits melanoma development through autophagy and apoptosis mechanisms. Cell Death Dis. 2011;2:e199.
Salani B, Maffioli S, Hamoudane M, et al. Caveolin-1 is essential for metformin inhibitory effect on igf1 action in non-small-cell lung cancer cells. FASEB J. 2012;26(2):788–98.
Quinn BJ, Dallos M, Kitagawa H, et al. Inhibition of lung tumorigenesis by metformin is associated with decreased plasma igf-i and diminished receptor tyrosine kinase signaling. Cancer Prev Res (Phila). 2013;6(8):801–10.
Feng Y, Ke C, Tang Q, et al. Metformin promotes autophagy and apoptosis in esophageal squamous cell carcinoma by downregulating stat3 signaling. Cell Death Dis. 2014;5:e1088.
Colquhoun AJ, Venier NA, Vandersluis AD, et al. Metformin enhances the antiproliferative and apoptotic effect of bicalutamide in prostate cancer. Prostate Cancer Prostatic Dis. 2012;15(4):346–52.
Li L, Han R, Xiao H, et al. Metformin sensitizes egfr-tki-resistant human lung cancer cells in vitro and in vivo through inhibition of il-6 signaling and emt reversal. Clin Cancer Res. 2014;20(10):2714–26.
Blandino G, Valerio M, Cioce M, et al. Metformin elicits anticancer effects through the sequential modulation of dicer and c-myc. Nat Commun. 2012;3:865.
Xu Y, Lu S. Metformin inhibits esophagus cancer proliferation through upregulation of usp7. Cell Physiol Biochem. 2013;32(5):1178–86.
Algire C, Amrein L, Zakikhani M, et al. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr Relat Cancer. 2010;17(2):351–60.
Niehr F, von Euw E, Attar N, et al. Combination therapy with vemurafenib (plx4032/rg7204) and metformin in melanoma cell lines with distinct driver mutations. J Transl Med. 2011;9:76.
Han G, Gong H, Wang Y, et al. Ampk/mtor-mediated inhibition of survivin partly contributes to metformin-induced apoptosis in human gastric cancer cell. Cancer Biol Ther. 2015;16:77–87.
Zhao D, Long XD, Lu TF, et al. Metformin decreases il-22 secretion to suppress tumor growth in an orthotopic mouse model of hepatocellular carcinoma. Int J Cancer. 2015;136(11):2556–65.
Zi FM, He JS, Li Y, et al. Metformin displays anti-myeloma activity and synergistic effect with dexamethasone in in vitro and in vivo xenograft models. Cancer Lett. 2014;356(2):443–53.
Qu C, Zhang W, Zheng G, et al. Metformin reverses multidrug resistance and epithelial-mesenchymal transition (emt) via activating amp-activated protein kinase (ampk) in human breast cancer cells. Mol Cell Biochem. 2014;386(1–2):63–71.
Lengyel E, Litchfield LM, Mitra AK, et al. Metformin inhibits ovarian cancer growth and increases sensitivity to paclitaxel in mouse models. Am J Obstet Gynecol. 2015;212(4):479.e1–10. doi:10.1016/j.ajog.2014.10.026.
Uehara T, Mitsuhashi A, Tsuruoka N, et al. Metformin potentiates the anticancer effects of cisplatin under normoxic conditions in vitro. Oncol Rep. 2015;33(2):744–50.
Fasih A, Elbaz HA, Huttemann M, et al. Radiosensitization of pancreatic cancer cells by metformin through the ampk pathway. Radiat Res. 2014;182(1):50–9.
Zhang Y, Storr SJ, Johnson K, et al. Involvement of metformin and ampk in the radioresponse and prognosis of luminal versus basal-like breast cancer treated with radiotherapy. Oncotarget. 2014;5(24):12936–49.
Nangia-Makker P, Yu Y, Vasudevan A, et al. Metformin: a potential therapeutic agent for recurrent colon cancer. PLoS ONE. 2014;9(1):e84369.
Barriere G, Tartary M, Rigaud M. Metformin: a rising star to fight the epithelial mesenchymal transition in oncology. Anticancer Agents Med Chem. 2013;13(2):333–40.
Shaw RJ, Lamia KA, Vasquez D, et al. The kinase lkb1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310(5754):1642–6.
Hardie DG. Amp-activated/snf1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8(10):774–85.
Vazquez-Martin A, Lopez-Bonet E, Oliveras-Ferraros C, et al. Mitotic kinase dynamics of the active form of ampk (phospho-ampkalphathr172) in human cancer cells. Cell Cycle. 2009;8(5):788–91.
Vakana E, Altman JK, Glaser H, et al. Antileukemic effects of ampk activators on bcr-abl-expressing cells. Blood. 2011;118(24):6399–402.
Algire C, Amrein L, Bazile M, et al. Diet and tumor lkb1 expression interact to determine sensitivity to anti-neoplastic effects of metformin in vivo. Oncogene. 2011;30(10):1174–82.
Grimaldi C, Chiarini F, Tabellini G, et al. Amp-dependent kinase/mammalian target of rapamycin complex 1 signaling in t-cell acute lymphoblastic leukemia: therapeutic implications. Leukemia. 2012;26(1):91–100.
Cazzaniga M, Bonanni B, Guerrieri-Gonzaga A, et al. Is it time to test metformin in breast cancer clinical trials? Cancer Epidemiol Biomarkers Prev. 2009;18(3):701–5.
Kisfalvi K, Eibl G, Sinnett-Smith J, et al. Metformin disrupts crosstalk between g protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res. 2009;69(16):6539–45.
Klubo-Gwiezdzinska J, Jensen K, Costello J, et al. Metformin inhibits growth and decreases resistance to anoikis in medullary thyroid cancer cells. Endocr Relat Cancer. 2012;19(3):447–56.
Shi WY, Xiao D, Wang L, et al. Therapeutic metformin/ampk activation blocked lymphoma cell growth via inhibition of mtor pathway and induction of autophagy. Cell Death Dis. 2012;3:e275.
Kim HG, Hien TT, Han EH, et al. Metformin inhibits p-glycoprotein expression via the nf-kappab pathway and cre transcriptional activity through ampk activation. Br J Pharmacol. 2011;162(5):1096–108.
Sahra IB, Laurent K, Loubat A, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin d1 level. Oncogene. 2008;27(25):3576–86.
Sahra IB, Regazzetti C, Robert G, et al. Metformin, independent of ampk, induces mtor inhibition and cell-cycle arrest through redd1. Cancer Res. 2011;71(13):4366–72.
Yasmeen A, Beauchamp MC, Piura E, et al. Induction of apoptosis by metformin in epithelial ovarian cancer: involvement of the bcl-2 family proteins. Gynecol Oncol. 2011;121(3):492–8.
Muaddi H, Chowdhury S, Vellanki R, et al. Contributions of ampk and p53 dependent signaling to radiation response in the presence of metformin. Radiother Oncol. 2013;108(3):446–50.
Arai M, Uchiba M, Komura H, et al. Metformin, an antidiabetic agent, suppresses the production of tumor necrosis factor and tissue factor by inhibiting early growth response factor-1 expression in human monocytes in vitro. J Pharmacol Exp Ther. 2010;334(1):206–13.
Gou S, Cui P, Li X, et al. Low concentrations of metformin selectively inhibit cd133(+) cell proliferation in pancreatic cancer and have anticancer action. PLoS ONE. 2013;8(5):e63969.
Zoncu R, Efeyan A, Sabatini DM. Mtor: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35.
Yecies JL, Manning BD. Mtor links oncogenic signaling to tumor cell metabolism. J Mol Med (Berl). 2011;89(3):221–8.
Cheaib B, Auguste A, Leary A. The pi3 k/akt/mtor pathway in ovarian cancer: therapeutic opportunities and challenges. Chin J Cancer. 2015;34(1):4–16.
Zhou W, Marcus AI, Vertino PM. Dysregulation of mtor activity through lkb1 inactivation. Chin J Cancer. 2013;32(8):427–33.
Hay N, Sonenberg N. Upstream and downstream of mtor. Genes Dev. 2004;18(16):1926–45.
Dancey J. Mtor signaling and drug development in cancer. Nat Rev Clin Oncol. 2010;7(4):209–19.
Klumpen HJ, Beijnen JH, Gurney H, et al. Inhibitors of mtor. Oncologist. 2010;15(12):1262–9.
Nahta R, O’Regan RM. Evolving strategies for overcoming resistance to her2-directed therapy: targeting the pi3k/akt/mtor pathway. Clin Breast Cancer. 2010;10(Suppl3):S72–8.
Sheri A, Martin LA, Johnston S. Targeting endocrine resistance: is there a role for mtor inhibition? Clin Breast Cancer. 2010;10(Suppl 3):S79–85.
Wullschleger S, Loewith R, Hall MN. Tor signaling in growth and metabolism. Cell. 2006;124(3):471–84.
Inoki K, Corradetti MN, Guan KL. Dysregulation of the tsc-mtor pathway in human disease. Nat Genet. 2005;37(1):19–24.
Shaw RJ, Cantley LC. Ras, pi(3)k and mtor signalling controls tumour cell growth. Nature. 2006;441(7092):424–30.
Holz MK, Ballif BA, Gygi SP, et al. Mtor and s6k1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123(4):569–80.
Zakikhani M, Dowling R, Fantus IG, et al. Metformin is an amp kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66(21):10269–73.
Dowling RJ, Zakikhani M, Fantus IG, et al. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67(22):10804–12.
Green AS, Chapuis N, Maciel TT, et al. The lkb1/ampk signaling pathway has tumor suppressor activity in acute myeloid leukemia through the repression of mtor-dependent oncogenic mrna translation. Blood. 2010;116(20):4262–73.
Pan J, Chen C, Jin Y, et al. Differential impact of structurally different anti-diabetic drugs on proliferation and chemosensitivity of acute lymphoblastic leukemia cells. Cell Cycle. 2012;11(12):2314–26.
Morgillo F, Sasso FC, Della Corte CM, et al. Synergistic effects of metformin treatment in combination with gefitinib, a selective egfr tyrosine kinase inhibitor, in lkb1 wild-type nsclc cell lines. Clin Cancer Res. 2013;19(13):3508–19.
Memmott RM, Mercado JR, Maier CR, et al. Metformin prevents tobacco carcinogen-induced lung tumorigenesis. Cancer Prev Res (Phila). 2010;3(9):1066–76.
Rocha GZ, Dias MM, Ropelle ER, et al. Metformin amplifies chemotherapy-induced ampk activation and antitumoral growth. Clin Cancer Res. 2011;17(12):3993–4005.
Deng XS, Wang S, Deng A, et al. Metformin targets stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle. 2012;11(2):367–76.
Vitale-Cross L, Molinolo AA, Martin D, et al. Metformin prevents the development of oral squamous cell carcinomas from carcinogen-induced premalignant lesions. Cancer Prev Res (Phila). 2012;5(4):562–73.
Cho SW, Yi KH, Han SK, et al. Therapeutic potential of metformin in papillary thyroid cancer in vitro and in vivo. Mol Cell Endocrinol. 2014;393(1–2):24–9.
Checkley LA, Rho O, Angel JM, et al. Metformin inhibits skin tumor promotion in overweight and obese mice. Cancer Prev Res (Phila). 2014;7(1):54–64.
Shaw RJ, Bardeesy N, Manning BD, et al. The lkb1 tumor suppressor negatively regulates mtor signaling. Cancer Cell. 2004;6(1):91–9.
Vazquez-Martin A, Oliveras-Ferraros C, del Barco S, et al. The antidiabetic drug metformin: a pharmaceutical ampk activator to overcome breast cancer resistance to her2 inhibitors while decreasing risk of cardiomyopathy. Ann Oncol. 2009;20(3):592–5.
Inoki K, Zhu T, Guan KL. Tsc2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–90.
Gwinn DM, Shackelford DB, Egan DF, et al. Ampk phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–26.
Engelman JA, Cantley LC. Chemoprevention meets glucose control. Cancer Prev Res (Phila). 2010;3(9):1049–52.
Memmott RM, Dennis PA. Lkb1 and mammalian target of rapamycin as predictive factors for the anticancer efficacy of metformin. J Clin Oncol. 2009;27(34):e226 (author reply e227).
Kalender A, Selvaraj A, Kim SY, et al. Metformin, independent of ampk, inhibits mtorc1 in a rag gtpase-dependent manner. Cell Metab. 2010;11(5):390–401.
Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8(12):915–28.
Baserga R, Peruzzi F, Reiss K. The igf-1 receptor in cancer biology. Int J Cancer. 2003;107(6):873–7.
Manning BD, Cantley LC. Akt/pkb signaling: navigating downstream. Cell. 2007;129(7):1261–74.
Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by erk, jnk, and p38 protein kinases. Science. 2002;298(5600):1911–2.
Lopez T, Hanahan D. Elevated levels of igf-1 receptor convey invasive and metastatic capability in a mouse model of pancreatic islet tumorigenesis. Cancer Cell. 2002;1(4):339–53.
Jones RA, Campbell CI, Gunther EJ, et al. Transgenic overexpression of igf-ir disrupts mammary ductal morphogenesis and induces tumor formation. Oncogene. 2007;26(11):1636–44.
McCampbell AS, Broaddus RR, Loose DS, et al. Overexpression of the insulin-like growth factor i receptor and activation of the akt pathway in hyperplastic endometrium. Clin Cancer Res. 2006;12(21):6373–8.
Wang CF, Zhang G, Zhao LJ, et al. Overexpression of the insulin receptor isoform a promotes endometrial carcinoma cell growth. PLoS ONE. 2013;8(8):e69001.
Clemmons DR, Maile LA, Ling Y, et al. Role of the integrin alphavbeta3 in mediating increased smooth muscle cell responsiveness to igf-i in response to hyperglycemic stress. Growth Horm IGF Res. 2007;17(4):265–70.
Malaguarnera R, Sacco A, Morcavallo A, et al. Metformin inhibits androgen-induced igf-ir up-regulation in prostate cancer cells by disrupting membrane-initiated androgen signaling. Endocrinology. 2014;155(4):1207–21.
Zakikhani M, Blouin MJ, Piura E, et al. Metformin and rapamycin have distinct effects on the akt pathway and proliferation in breast cancer cells. Breast Cancer Res Treat. 2010;123(1):271–9.
Ning J, Clemmons DR. Amp-activated protein kinase inhibits igf-i signaling and protein synthesis in vascular smooth muscle cells via stimulation of insulin receptor substrate 1 s794 and tuberous sclerosis 2 s1345 phosphorylation. Mol Endocrinol. 2010;24(6):1218–29.
Karnevi E, Said K, Andersson R, et al. Metformin-mediated growth inhibition involves suppression of the igf-i receptor signalling pathway in human pancreatic cancer cells. BMC Cancer. 2013;13:235.
Rozengurt E, Sinnett-Smith J, Kisfalvi K. Crosstalk between insulin/insulin-like growth factor-1 receptors and g protein-coupled receptor signaling systems: a novel target for the antidiabetic drug metformin in pancreatic cancer. Clin Cancer Res. 2010;16(9):2505–11.
Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81(2):807–69.
McCubrey JA, Steelman LS, Chappell WH, et al. Roles of the raf/mek/erk pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773(8):1263–84.
Sharma A, Tran MA, Liang S, et al. Targeting mitogen-activated protein kinase/extracellular signal-regulated kinase kinase in the mutant (v600e) b-raf signaling cascade effectively inhibits melanoma lung metastases. Cancer Res. 2006;66(16):8200–9.
Panka DJ, Atkins MB, Mier JW. Targeting the mitogen-activated protein kinase pathway in the treatment of malignant melanoma. Clin Cancer Res. 2006;12(7 Pt 2):2371s–5s.
Tseng SC, Huang YC, Chen HJ, et al. Metformin-mediated downregulation of p38 mitogen-activated protein kinase-dependent excision repair cross-complementing 1 decreases DNA repair capacity and sensitizes human lung cancer cells to paclitaxel. Biochem Pharmacol. 2013;85(4):583–94.
Hwang YP, Jeong HG. Metformin blocks migration and invasion of tumour cells by inhibition of matrix metalloproteinase-9 activation through a calcium and protein kinase calpha-dependent pathway: phorbol-12-myristate-13-acetate-induced/extracellular signal-regulated kinase/activator protein-1. Br J Pharmacol. 2010;160(5):1195–211.
Liu B, Fan Z, Edgerton SM, et al. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle. 2009;8(13):2031–40.
Alimova IN, Liu B, Fan Z, et al. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle. 2009;8(6):909–15.
Zordoky BN, Bark D, Soltys CL, et al. The anti-proliferative effect of metformin in triple-negative mda-mb-231 breast cancer cells is highly dependent on glucose concentration: implications for cancer therapy and prevention. Biochim Biophys Acta. 2014;1840(6):1943–57.
Queiroz EA, Puukila S, Eichler R, et al. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, ampk and foxo3a in mcf-7 breast cancer cells. PLoS ONE. 2014;9(5):e98207.
Monteagudo S, Perez-Martinez FC, Perez-Carrion MD, et al. Inhibition of p42 mapk using a nonviral vector-delivered sirna potentiates the anti-tumor effect of metformin in prostate cancer cells. Nanomedicine (Lond). 2012;7(4):493–506.
Zhang Q, Celestino J, Schmandt R, et al. Chemopreventive effects of metformin on obesity-associated endometrial proliferation. Am J Obstet Gynecol. 2013;209(1):24.e1–24.e12. doi:10.1016/j.ajog.2013.03.008
Chaudhary SC, Kurundkar D, Elmets CA, et al. Metformin, an antidiabetic agent reduces growth of cutaneous squamous cell carcinoma by targeting mtor signaling pathway. Photochem Photobiol. 2012;88(5):1149–56.
Wu N, Gu C, Gu H, et al. Metformin induces apoptosis of lung cancer cells through activating jnk/p38 mapk pathway and gadd153. Neoplasma. 2011;58(6):482–90.
Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The antidiabetic drug metformin suppresses her2 (erbb-2) oncoprotein overexpression via inhibition of the mtor effector p70s6k1 in human breast carcinoma cells. Cell Cycle. 2009;8(1):88–96.
Vazquez-Martin A, Oliveras-Ferraros C, Del Barco S, et al. The anti-diabetic drug metformin suppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. Breast Cancer Res Treat. 2011;126(2):355–64.
Rayet B, Gelinas C. Aberrant rel/nfkb genes and activity in human cancer. Oncogene. 1999;18(49):6938–47.
Wang CY, Cusack JC Jr, Liu R, et al. Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of nf-kappab. Nat Med. 1999;5(4):412–7.
Jung M, Dritschilo A. Nf-kappa b signaling pathway as a target for human tumor radiosensitization. Semin Radiat Oncol. 2001;11(4):346–51.
Aggarwal BB. Nuclear factor-kappab: the enemy within. Cancer Cell. 2004;6(3):203–8.
Escarcega RO, Fuentes-Alexandro S, Garcia-Carrasco M, et al. The transcription factor nuclear factor-kappa b and cancer. Clin Oncol (R Coll Radiol). 2007;19(2):154–61.
Zheng L, Yang W, Wu F, et al. Prognostic significance of ampk activation and therapeutic effects of metformin in hepatocellular carcinoma. Clin Cancer Res. 2013;19(19):5372–80.
Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–11.
Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58:267–84.
Lou H, Dean M. Targeted therapy for cancer stem cells: the patched pathway and abc transporters. Oncogene. 2007;26(9):1357–60.
Ma S, Lee TK, Zheng BJ, et al. Cd133+ hcc cancer stem cells confer chemoresistance by preferential expression of the akt/pkb survival pathway. Oncogene. 2008;27(12):1749–58.
Zhou BB, Zhang H, Damelin M, et al. Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nat Rev Drug Discov. 2009;8(10):806–23.
Frosina G. DNA repair and resistance of gliomas to chemotherapy and radiotherapy. Mol Cancer Res. 2009;7(7):989–99.
Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–60.
Phillips TM, McBride WH, Pajonk F. The response of cd24(-/low)/cd44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;98(24):1777–85.
Baumann M, Krause M, Hill R. Exploring the role of cancer stem cells in radioresistance. Nat Rev Cancer. 2008;8(7):545–54.
Alvero AB, Chen R, Fu HH, et al. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle. 2009;8(1):158–66.
Hirsch HA, Iliopoulos D, Tsichlis PN, et al. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69(19):7507–11.
Iliopoulos D, Hirsch HA, Struhl K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 2011;71(9):3196–201.
Bao B, Wang Z, Ali S, et al. Metformin inhibits cell proliferation, migration and invasion by attenuating csc function mediated by deregulating mirnas in pancreatic cancer cells. Cancer Prev Res (Phila). 2012;5(3):355–64.
Mohammed A, Janakiram NB, Brewer M, et al. Antidiabetic drug metformin prevents progression of pancreatic cancer by targeting in part cancer stem cells and mtor signaling. Transl Oncol. 2013;6(6):649–59.
Honjo S, Ajani JA, Scott AW, et al. Metformin sensitizes chemotherapy by targeting cancer stem cells and the mtor pathway in esophageal cancer. Int J Oncol. 2014;45(2):567–74.
Vazquez-Martin A, Oliveras-Ferraros C, Cufi S, et al. The anti-diabetic drug metformin suppresses the metastasis-associated protein cd24 in mda-mb-468 triple-negative breast cancer cells. Oncol Rep. 2011;25(1):135–40.
Song CW, Lee H, Dings RP, et al. Metformin kills and radiosensitizes cancer cells and preferentially kills cancer stem cells. Sci Rep. 2012;2:362.
Hirsch HA, Iliopoulos D, Struhl K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc Natl Acad Sci USA. 2013;110(3):972–7.
Authors’ contributions
YL and YY designed and wrote the manuscript; YL and XL designed the figure and table; EK and CQ revised the manuscript; YL and JZ co-designed, reviewed, and finalized the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by National Natural Science Foundation of China (NSFC) Key Project 81130046 (to JZ); NSFC81171993 (to YL) and NSFC81272415 (to YL); Guangxi Key Projects 2013GXNSFEA053004 (to JZ); Guangxi Projects 1355004-5 (to JZ) and 2012GXNSFCB053004 (to YL); Guangxi Ministry of Education 201202ZD022 (to YL) and 201201ZD004 (to JZ). The authors thank Ms. Xin Huang and Ms. Xiaolin Zhou for editing.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Yong Lei and Yanhua Yi contributed equally to this work
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lei, Y., Yi, Y., Liu, Y. et al. Metformin targets multiple signaling pathways in cancer. Chin J Cancer 36, 17 (2017). https://doi.org/10.1186/s40880-017-0184-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40880-017-0184-9