
Abstract
Collembola are soil-dwelling arthropods that play a key role in the soil ecosystem. Allonychiurus kimi (Lee) (Collembola: Onychiuridae) was isolated from the natural environment and has been maintained for 20 years under laboratory conditions. Though the morphological and physiological features of A. kimi are being widely used to evaluate the impact of pesticides and heavy metals on the soil ecosystem, variations observed in these features might be on account of its microbiota. However, the microbiota composition of the laboratory-maintained A. kimi is undetermined and how the community structure is changing in response to soil environments or interacting with the soil microbiota are still unknown. In this study, we determined the microbiota of laboratory-maintained A. kimi at both adult and juvenile stages and examined how the microbiota of A. kimi is affected by the microbial community in the soil environments. Chryseobacterium, Pandoraea, Sphingomonas, Escherichia–Shigella, and Acinetobacter were the core microbiota of A. kimi. Exposure of the laboratory-maintained A. kimi to different soil microbial communities drove dynamic shifts in the composition of A. kimi microbiota. Microbial association network analysis suggested that gut microbiota of lab-grown A. kimi was affected by exposing to soil microbial community. This study implies that shifts in the bacterial community of adult A. kimi can be utilized as an indicator to evaluate the soil ecosystem.
Similar content being viewed by others

Introduction
Soil ecosystems are rich habitats for a wide range of microbial and fauna communities. Interactions between these two communities vary from simple predator–prey interactions to shaping the health and fertility of the soil ecosystem [23, 32]. The soil fauna plays a key role in the dynamics of the soil microbial community and soil biomass [10, 24]. Soil microbial and faunal communities together mediate soil formation, microclimate regulation, and disease control [14, 21, 33]. Although many of these functions are aided by the diverse microbial communities colonized in the soil fauna [17], the ecological role of soil inhabitants and their microbiota as a single unit is less well studied.
Collembola are highly abundant, soil-dwelling microarthropods, forming an important part of the soil fauna community [41]. Owing to their small size, diverse ecological preferences, and ease of sampling, Collembola serve as bioindicators of ecosystem health and soil quality [22]. Among the many Collembola species studied so far, the European parthenogenetic Collembola species, Folsomia candida is widely being used as an internationally standardized test species to monitor the soil quality [20]. Bacterial 16S rRNA and fungal ITS amplicon sequencing has produced a comprehensive overview on the microbiome of F. candida [1]. However, it has very little ecological relevance to soil ecosystems in the Korean peninsula, due to its low abundance [43]. Allonychiurus kimi (Lee) is a common Collembolan species native to Korean soils and is also known to be abundant in paddy fields of Korea [26]. A. kimi was listed as an alternative to F. candida for toxicity tests in Organization for Economic Co-operation and Development (OECD) 232 guidelines for testing chemicals [37] and it is used as an ecotoxicological test animal in accordance with the International Organization for Standardization (ISO) guidelines [45, 46]. The pure-bred model organism, A. kimi, has been maintained under constant laboratory conditions with controlled feeding for 20 years, ever since the species was isolated from a natural soil environment [26].
Several studies have been conducted on the environmental factors (such as temperature, humidity, nutrients, and soil contaminants), which affect the viability and reproduction of A. kimi [44,45,46,47]. The changes in individual features of A. kimi have widely been used for monitoring the soil quality in those studies. However, the composition of A. kimi microbiota and its interactions with the soil environment are largely elusive. Owing to constant rearing under laboratory conditions, A. kimi is likely to have a stable microbiota. We assume that the microbiota of lab‐grown A. kimi may change once it is exposed to different field soils. Thus, the microbiota of A. kimi can be used as a model system to assess the quality and the health of the soil ecosystem.
In the present study, we report a comprehensive overview of the bacterial community composition of a model Collembola, A. kimi, and lay the groundwork for future ecotoxicological studies supported by bacterial community data. We introduced a laboratory-maintained population of A. kimi into the soil with different physicochemical properties (e.g., sandy or clay loam) collected from two distinct geographical locations in the Republic of Korea. Temporal changes in the bacterial community structure of both the A. kimi and the soil were characterized using amplicon sequencing. Microbial interactions and putative keystone taxa were identified using the co-occurrence microbial interaction analysis. Further, by allowing the adult individuals to reproduce under the same conditions, we examined the microbiota of the first-generation juveniles and compared it with that of the adults. Thereby, we assessed the contribution of vertical transmission of the parental microbiota and acquisition of microbiota from the environment during the development of A. kimi.
Materials and methods
Test species and culture conditions.
Allonychiurus kimi (Lee) (formerly known as Paronychiurus kimi) was originally isolated from the paddy soil in the Republic of Korea and has been reared for 20 years under defined feeding conditions [26]. The organisms were grown on a moist substrate consisting of plaster of Paris, activated charcoal, and distilled water in plastic petri dishes (9.5 cm in diameter, 1.5 cm in height) that were filled up to ~ 0.5 cm of the height with the media. Allonychiurus kimi cultures were maintained at 20 ± 1 °C under continuous darkness and fed with Brewer’s yeast [26]. To obtain age synchronized adult A. kimi populations, 100 adults were separately introduced into several breeding substrates and allowed to lay eggs and then a cohort of eggs was transferred to a new substrate. After the eggs were hatched, the juveniles were cultured under the same conditions. A homogenous, age synchronized (42–46 days old) population of A. kimi, prepared using this procedure, was used in this study.
Soil sampling and preparation
The soil used in this experiment was sampled at a depth of 10 cm from Deokso (N 37° 34′ 56″, E 127° 14′ 8″) and Jinju (N 35° 06′ 28″, E 128° 07′ 09″) in October 2017. The two sampling sites were approximately 290 km apart (Fig. 1a). In each sampling site, ten soil samples at a distance of at least 3 m apart were taken using a soil core sampler (diameter: 5 cm, height: 10 cm) at a depth of 10 cm except for the organic layer. The sampled soil was mixed thoroughly and was sieved through a 2 mm mesh to remove stones, debris, plant materials and large soil animals such as ground beetles of the Carabidae family. To preserve the soil microbial community while removing the soil fauna, the sieved soil was frozen at − 20 °C for 48 h and then allowed to be thawed for 24 h at room temperature in the dark. This procedure was repeated three times. This method has been used for defaunation with a minimal impact on the microbial community [39, 50]. For each sampling site, four polystyrene vessels (for four sampling times) each containing 100 g of defaunated soil were prepared immediately before inoculating A. kimi. An additional soil sample, which represents day0, from each site was stored at − 80 °C until DNA extraction.

Overview of experimental design. a The soil collection sites, Deokso and Jinju are indicated on a map of the Republic of Korea. b Schematic representation of experimental design. Experiment was started with an age-synchronized homogenous population of adult A. kimi. Sampling time and age of adult and juvenile A. kimi and the type of the sample collected at each sampling time point are indicated
Experimental setup
On the day0, a homogenous population of 30 A. kimi adults (42–46 days old) were introduced into each vessel containing soil from two sampling sites, Deokso and Jinju. The test vessels were kept at 20 ± 1 °C under continuous darkness. The test vessels were aerated and weighed weekly to replenish the moisture loss by the addition of sterile deionized water, if needed. After 1, 2, 4, and 8 weeks, the surviving adults and juveniles produced in each vessel were floated with adding sterile deionized water. Adults and juveniles were separated based on their morphological differences [46] and were transferred into separate 2 ml tubes for DNA extraction. Juveniles were found only in sampling time points at 4 and 8 weeks. Additionally, 10 g of soil was collected from each vessel for DNA extraction. As the control group, a pool of 30 A. kimi adults (42–46 days old) were introduced into a fresh moist substrate used for culture. The moist substrates in the Petri dishes were maintained at 20 ± 1 °C in continuous darkness and provided with Brewer’s yeast as food weekly. After that, the surviving adults and juveniles were sampled at the same time points mentioned above. Additionally, we used the starting homogenous population of A. kimi as day zero. All samples were preserved at − 80 °C until DNA extraction. All together, we collected 13 adult A. kimi samples (3 culture conditions × 4 sampling times, and adults at day zero), 6 juvenile A. kimi samples (3 culture conditions × 2 sampling times), and 10 soil samples (2 culture conditions × 5 sampling times including day zero) for sequencing (Fig. 1b).
DNA extraction from A. kimi and soil.
A pool of 30 adult individuals or 60 juvenile individuals was collected at each time point from each culture condition and used for the extraction of total DNA. Before the DNA extraction, A. kimi samples were washed twice for 2 min each with 70% ethanol and subsequently washed with sterile phosphate buffered saline, to remove the microorganisms attached to the body surface [18]. From each plate, 200 mg of soil was sampled and used as the starting material for DNA extraction. Total DNA of A. kimi and soil were extracted using the Quick DNA Fecal/Soil microbe mini prep kit (Zymo Research, Irvine, CA, USA), according to the manufacturer’s protocol.
Bacterial 16S rRNA gene sequencing
Bacterial 16S rRNA gene sequencing was used to analyze the microbial community structure. All the primers were synthesized by Integrated DNA Technologies (IDT, Singapore). The V3–V4 regions of 16S rRNA gene were amplified using the following universal primer set (forward 5′-CCTACGGGNGGCWGCAG-3′, reverse 5′-GACTACHVGGGTATCTAATCC-3′) [29], and Q5 high fidelity DNA polymerase (NEB, Ipswich, MA, USA). Illumina overhang adapter sequences were added to the above universal primer set (forward overhang 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐[locus specific sequence], reverse overhang: 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGAC AG‐[locus specific sequence]. The thermocycler parameters were as follows: initial denaturation 3 min at 98 °C, followed by 20 amplification cycles (30 s at 98 °C, 30 s at 55 °C, 1 min at 72 °C), final extension 5 min at 72 °C. The resulting DNA amplicons were purified using the Agencourt AMPure XP beads (Beckman Coulter, Brea, CA, USA). A second PCR was performed to attach Illumina universal p5/p7 overhang sequences and sample specific barcodes (forward 5′-CAAGCAGAAGACGGCATACGAGAT[i7]GTCTCGTGGGCTCGG, reverse 5′-AATGATACGGCGACCACCGAGATCTACAC[i5]TCGTCGGCAGCGTC; where [i7] and [i5] are 6 base-pair sample specific barcodes) The thermocycler parameters for the second PCR were as follows: initial denaturation 1 min at 98 °C, followed by 12 amplification cycles (15 s at 98 °C, 15 s at 55 °C, 30 s at 72 °C), final extension 3 min at 72 °C. Amplification was performed using Q5 high fidelity DNA polymerase (NEB, Ipswich, MA, USA). Sequencing ready libraries were purified using the Agencourt AMPure XP beads (Beckman Coulter, Brea, CA, USA). Sequencing was conducted using the Illumina MiSeq Platform (Illumina, Diego, CA, USA) with MiSeq Reagent kit V3 (2 × 300 PE) (Illumina, Diego, CA, USA).
Control experiments for DNA extraction, library preparation and sequencing were conducted as follows. A mock DNA library was prepared with the ZymoBiomics microbial community DNA standard (Zymo Research, Irvine, CA, USA), using the amplicon library preparation procedure described above. This control library allowed us to assess the potential bias and errors associated with the amplification and sequencing steps. DNA extraction negative control was carried out by following the same protocol without soil or A. kimi. The negative control for PCR was carried out using the DNA extraction negative control as the template and following the same PCR conditions. We confirmed that the samples were not contaminated during DNA extraction or library preparation.
Bioinformatic processing of sequencing data
Quality control of raw Illumina sequence reads was performed using Trim Galore ver. 0.5.0 (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Illumina universal adapter sequences were trimmed, reads shorter than 40 bp were removed and bases with Q < 20 were trimmed from the 3′ and 5′ ends of reads. Forward and reverse reads were merged into concatenated reads using PEAR ver. 0.9.8 [55]. The merged reads were clustered into Operational Taxonomic Units (OTUs) at 97% sequence similarity, using QIIME ver. 1.9.1 [13], which is an open-source bioinformatics pipeline for performing microbiome analysis. The SILVA database release (ver. 132) was used as the reference database for taxonomic assignment in the QIIME workflow [40]. Statistical analysis and visualization were performed using R version 3.6.0 [49] via RStudio version 1.2.1335 [48]. The R packages, “phyloseq” [36], “DEseq” [52] and “microbiome” [31] were used to analyze the diversity of the microbial community.
Alpha diversity was assessed based on the observed OTUs, Chao1, Shannon and Simpson diversity indices. As the alpha diversity indices did not meet the assumption of equal variance, the differences in alpha diversity between samples were calculated using the non-parametric Wilcoxon rank-sum test (Mann–Whitney). Beta diversity was analyzed using the Bray–Curtis dissimilarity and sample ordination was visualized using the multi-dimensional scaling (MDS). Permutational Multivariate Analysis of Variance (PERMANOVA) test was performed to test whether the sample groups differed significantly.
Construction of ecological networks
OTUs with a relative abundance of at least 0.01% were used to construct a bacterial co-occurrence network for adult A. kimi using the SParse InversE Covariance estimation for Ecological Association Inference (SPIEC-EASI) method [30]. This method is robust to the challenges encountered when using compositional data with low sample counts. We then transformed the OTU count data, and network analysis was performed using the neighborhood selection (MB method) with a minimum lambda threshold of 0.01 for graphical model inference. All calculations were made using the SpiecEasi R package (version 1.0.7) [30]. We used the “igraph” R package [15] for generating the network plots. The network plots were then exported to Cytoscape (version 3.7.2) [42] for visualization. The ‘Network analyzer’ tool in Cytoscape was used to determine the network topology parameters. We used three parameters, (1) degree, (2) closeness centrality (3) betweenness centrality, to predict putative keystone taxa. Degree is defined as the number of edges connected to a node. Closeness centrality depends on the average shortest path and represents the central importance of a node. Betweenness centrality defines the role of a node as a bridge in the network. OTUs with highest degree, highest closeness of centrality and lowest betweenness centrality score were predicted as putative keystone taxa as defined in previous studies [8, 12]
Results
Physicochemical properties of the soil environments exposed to the laboratory-maintained A. kimi
To investigate the effects of soil environments for the microbiota of the laboratory-maintained A. kimi, we selected two geologically distant soil sampling sites in the Republic of Korea: Deokso and Jinju (Fig. 1a) and analyzed the physicochemical properties of the soils (Table 1). The soil sampled from Deokso was sandy loam, containing 64.5% sand, 16.7% slit, and 18.8% clay. It is classified as Fragiudults with a pH of 6.55 ± 0.02, and soil and water at a ratio of 1:5, w/w. The soil sampled in Jinju was clay loam containing 32.4% sand, 35.3% slit, and 32.3% clay, classified as Paleudults with a pH of 6.16 ± 0.09 and soil and water at a ratio of 1:5, w/w. Deokso and Jinju soils had similar organic matter contents, of 1.81% and 1.60%, respectively.
Overall bacterial community in the laboratory-maintained A. kimi and the soil samples
We sequenced 13 adult A. kimi samples (3 culture conditions × 4 sampling times, and adults at day zero) and 10 soil samples (2 culture conditions × 5 sampling times) including day zero. Juveniles were found only in sampling time points at 4 and 8 weeks (Fig. 1b) and 6 juvenile A. kimi samples (3 culture conditions × 2 sampling times) were sequenced. Adult and juvenile individuals were distinguished based on their morphological characteristics (Additional file 1: Fig. S1). We obtained a total of 2,712,257 reads for A. kimi with an average of 142,750 (± 92,271) (mean ± SD) reads per sample, and 1,407,805 reads for soil, with an average of 140,780 (± 64,857) (mean ± SD) reads per sample. The mock community generated 58,091 reads, and we identified all the bacterial strains in the mock community at the expected proportions. The prevalence of key phyla in A. kimi adults and juveniles and soil samples provided the first insight into the abundant taxa in our data set (Fig. 2a, b). Proteobacteria, Actinobacteria and Bacteroidetes, and Firmicutes were the dominant bacteria phyla common to both A. kimi and soil. Additional bacterial phyla, including Acidobacteria, Saccharibacteria, Chloroflexi, and Planctomycetes, were also detected in soil samples at relatively high abundance. We analyzed the shared and unique OTUs with > 0.01% relative abundance within and between the different groups of samples (Fig. 2c–e). The majority of the OTUs (392 OTUs) found in A. kimi were shared not only between adults and juveniles, but also across the culture conditions: laboratory-grown and two soil environments (Deokso and Jinju).

Taxonomic composition of the A. kimi and soil microbiota. Taxa prevalence vs total abundance graph for bacterial phyla detected in a A. kimi and b soil after filtering the low abundant taxa. Bacterial OTUs with > 0.01% relative abundance that were unique and shared between a adult and juvenile A. kimi samples, b among lab-grown and soil-grown adult A. kimi samples, and c among lab-grown and soil-grown juvenile A. kimi samples
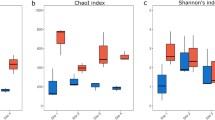
We estimated the alpha diversity of A. kimi and soil samples using the observed OTUs, Chao1, Shannon, and Inverse Simpson indices (Additional file 1: Fig. S2). Alpha diversity matrices were not significantly different among A. kimi samples. The alpha diversity of A. kimi, either belonging to different developmental stages or grown in different culture environments, showed no differences (p > 0.1) (Additional file 1: Figs. S3 and S4). However, the alpha diversity matrices were varied between soil environments. The observed OTU and Chao1 indices of the Deokso soil was significantly higher than that of the Jinju soil (p < 0.01) while the Shannon and Inverse Simpson indices showed no significant difference (Additional file 1: Fig. S5). The alpha diversity of the soil in terms of Shannon and Inverse Simpson indices was significantly higher than that of A. kimi (p < 0.001) but observed OTU and Chao1 indices showed no significant difference (Additional file 1: Fig. S6).
Comparison of bacterial community of adult and juvenile A. kimi: a shift during the growth
While the dominant phyla in A. kimi were identified as Actinobacteria, Proteobacteria and Bacteroidetes representing > 97% of the whole A. kimi microbiota (Fig. 3a), the relative proportion of these three phyla was varied between the microbiota of juvenile and adult A. kimi. The relative proportions of the three abundant phyla were mostly consistent in the adult microbiota in various sampling conditions, revealing Bacteroidetes as the dominant phylum in general. The microbiota of juveniles showed a similar community composition in all samples (lab, Deokso and Jinju) at the time of first sampling of the juveniles in the fourth week. However, at the second sampling in the eighth week, the dominant phylum was changed in lab-grown juveniles. Proteobacteria was dominant in all soil-grown juveniles whereas Actinobacteria was most abundant in lab-grown juveniles in the eighth week sample (Fig. 3a). When the taxonomic ranks of the dominant OTUs were resolved at the genus level, Chryseobacterium, Streptomyces, Comamonas, and Acinetobacter were appeared as the most abundant genera in the microbiota of adult A. kimi (Fig. 3b). Similarly, Pandoraea, Sphingomonas, Escherichia–Shigella, and Acinetobacter were identified as the most abundant genera in juvenile A. kimi (Fig. 3c).

Microbial community composition in adult and juvenile A. kimi. a Relative abundance of different phyla (relative abundance > 0.001%) in adult and juvenile A. kimi. Relative abundance of the 10 most abundant OTUs resolved at the genus level in, b adult A. kimi and c juvenile A. kimi
The core microbiota, which is stable and abundant members of the bacterial community [5, 16, 56], of adult and juvenile A. kimi consisted of 13 OTUs belonging to Chryseobacterium, Pandoraea, Sphingomonas, Escherichia–Shigella, and Acinetobacter (at 0.1% relative abundance and 50% prevalence threshold) (Fig. 4a). Ordination analysis of adults and juveniles, using the Bray–Curtis dissimilarity distance, visualized that the adult and juvenile groups clustered separately except that lab-grown 4-week-old juveniles that came into a cluster with lab-grown adult samples (Fig. 4b). The changes in the bacterial composition of adults and juveniles were tested using the Permutational Multivariate Analysis of Variance (PERMANOVA) on the Bray–Curtis dissimilarity matrix. The proportions of three dominant OTUs, belonging to Sediminibacterium and Propionibacterium, were differentially increased in juvenile samples compared to adults (p < 0.001, R2 = 0.155) (Fig. 4c). Similarly, we observed that 11 OTUs belonging to 6 genera (Chryseobacterium, Acinetobacter, Glutamicibacter, Leucobacter Ensifer, and Kocuria) were differentially abundant (p < 0.001) in adults compared to the juveniles.

The core microbiota and comparison of the adult and juvenile A. kimi microbiota. a The core microbiota of adult and juvenile A. kimi with over 0.1% relative abundance and over 50% prevalence thresholds consisted of 13 OTUs. b Ordination analysis to visualize the differences between adult and juvenile microbiota. The multidimensional scaling plot was based on the Bray–Curtis dissimilarity matrix. c Differentially abundant bacterial taxa in adult and juvenile samples. Differentially abundant genera were shown as the log2 fold change. Bacterial taxa showing significantly high abundance (p < 0.001) in adult or juvenile A. kimi are indicated
We identified 283 OTUs (with > 0.01% relative abundance) which were shared between the microbiota of adult and juvenile A. kimi. In addition, 95 OTUs were unique to adults, and 52 were unique to juveniles, suggesting the existence of specific microbiomes at different developmental stages (Additional file 1: Fig. S7a). OTUs belonging to Acidobacteria, Chloroflexi, Saccharibacteria and Verrucomicrobia were found exclusively in adult A. kimi. In contrast, Fusobacteria were only found in juveniles (Additional file 1: Fig. S7b). OTUs belonging to the orders Acidimicrobiales, Catenullisporales, Frankiales, Gaiellales, Solirubrobacterales and Streptosporangiales of the phylum Actinobacteria; Desulfurellales, Rhodospirillales and Myxococcales of the phylum Proteobacteria; and Cytophagales of the phylum Bacteroidetes were found only in adult A. kimi. Actinomycetales of the phylum Actinobacteria was found exclusively in juvenile A. kimi (Additional file 1: Fig. S7c).
Community shift in the adult A. kimi microbiota under laboratory and different soil environments
The dominant bacterial taxa in the microbiota of lab-grown adult A. kimi were different from that of the soil-grown adults. At the genus level, Leucobacter, Chryseobacterium, Acinetobacter, Glutamicibacter, and Comamonas were among the 10 most abundant taxa in the microbiota of lab-grown adult A. kimi (Fig. 5a). Chryseobacterium, Streptomyces, Ensifer, Acinetobacter, and Kitasatospora were detected in high abundance in the microbiota of adult A. kimi grown in Deokso soil (Fig. 5b). Chryseobacterium, Streptomyces, Comamonas, Kitasatospora, and Sphingomonas were among the most abundant taxa in adult A. kimi grown in Jinju soil (Fig. 5c).

Microbial community composition in adult A. kimi grown under different environmental conditions. Relative abundance of the 10 most abundant bacterial OTUs resolved at the genus level in, a lab grown adult A. kimi, b adult A. kimi grown in Deokso soil and c adult A. kimi grown in Jinju soil
Ordination analysis using the Bray–Curtis dissimilarity distances indicated that soil-grown adult A. kimi clustered separately from lab-grown adults (Fig. 6a). A shift in the bacterial community composition between soil- and lab-grown samples was observed in the PERMANOVA based on the Bray–Curtis dissimilarity matrix (p < 0.01, R2 = 0.183). The changes in abundant taxa in soil- and lab-grown adult A. kimi were identified. We observed 9 OTUs, including Ensifer, Streptomyces, Pseudomonas, and Bacillus, that showed significantly high differential abundance (p < 0.001) when A. kimi were grown in soil. Similarly, 32 OTUs have significantly high differential abundance (p < 0.001) in lab-grown A. kimi, including Acinetobacter, Glutamicibacter, and Leucobacter and Nocardioides (Fig. 6b).

The comparison of adult A. kimi grown under different environmental conditions. a Ordination analysis to visualize the difference between lab-grown and soil-grown adult A. kimi. The multidiamentioanl scaling plot was based on the Bray–Curtis dissimilarity matrix. b Differentially abundant bacterial taxa in lab-grown and soil-grown adult A. kimi groups. Differentially abundant genera were shown as the log2 fold change. Bacterial taxa showing significantly high abundance (p < 0.001) in soil- or lab-grown A. kimi are indicated
To evaluate the effect of soil environments on A. kimi, the bacterial community in the soil environments were compared to the microbiota of A. kimi grown in the given soils. Proteobacteria was the dominant phyla in both Deokso and Jinju soils, followed by Actinobacteria and Bacteroidetes (Additional file 1: Fig. S8a). The core microbiota of two soil environments (Deokso and Jinju) consisted of 20 OTUs (with 0.1% relative abundance and 50% prevalence thresholds) (Additional file 1: Fig. S8b). The statistically significant difference in the community composition was not found between the two groups of soil samples in the PERMANOVA based on the Bray–Curtis dissimilarity matrix (Additional file 1: Fig. S8c).
We investigated the bacterial taxa that were shared between the given soil environment and soil-grown A. kimi but not shared with lab-grown A. kimi. The presence of such taxa might suggest bacterial transfers from the soil to the A. kimi microbiota. Bacterial taxa belonging to the Acidobacteria, Chloroflexi, Planctomycetes, and Verrucomicrobia were exclusively found in soil-grown adult A. kimi (Additional file 1: Fig. S9a). OTUs belonging to the orders Acidimicrobiales, Micromonosporales, Streptosporangiales, and Solirubrobacterales of the phylum Actinobacteria; Caulobacterales and Myxococcales of the phylum Proteobacteria; and Cytophagales of the phylum Bacteroidetes were also found in soil-grown adult A. kimi (Additional file 1: Fig. S9b). We identified 118 OTUs (> 0.01% relative abundance) that were likely transferred from Deokso soil to Deokso soil-grown adult A. kimi microbiota. Ensifer was the most abundant genus having 6.7% mean relative abundance in A. kimi microbiota, followed by 11 OTUs, including Bacillus, Streptomyces, Lentzea and Mycobacterium, having > 0.1% mean relative abundance. Similar to Deokso soil-grown samples, 122 OTUs (> 0.01% relative abundance) were identified to be transferred from Jinju soil to Jinju soil-grown adult A. kimi. The 12 OTUs (> 0.1% mean relative abundance) belonged to Bacillus (3.7% relative abundance), Enterobacter, Acinetobacter, Ensifer and Mycobacterium. The absence of these soil-originated bacterial OTUs in the microbiota of lab-grown A. kimi implies that these bacteria were transferred to the A. kimi after they were exposed to the soil environment.
Microbial co-occurrence network analysis in the microbiota of adult A. kimi
To search the patterns of co-occurrence of bacterial taxa in the adult A. kimi microbiota, we employed the SParse InversE Covariance estimation for Ecological Association Inference (SPIEC-EASI) analysis [30]. The overall bacterial co-occurrence network in adult A. kimi (lab-grown and soil-grown combined) consisted of 146 nodes (with > 0.01% relative abundance) and 182 edges (Fig. 7a). The overall network structure showed more positive than negative co-associations. To examine the structure and connectivity of the microbial network, we calculated the network density (defined as the ratio of realized to possible number of edges), the clustering coefficient (defined as the probability that nodes close to a given node are connected), and the average number of neighbors (defined as the average number of edges per node) [28]. The co-occurrence network for adult A. kimi microbiota has a network density of 0.017, a clustering coefficient of 0.089 and 2.493 neighbors on average. We selected six OTUs with the highest degree, high closeness centrality and low betweenness centrality in the overall network as putative keystone taxa. These putative keystone taxa identified in the co-occurrence network included both high abundance OTUs (Chryseobacterium and Streptomyces) and low abundance OTUs (Mycobacterium and Lysinimonas). This observation suggested the importance of low abundance taxa to the structure of the microbial network.

Network analysis of the bacterial taxa in the adult A. kimi microbiome. Bacterial network in the adult A. kimi microbiome was constructed using the SParse InversE Covariance estimation for Ecological Association Inference (SPIEC-EASI). a The overall bacterial co-occurrence network in lab-grown, and soil-grown adult A. kimi. b Bacterial co-occurrence network in lab-grown adult A. kimi, c bacterial co-occurrence network in soil-grown adult A. kimi. Nodes representing corresponding OTUs and two OTUs with significant co-association were linked by an edge. Node color represents the phylum to which the OTU belongs. Positive associations are colored with a blue edge and negative associations with red. Large circles indicate the putative keystone taxa in each network. OTUs with the highest degree, high closeness centrality, and low betweenness centrality were selected as putative keystone taxa
Since the bacterial community compositions of lab-grown and soil-grown adult A. kimi were different, we built the bacterial co-occurrence networks using lab-grown and soil-grown adult A. kimi microbiota independently. The co-occurrence network of lab-grown adult A. kimi microbiota consisted of 120 nodes and 152 edges (Fig. 7b), whereas that of soil-grown adult A. kimi had 137 nodes and 114 edges (Fig. 7c). The average number of neighbors was 2.5 for the network of lab-grown adult A. kimi microbiota, and 1.7 for that of the soil-grown. The clustering coefficient of the network of lab-grown adult A. kimi microbiota was 0.043 while that of soil-grown adult A. kimi microbiota was 0.005. We also observed that the network density of lab-grown adult A. kimi microbiota (0.021) was higher than that of soil-grown (0.012). Chryseobacterium and Glutamicibacter were common keystone taxa in both networks. The degree of the keystone taxa in soil-grown adult A. kimi microbiota was lower than that of lab-grown, indicating that the established microbial network in lab-grown adult A. kimi was perturbed by the soil bacteria that colonized in A. kimi and altered the previously established bacterial network structure.
Discussion
Allonychiurus kimi (Lee) is widely being used in the Republic of Korea as a test species in ecotoxicological studies. Its adaptation to the physicochemical properties of Korean soil makes A. kimi a better candidate in the Republic of Korea than F. candida, which is widely being used worldwide as a test organism. Although several studies have been conducted into the microbiota of F. candida and other Collembola species, including Orchesella cincta and Orchesellides sinensis [1, 2, 35], the composition of the microbiota of A. kimi is still poorly understood. Here, we focused on the microbiota of a laboratory-maintained A. kimi population, and the way in which the microbiota changes across developmental stages and when exposed to new environmental conditions. The bacterial community composition of A. kimi is similar to that of other Collembola species reported in the literature [1, 2, 35]. The microbiota of laboratory-maintained F. candida consists of four main phyla: Actinobacteria, Bacteroidetes, Proteobacteria, and Firmicutes [57], which is similar to the microbiota of A. kimi but relative abundance of Firmicutes was lower than that of F. candida, O. cincta or O. sinensis. These four main phyla are also known as the core microbiota of other soil-dwelling animals, such as earthworms and nematodes [9, 38]. The genus Chryseobacterium found as a dominant genus in A. kimi was previously reported in another Collembola species, Orchesella cincta, at high abundance across body sites but was not found in F. candida [6].
Wolbachia is known as a common symbiont in certain Collembola species [1]. It is noteworthy that, in our study, the microbiota of the laboratory-maintained A. kimi has very low abundance of Wolbachia. Since we did not compare the microbiota of wild A. kimi with those of laboratory-maintained organisms, we were unable to determine whether the low abundance of Wolbachia is a consequence of continuous maintenance under laboratory conditions. However, it has been reported that certain other Collembola species also have a low abundance of Wolbachia in their microbiota [51].
Comparison of adult and juvenile A. kimi microbiota suggested that both hereditary and environmental factors affect the A. kimi microbiota. Spatial and temporal variation in the insect gut microbiome across host developmental stages has previously been reported [3, 4]. However, microbiome studies on Collembola have been mainly confined to adults. We showed that the microbiota of adult and juvenile A. kimi had different compositions depending on the developmental stage (Fig. 2). The juvenile microbiota was dominated by Proteobacteria, whereas Bacteroidetes were dominant in adults. We suggest that the similar bacterial community composition of A. kimi juveniles at the birth, irrespective of the growth environment, might be a consequence of vertical transmission of the parental A. kimi microbiota in establishing the juvenile microbiota. Later, environmental factors such as nutrients, humidity, temperature, chemical/physical composition of soil played key role in shaping the juvenile microbiota into its adult composition. We assume that the complex process of transforming the juvenile microbiota into adult microbiota is similar to what is described in the literature [11, 17].
The microbiota of soil dwelling animals can therefore be affected not only by inherent host related factors, but also by environmental factors [3, 16]. The effects of environmental factors on the microbiota are transient and subject to dynamic change [4]. We observed that the bacterial community composition of adult A. kimi gradually changed when they were grown in soil. Actinobacteria were abundant in lab-grown adult A. kimi, whereas Bacteroidetes dominated in soil-grown adults. These observations suggest that the complex environmental factors play a pivotal role in altering the host microbiota. The members of the soil microbiota may alter the host microbiota by colonizing the host. In addition, different dietary patterns in the two groups can also affect the microbiota, since the diet is a key factor in altering the Collembola microbiota [53]. These were supported by previous studies which emphasized the effects of environmental factors in altering the host-associated microbiota in Collembola [27, 54].
The A. kimi test species, that we used in this study, was reared under consistent laboratory conditions (diet, humidity, temperature etc.) for 20 years. Therefore, it can be speculated that the native microbiota, which was present in the wild A. kimi, has changed throughout this period, and the laboratory-maintained species has a well-established microbial network. And this microbial network could be different from that of the native soil-dwelling wild species. We hypothesized that the established microbial network of laboratory-maintained A. kimi could be perturbed once the organisms were introduced into the soil. Analysis of the microbial networks can provide insights about how bacterial community structure changes in response to environmental factors [19]. Several bacterial taxa were found exclusively in soil-grown A. kimi, suggesting that these bacteria were transferred from the soil to A. kimi and colonized. This colonization may perturb the existing microbial network in A. kimi for a certain period until it re-stabilizes. Distortion of the existing microbial network of lab-grown A. kimi was explained by the network parameters such as network density, clustering coefficient and average number of neighbors. All three parameters obtained for the microbial network in soil grown A. kimi were lower than that of lab-grown A. kimi. This evidence suggested that soil microbiota drive structural changes in the established microbial network of lab-grown A. kimi.
The keystone taxa are defined as taxa that are highly connected and contribute individually or in association with other taxa to the structure and function of the microbiome irrespective of their abundance [7]. We identified the keystone taxa such as Chryseobacterium and Glutamicibacter in both lab-grown and soil-grown A. kimi microbial networks (Fig. 6). Chryseobacterium was recognized as a potential pathogen in insects, including bark beetles [25]. Glutamicibacter was suggested to aid host metabolism [34]. Although Kitasatospora and Pandoraea were not found in high abundance, we identified them as putative keystone taxa in the microbial network of the soil-grown A. kimi. This finding suggested that taxa of low abundance may be significant in shaping the network structure of the microbiota. However, empirical evidence emphasizing the importance of these keystone taxa in Collembola were hardly found in the literature and further study is required to validate the importance of the low abundance keystone taxa.
The lack of microbiome data for A. kimi has been a long-lasting gap in the assessment of the role of the A. kimi microbiota in ecotoxicological studies. Since the bacterial community composition was characterized in this study, A. kimi can be used not only as a model organism for ecotoxicological studies, but also as a gnotobiotic-type model organism with which to study the contribution of the microbiota to its ecotoxicological effects. Our study provided useful insights into the bacterial community composition of laboratory-maintained A. kimi and the potential spatio-temporal changes in the microbiota under field conditions. Allonychiurus kimi microbiota is permissive, allowing members of the soil microbiota to colonize in A. kimi and form dynamic associations with the soil microbiota. Therefore, the microbiota restructuring patterns in A. kimi, upon exposure to different environmental conditions, can also be used as an indicator of ecotoxicological effects. Since the A. kimi microbiota is amenable to both laboratory and field conditions, our study will pioneer the role of the host-associated microbiota in the ecotoxicological effects exerted by this tiny soil dwelling arthropod, A. kimi.
Availability of data and materials
The raw reads generated and analyzed in this study are available in the NCBI database under the BioProject accession number PRJNA752221. Raw sequence reads from soil samples are available in the NCBI Sequence Read Archive (SRA) under accession numbers SRR15352802–SRR15352811 and raw sequence reads from A. kimi samples are available under accession numbers SRR15355258–SRR15355276.
Change history
15 April 2022
Following the original publication of this article, the authors flagged that the affiliations information of the corresponding authors was incorrect; please see the linked correction for further details.
25 April 2022
A Correction to this paper has been published: https://doi.org/10.1186/s40793-022-00414-4
References
Agamennone V, Jakupović D, Weedon JT, Suring WJ, van Straalen NM, Roelofs D, Röling WF. The microbiome of Folsomia candida: an assessment of bacterial diversity in a Wolbachia-containing animal. FEMS Microbiol Ecol. 2015;91(11):fiv128.
Agamennone V, Roelofs D, van Straalen N, Janssens T. Antimicrobial activity in culturable gut microbial communities of springtails. J Appl Microbiol. 2018;125(3):740–52.
Anslan S, Bahram M, Tedersoo L. Temporal changes in fungal communities associated with guts and appendages of Collembola as based on culturing and high-throughput sequencing. Soil Biol Biochem. 2016;96:152–9.
Anslan S, Bahram M, Tedersoo L. Seasonal and annual variation in fungal communities associated with epigeic springtails (Collembola spp.) in boreal forests. Soil Biol Biochem. 2018;116:245–52.
Astudillo-García C, Bell JJ, Webster NS, Glasl B, Jompa J, Montoya JM, Taylor MW. Evaluating the core microbiota in complex communities: a systematic investigation. Environ Microbiol. 2017;19(4):1450–62.
Bahrndorff S, de Jonge N, Hansen JK, Lauritzen JMS, Spanggaard LH, Sørensen MH, Yde M, Nielsen JL. Diversity and metabolic potential of the microbiota associated with a soil arthropod. Sci Rep. 2018;8(1):2491.
Banerjee S, Schlaeppi K, van der Heijden MG. Keystone taxa as drivers of microbiome structure and functioning. Nat Rev Microbiol. 2018;16(9):567–76.
Banerjee S, Walder F, Büchi L, Meyer M, Held AY, Gattinger A, Keller T, Charles R, van der Heijden MG. Agricultural intensification reduces microbial network complexity and the abundance of keystone taxa in roots. ISME J. 2019;13(7):1722–36.
Berg M, Stenuit B, Ho J, Wang A, Parke C, Knight M, Alvarez-Cohen L, Shapira M. Assembly of the Caenorhabditis elegans gut microbiota from diverse soil microbial environments. ISME J. 2016;10(8):1998–2009.
Bernard L, Chapuis-Lardy L, Razafimbelo T, Razafindrakoto M, Pablo A-L, Legname E, Poulain J, Brüls T, O’donohue M, Chotte JL, Brauman A. Endogeic earthworms shape bacterial functional communities and affect organic matter mineralization in a tropical soil. ISME J. 2012;6(1):213.
Bright M, Bulgheresi S. A complex journey: transmission of microbial symbionts. Nat Rev Microbiol. 2010;8(3):218–30.
Berry D, Widder S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front Microbiol. 2014;5:219.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Huttley GA, Gordon JI. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335.
Crowther TW, Thomas SM, Maynard DS, Baldrian P, Covey K, Frey SD, van Diepen LT, Bradford MA. Biotic interactions mediate soil microbial feedbacks to climate change. Proc Natl Acad Sci. 2015;112:7033–8.
Csardi G, Nepusz T. The igraph software package for complex network research. InterJ Complex Syst. 2006;1695(5):1–9.
Ding J, Zhu D, Li H, Ding K, Chen QL, Lassen SB, Ke X, O’Connor P, Zhu YG. The gut microbiota of soil organisms show species-specific responses to liming. Sci Total Environ. 2019;659:715–23.
Engel P, Moran NA. The gut microbiota of insects–diversity in structure and function. FEMS Microbiol Rev. 2013;37(5):699–735.
Faddeeva-Vakhrusheva A, Kraaijeveld K, Derks MF, Anvar SY, Agamennone V, Suring W, Kampfraath AA, Ellers J, Le Ngoc G, Mariën J, Gestel CA. Coping with living in the soil: the genome of the parthenogenetic springtail Folsomia candida. BMC Genom. 2017;18(1):493.
Faust K, Raes J. Microbial interactions: from networks to models. Nat Rev Microbiol. 2012;10(8):538–50.
Fountain MT, Hopkin SP. Folsomia candida (Collembola): a “standard” soil arthropod. Annu Rev Entomol. 2005;50:201–22.
Friberg H, Lagerlöf J, Rämert B. Influence of soil fauna on fungal plant pathogens in agricultural and horticultural systems. Biocontrol Sci Technol. 2005;15(7):641–58.
Gerlach J, Samways M, Pryke J. Terrestrial invertebrates as bioindicators: an overview of available taxonomic groups. J Insect Conserv. 2013;17(4):831–50.
Hassall M, Adl S, Berg M, Griffiths B, Scheu S. Soil fauna–microbe interactions: towards a conceptual framework for research. Eur J Soil Biol. 2006;42:S54–60.
Héry M, Singer AC, Kumaresan D, Bodrossy L, Stralis-Pavese N, Prosser JI, Thompson IP, Murrell JC. Effect of earthworms on the community structure of active methanotrophic bacteria in a landfill cover soil. ISME J. 2008;2(1):92.
Hofstetter RW, Dinkins-Bookwalter J, Davis TS, Klepzig KD. Symbiotic associations of bark beetles. In: Vega FE, Hofstetter RW, editors. Bark beetles. Amsterdam: Elsevier; 2015. p. 209–45.
Kang S, Choi WI, Ryoo MI. Demography of Paronychiurus kimi (Lee) (Collembola: Onychiuridae) under the influence of glufosinate-ammonium on plaster charcoal substrate and in artificial soil. Appl Soil Ecol. 2001;18(1):39–45.
Kikuchi Y, Hosokawa T, Fukatsu T. Insect-microbe mutualism without vertical transmission: a stinkbug acquires a beneficial gut symbiont from the environment every generation. Appl Environ Microbiol. 2007;73(13):4308–16.
Kim HJ, Kim H, Kim JJ, Myeong NR, Kim T, Park T, Kim E, Choi JY, Lee J, An S, Sul WJ. Fragile skin microbiomes in megacities are assembled by a predominantly niche-based process. Sci Adv. 2018;4(3):e1701581.
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucl Acids Res. 2013;41(1):e1–e1.
Kurtz ZD, Müller CL, Miraldi ER, Littman DR, Blaser MJ, Bonneau RA. Sparse and compositionally robust inference of microbial ecological networks. PLoS Comput Biol. 2015;11(5):e1004226.
Lahti L, Shetty S, Blake T, Salojarvi J. Microbiome R package. Tools Microbiome Anal R. 2017.
Lal R. Soil health and carbon management. Food Energy Secur. 2016;5(4):212–22.
Lavelle P, Decaëns T, Aubert M, Barot S, Blouin M, Bureau F, Margerie P, Mora P, Rossi JP. Soil invertebrates and ecosystem services. Eur J Soil Biol. 2006;42:S3–15.
Liu W, Zhang X, Wu N, Ren Y, Wang X. High diversity and functional complementation of alimentary canal microbiota ensure small brown planthopper to adapt different biogeographic environments. Front Microbiol. 2020;10:2953.
Ma Y, Chen WJ, Li ZH, Zhang F, Gao Y, Luan YX. Revisiting the phylogeny of Wolbachia in Collembola. Ecol Evol. 2017;7(7):2009–17.
McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8(4):e61217.
OECD. Test no. 232: collembolan reproduction test in soil. Organisation for economic co-operation and development (OECD), OECD guidelines for the testing of chemicals, section 2. Berlin: OECD Publishing; 2016.
Pass DA, Morgan AJ, Read DS, Field D, Weightman AJ, Kille P. The effect of anthropogenic arsenic contamination on the earthworm microbiome. Environ Microbiol. 2015;17(6):1884–96.
Poll J, Marhan S, Haase S, Hallmann J, Kandeler E, Ruess L. Low amounts of herbivory by root-knot nematodes affect microbial community dynamics and carbon allocation in the rhizosphere. FEMS Microbiol Ecol. 2007;62(3):268–79.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucl Acids Res. 2012;41(D1):D590–6.
Rusek J. Biodiversity of Collembola and their functional role in the ecosystem. Biodivers Conserv. 1998;7(9):1207–19.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Shin Y, Yoon I. Check list of insects from Korea. Seoul: Kon-Kuk University Press; 1994. p. 362–3.
Son J, Lee Y-S, Kim Y, Shin K-I, Hyun S, Cho K. Joint toxic action of binary metal mixtures of copper, manganese and nickel to Paronychiurus kimi (Collembola). Ecotoxicol Environ Saf. 2016;132:164–9.
Son J, Mo H-H, Shin K-I, Ryoo MI, Cho K. Combined effects of organic matter and pH on acute toxicity of cadmium to Paronychiurus kimi (Collembola): development of response surface model. Soil Res. 2009;47(6):549–54.
Son J, Mo H-H, Yang N-H, Shin K-I, Cho K. Determination of Paronychiurus kimi (Collembola: Onychiuridae) age structures by head width measurements with reference to cadmium toxicity. Appl Soil Ecol. 2009;43(1):47–52.
Son J, Shin K-I, Cho K. Response surface model for predicting chronic toxicity of cadmium to Paronychiurus kimi (Collembola), with a special emphasis on the importance of soil characteristics in the reproduction test. Chemosphere. 2009;77(7):889–94.
Team R. RStudio: integrated development for R. Boston: RStudio, Inc.; 2015. http://www.rstudio.com, 42, 14.
Team RC. R: A language and environment for statistical computing. 2013.
Thakur MP, Herrmann M, Steinauer K, Rennoch S, Cesarz S, Eisenhauer N. Cascading effects of belowground predators on plant communities are density-dependent. Ecol Evol. 2015;5(19):4300–14.
Timmermans MJ, Mariën J, Roelofs D, van Straalen NM, Ellers J. Evidence for multiple origins of Wolbachia infection in springtails. Pedobiologia. 2004;48(5–6):469–75.
Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26(1):136–8.
Xiang Q, Zhu D, Chen QL, Delgado-Baquerizo M, Su JQ, Qiao M, Yang XR, Zhu YG. Effects of diet on gut microbiota of soil collembolans. Sci Total Environ. 2019;676:197–205.
Yun JH, Roh SW, Whon TW, Jung MJ, Kim MS, Park DS, Yoon C, Nam YD, Kim YJ, Kim JY, Choi JH. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl Environ Microbiol. 2014;80(17):5254–64.
Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 2013;30(5):614–20.
Zhang Q, Zhang Z, Lu T, Yu Y, Penuelas J, Zhu YG, Qian H. Gammaproteobacteria, a core taxon in the guts of soil fauna, are potential responders to environmental concentrations of soil pollutants. Microbiome. 2021;9(1):1–17.
Zhu D, Chen QL, An XL, Yang XR, Christie P, Ke X, Wu LH, Zhu YG. Exposure of soil collembolans to microplastics perturbs their gut microbiota and alters their isotopic composition. Soil Biol Biochem. 2018;116:302–10.
Acknowledgements
This work was supported by Korea Institute of Planning and Evaluation for Technology in Food, Agriculture and Forestry (IPET) through High Value-added Food Technology Development Program, funded by Ministry of Agriculture, Food and Rural Affairs (MAFRA)(321034052HD020), National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. NRF-2019R1A2C1089704), National Research Foundation of Korea (NRF) grant funded by the Korea government (Ministry of Science and ICT) (NRF-2022R1A2C1011508). This study was supported by a Korea University grant.
Author information
Authors and Affiliations
Contributions
DP, JW, KC and IGC contributed to the study design. KC and IGC received the funds and supervised the study. JW carried out the field experiments, culturing, and sample collection. DP carried out DNA extraction, library preparation, sequencing, and bioinformatics analysis. DP and JW wrote the initial draft of the manuscript. DP, JW, KC and IGC contributed to the proofreading redrafting of the manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that there is no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Supplementary figures, Fig S1–S9. Fig S1. Graphical representation of the life cycle of A. kimi; Fig S2. Alpha diversity analysis; Fig S3–S6. Comparison of alpha diversity matrices; Fig S7. Shared and unique OTUs between adult and juvenile A. kimi; Fig S8. Microbial community composition in Deokso and Jinju soil; Fig S9. Shared and unique OTUs between adult A. kimi.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pathiraja, D., Wee, J., Cho, K. et al. Soil environment reshapes microbiota of laboratory-maintained Collembola during host development. Environmental Microbiome 17, 16 (2022). https://doi.org/10.1186/s40793-022-00411-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40793-022-00411-7