
Abstract
Bacteroides barnesiae Lan et al. 2006 is a species of the genus Bacteroides, which belongs to the family Bacteroidaceae. Strain BL2T is of interest because it was isolated from the gut of a chicken and the growing awareness that the anaerobic microbiota of the caecum is of benefit for the host and may impact poultry farming. The 3,621,509 bp long genome with its 3,059 protein-coding and 97 RNA genes is a part of the Genomic Encyclopedia of Type Strains, Phase I: the one thousand microbial genomes (KMG) project.
Similar content being viewed by others


Introduction
Strain BL2T (= DSM 18169 = CCUG 54636 = JCM 13652) is the type strain of Bacteroides barnesiae which belongs to the genus Bacteroides [1]. The species epithet is derived from the name of Ella M. Barnes, a British microbiologist, who has contributed much to our knowledge of intestinal bacteriology and anaerobic bacteriology in general. B. barnesiae strain BL2T was isolated from caecum of a healthy chicken. Four other strains belonging to the same species have been isolated from the same source [1]. The genus Bacteroides represents one of the predominant anaerobic genera found in chicken caecum [2–4]. Bacteroides species are thought to play a fundamental role in the breakdown of complex molecules (such as polysaccharides) into simpler compounds that are used by the animal host as well as the microorganisms themselves [5, 6], in the utilization of nitrogenous substances and in the biotransformation of bile acids and other steroids [7]. They also play a role as beneficent protectors of the gut against pathogenic microorganisms [8]. Here we present a summary classification and set of features for B. barnesiae strain BL2T, together with the description of the complete genomic sequencing and annotation.
Organism information
Classification and features
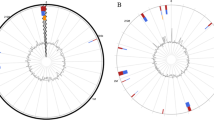
A 1301 bp long contig contained the most complete 16S rRNA gene copy in the draft genome. This partial gene differed by 7 nucleotides (0.5 %) from the 16S rRNA reference sequence (AB253726) generated for the original description of B. barnesiae [1]. Such a difference is not unusual when comparing original sequences from the time organisms were initially described with sequences of type strain genomes sequenced in the KMG project [9], a problem that was only partially resolved in the sequencing orphan species initiative (SOS) [10]. A representative 16S rRNA gene sequence of strain BL2T was compared with GenBank using NCBI BLAST. The single most frequent genus found was Bacteroides . The highest-scoring environmental sequences (up to 99.8 % sequence identity), including HQ784912 (‘gastrointestinal specimens clone ELU0102-T240-S-NI_000093’), were all from a study on gastrointestinal specimens linked to inflammatory bowel diseases phenotype in human ileum [11] and indicate that close relatives of strain BL2T and representatives of B. barnesiae are also relevant to human health. Fig. 1 shows the phylogenetic position of B. barnesiae in a 16S rRNA gene sequence-based tree.

Phylogenetic tree based on the 16S rRNA gene sequences showing the relationship of Bacteroides barnesiae strain BL2T among the genus Bacteroides . The tree was constructed by the neighbor-joining method. Numbers at nodes indicate the percentage bootstrap values of 1000 replicates. Bars, 0.01 substitutions per nucleotide position. Accession numbers are given for each strain
The cells of B. barnesiae are pleomorphic rods (0.5-1.4 × 0.8-10.6 μm) (Fig. 2). The cells are usually arranged singly or in pairs [1]. B. barnesiae is a Gram-negative, non-sporeforming bacterium (Table 1) that is described as non-motile, with only seven genes associated with motility having been found in the genome (see below). The optimum temperature for growth of strain BL2T is 37 °C. B. barnesiae is a strictly anaerobic chemoorganotroph and is able to ferment glucose, lactose, sucrose, maltose, salicin, xylose, cellobiose, mannose and raffinose [1]. The organism hydrolyzes esculin but does not liquefy gelatin, and neither reduces nitrate nor produces indole from tryptophan [1]. B. barnesiae does not utilize mannitol, arabinose, glycerol, melezitose, sorbitol, rhamnose or trehalose [1]. Growth is possible in the presence of bile [1]. Major fermentation products from broth (1 % peptone, 1 % yeast extract, and 1 % glucose each (w/v)) are acetic acid and succinic acid, whereas isovaleric acid is produced in small amounts [1]. B. barnesiae shows activity for α-galactosidase, β-galactosidase, α-glucosidase, β-glucosidase, α-arabinosidase, N-acetyl-β-glucosaminidase, α-fucosidase, alkaline phosphatase, leucyl glycine arylamidase, alanine arylamidase and glutamyl glutamic acid arylamidase but no activity urease, catalase, arginine dihydrolase, β-galactosidase 6-phosphate, β-glucuronidase, glutamic acid decarboxylase and arginine, proline, phenylalanine, leucine, pyroglutamic acid, tyrosine, glycine, histidine and serine arylamidase [1].

Light microscope image of strain BL2T
B. barnesiae strain BL2T contains menaquinones MK-10 (58 %) and MK-11 (34 %) as principal respiratory quinones, small amounts of MK-8, MK-9 and MK-12 (2 % each) are found as minor components [1]. The major fatty acids found were anteiso-C15:0 (32 %), iso-C15:0 (15 %), 3-hydroxy C16:0 (10 %) and C16:0 (10 %). Fatty acids C14:0 (4 %), C15:0 (2 %), C18:1 ω9c (4 %), C18:2 ω6,9c (2 %) and 3-hydroxy iso-C17:0 (7 %) were found in minor amounts [1]. Chemotaxonomic features are in line with known features from other representatives of the genus [1].
Genome sequencing information
Genome project history
The organism was selected for sequencing on the basis of its phylogenetic position [12–14]. Sequencing of B. barnesiae strain BL2T is part of Genomic Encyclopedia of Type Strains, Phase I: the one thousand microbial genomes project [9] which aims not only to increase the sequencing coverage of key reference microbial genomes [15], but also to generate a large genomic basis for the discovery of genes encoding novel enzymes [16]. The genome project is deposited in the Genomes OnLine Database [17] and the permanent draft genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute using state of the art sequencing technology [18]. A summary of the project information is shown in Table 2.
Growth conditions and genomic DNA preparation
B. barnesiae strain BL2T, DSM 18169, was grown anaerobically in DSMZ medium 429 (Columbia Blood Agar) at 37 °C [19]. DNA was isolated from 0.5-1 g of cell paste using JetFlex genomic DNA purification (GENOMED) following the standard protocol as recommended by the manufacturer with and additional protease K (50 μl; 21 mg/ml) digest for 60 min. at 58 °C followed by addition of 200 μl Protein Precipitation Buffer after protein precipitation and overnight incubation on ice. DNA is available through the DNA Bank Network [20].
Genome sequencing and assembly
The permanent draft genome of B. barnesiae strain BL2T was generated using Illumina technology [18, 21]. An Illumina Standard shotgun library was constructed and sequenced using the Illumina HiSeq 2000 platform which generated 11,109,700 reads totaling 1,666.5 Mb. All general aspects of library construction and sequencing performed at the DOE-JGI can be found at [22]. All raw Illumina sequence data was passed through DUK, a filtering program developed at JGI, which removes known Illumina sequencing and library preparation artifacts [23]. Following steps were then performed for assembly: (1) filtered Illumina reads were assembled using Velvet [24], (2) 1–3 kb simulated paired end reads were created from Velvet Contigs using wgsim [25], (3) Illumina reads were assembled with simulated read pairs using Allpaths–LG (version r41043) [26]. Parameters for assembly steps were: 1) Velvet (velveth: 63 –shortPaired and velvetg: −very clean yes –export- Filtered yes –min contig lgth 500 –scaffolding no –cov cutoff 10) 2) wgsim (−e 0 –1 100 –2 100 –r 0 –R 0 –X 0) 3) Allpaths–LG (PrepareAllpathsInputs: PHRED 64 = 1 PLOIDY = 1 FRAG COVERAGE = 125 JUMP COVERAGE = 25 LONG JUMP COV = 50, RunAllpathsLG: THREADS = 8 RUN = std shredpairs TARGETS = standard VAPI WARN ONLY = True OVERWRITE = True). The final draft assembly contained 47 contigs in 43 scaffolds. The total size of the genome is 3.6 Mb and the final assembly is based on 443.6 Mb of Illumina data, which provides an average 122.7 × coverage of the genome.
Genome annotation
Genes were identified using Prodigal [27] as part of the DOE-JGI genome annotation pipeline [28, 29], following by a round of manual curation using the JGI GenePRIMP pipeline [30]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information non-redundant database, UniProt, TIGR-Fam, Pfam, PRIAM, KEGG, COG, and InterPro database. These data sources were combined to assert a product description for each predicted protein. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes-Expert Review platform [31].
Genome properties
The assembly of the draft genome sequence consists of 43 scaffolds amounting to 3,621,509 bp, and the G + C content is 46.8 % (Table 3). Of the 3,156 genes predicted, 3,059 were protein-coding genes, and 97 RNAs. The majority of the protein-coding genes (71.7 %) were assigned a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.
Insights from the genome sequence
B. barnesiae strain BL2T, Bacteroides salanitronis strain BL78T and Bacteroides gallinarum strain C35T were isolated from the cecum of the same healthy chicken [1]. The GGDC (Genome-to-Genome Distance Calculator) web server (GGDC 2.0) [32] was used for the estimation of the overall similarity between the three Bacteroides genomes. The comparison of B. barnesiae with B. salanitronis and B. gallinarum revealed that 11.1 % and 5.2 %, respectively, of the average of the genome lengths are covered with HSPs (high-scoring segment pairs). The identity within the HSPs was 83.6 % and 84.6 %, respectively, whereas the identity over the whole genome was 9.3 % and 4.4 %, respectively. The comparison of B. gallinarum with B. salanitronis revealed that 5.4 % of the genome is covered with HSPs, with an identity within in the HSPs of 84.1 % and an identity over the whole genome of 4.6 %. According to these calculations the similarity between B. barnesiae and B. salanitronis is higher than the similarity between B. barnesiae and B. gallinarum as well as the similarity between B. gallinarum and B. salanitronis .
The genome size of B. barnesiae (3.6 Mb) is significantly smaller than those of B. salanitronis (4.3 Mb) and B. gallinarum (4.9 Mb).
Conclusions
B. barnesiae strain BL2T genome consists of a single chromosome of 3.6 Mb predicted to encode 3,156 genes. Strain BL2T has a relatively small genome in comparison to other sequenced Bacteroides species isolated from the same chicken (4.3-4.9 Mb). These differences of genome size may be the results of adaptation in this niche. Further study will be necessary for elucidation of this idea.
Abbreviations
- KMG:
-
One thousand microbial genomes
- JGI:
-
Joint genome institute
- SOS:
-
Sequencing orphan species
- GGDC:
-
Genome-to-genome distance calculator
References
Lan PTN, Sakamoto M, Sakata S, Benno Y. Bacteroides barnesiae sp. nov., Bacteroides salanitronis sp. nov. and Bacteroides gallinarum sp. nov., isolated from chicken caecum. Int J Syst Evol Microbiol. 2006;56:2853–9. doi:10.1099/ijs.0.64517-0.
Barnes EM, Mead GC, Impey CS, Adams BW. Analysis of the avian intestinal flora. In: Lovelock DW, Davies R, editors. Techniques for the study of mixed populations, Society for Applied Bacteriology Technical Series no. 11. New York: Academic; 1978. p. 89–105.
Salanitro JP, Fairchilds IG, Zgornicki YD. Isolation, culture characteristics, and identification of anaerobic bacteria from the chicken caecum. Appl Microbiol. 1974;27:678–87. PubMed.
Salanitro JP, Blake IG, Muirhead PA. Studies on the cecal microflora of commercial broiler chickens. Appl Microbiol. 1974;28:439–47. PubMed.
Degnan BA, Macfarlane S, Quigley ME, Macfarlane GT. Starch utilization by Bacteroides ovatus isolated from the human large intestine. Curr Microbiol. 1997;34:290–6. doi:10.1007/s002849900184.
Reeves AR, Wang GR, Salyers AA. Characterization of four outer membrane proteins that plays a role in utilization of starch by Bacteroides thetaiotaomicron. J Bacteriol. 1997;179:643–9. PubMed.
Hentges DJ. Anaerobes as normal flora. In: Finegold SM, George WL, editors. Anaerobic infections in humans. San Diego: Academic; 1989. p. 37–53.
Hentges DJ. Role of the intestinal flora in host defense against infection. In: Hentges DJ, editor. Human intestinal microflora in health and disease. New York: Academic; 1983. p. 311–31.
Kyrpides NC, Woyke T, Eisen JA, Garrity G, Lilburn TG, Beck BJ, et al. Genomic Encyclopedia of Type Strains, Phase I: the one thousand microbial genomes (KMG-I) project. Stand Genomic Sci. 2013;9:628–34.
Yarza P, Spröer C, Swiderski J, Mrozek N, Spring S, Tindall BJ, et al. Sequencing orphan species initiative (SOS): filling the gaps in the 16S rRNA gene sequence database for all species with validly published names. Syst Appl Microbiol. 2013;36:69–73. doi:10.1016/j.syapm.2012.12.006.
Li E, Hamm CM, Gulati AS, Sartor RB, Chen H, Wu X, et al. Inflammatory bowel diseases phenotype, C. difficile and NOD2 genotype are associated with shifts in human ileum associated microbial composition. PLoS One. 2012;7:e26284. doi:10.1371/journal.pone.0026284.
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature. 2009;462:1056–60. doi:10.1038/nature08656.
Klenk HP, Göker M. En route to a genome - based classification of Archaea and Bacteria? Syst Appl Microbiol. 2010;33:175–82. doi:10.1016/j.syapm.2010.03.003.
Göker M, Klenk HP. Phylogeny-driven target selection for large-scale genome-sequencing (and other) projects. Stand Genomic Sci. 2013;8:360–74. doi:10.4056/sigs.3446951.
Kyrpides NC, Hugenholtz P, Eisen JA, Woyke T, Göker M, Parker CT, et al. Genomic encyclopedia of Bacteria and Archaea: sequencing a myriad of type strains. PLoS Biol. 2014;8:e1001920. doi:10.1371/journal.pbio.1001920.
Piao H, Froula J, Du C, Kim TW, Hawley E, Bauer S, et al. Identification of novel biomass-degrading enzymes from microbial dark matter: populating genome sequence space with functional annotation. Biotechnol Bioeng. 2014;111:1550–65. doi:10.1002/bit.25250.
Pagani I, Liolios K, Jansson J, Chen IM, Smirnova T, Nosrat B, et al. The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2012;40:D571–9. doi:10.1093/nar/gkr1100.
Mavromatis K, Land ML, Brettin TS, Quest DJ, Copeland A, Clum A, et al. The fast changing landscape of sequencing technologies and their impact on microbial genome assemblies and annotation. PLoS One. 2012;7:e48837. doi:10.1371/journal.pone.0048837.
List of growth media used at DSMZ: http://www.dsmz.de/catalogues/catalogue-microorganisms/culture-technology/list-of-media-for-microorganisms.html.
Gemeinholzer B, Dröge G, Zetzsche H, Haszprunar G, Klenk HP, Güntsch A, et al. The DNA Bank Network: the start from a German initiative. Biopreserv Biobank. 2011;9:51–5. doi:10.1089/bio.2010.0029.
Bennett S. Solexa Ltd. Pharmacogenomics. 2004;5:433–8. doi:10.1517/14622416.5.4.433.
JGI web page http://www.jgi.doe.gov.
Mingkun L, Copeland A, Han J. DUK – a fast and efficient kmer based sequence matching tool (DOE JGI User Meeting 2011). https://isswprod.lbl.gov/library/view-docs/public/output/rpt80221.PDF.
Zerbino D, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9. doi:10.1101/gr.074492.107.
https://github.com/lh3/wgsim
Gnerre S, MacCallum I. High–quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A. 2011;108:1513–8. doi:10.1073/pnas.1017351108.
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiati on site identification. BMC Bioinformatics. 2010;11:119. doi:10.1186/1471-2105-11-119.
Mavromatis K, Ivanova NN, Chen IM, Szeto E, Markowitz VM, Kyrpides NC. The DOE-JGI Standard operating procedure for the annotations of microbial genomes. Stand Genomic Sci. 2009;1:63–7. doi:10.4056/sigs.632.
Chen IM, Markowitz VM, Chu K, Anderson I, Mavromatis K, Kyrpides NC, et al. Improving microbial genome annotations in an integrated database context. PLoS One. 2013;8:e54859. doi:10.1371/journal.pone.0054859.
Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, et al. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods. 2010;7:455–7. doi:10.1038/nmeth.1457.
Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–8. doi:10.1093/bioinformatics/btp393.
Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. 2013;14:60. doi:10.1186/1471-2105-14-60.
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26:541–7. doi:10.1038/nbt1360.
Field D, Amaral-Zettler L, Cochrane G, Cole JR, Dawyndt P, Garrity GM, et al. The Genomic Standards Consortium. PLoS Biol. 2011;9:e1001088. doi:10.1371/journal.pbio.1001088.
Garrity G. NamesforLife BrowserTool takes expertise out of the database and puts it right in the browser. Microbiol Today. 2010;37:9.
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–9. doi:10.1073/pnas.87.12.4576.
Krieg NR, Ludwig W, Euzéby J, Whitman WB. Phylum XIV. Bacteroidetes phyl. nov. In: Krieg NR, Staley JT, Brown DR, Hedlund BP, Paster BJ, Ward NL, Ludwig W, Whitman WB, editors. Bergey’s Manual of Systematic Bacteriology, vol. 4. 2nd ed. New York: Springer; 2011. p. 25.
Euzéby J. Validation List no. 143. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol. 2012;62:1–4. doi:10.1099/ijs.0.039487-0.
Krieg NR. Class I. Bacteroidia class nov. In: Krieg NR, Staley JT, Brown DR, Hedlund BP, Paster BJ, Ward NL, Ludwig W, Whitman WB, editors. Bergey’s Manual of Systematic Bacteriology, vol. 4. 2nd ed. New York: Springer; 2011. p. 25.
Krieg NR. Order I. Bacteroidales ord. nov. In: Krieg NR, Staley JT, Brown DR, Hedlund BP, Paster BJ, Ward NL, Ludwig W, Whitman WB, editors. Bergey’s Manual of Systematic Bacteriology, vol. 4. 2nd ed. New York: Springer; 2011. p. 25.
Pribram E. Klassification der Schizomyceten. Leipzig: F. Deuticke; 1933. p. 1–143.
Skerman VBD, McGowan V, Sneath PHA. Approved lists of bacterial names. Int J Syst Bacteriol. 1980;30:225–420. doi:10.1099/00207713-30-1-225.
Castellani A, Chalmers AJ. Genus Bacteroides Castellani and Chalmers, 1918. Manual of Tropical Medicine. 3rd ed. New York: Williams Wood and Co; 1919. p. 959–60.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9. doi:10.1038/75556.
Acknowledgements
We would like to gratefully acknowledge the help of Iljana Schröder for growing B. barnesiae cultures, and Evelyne-Marie Brambilla for DNA extraction and quality control (both at DSMZ). This work was performed under the auspices of the US Department of Energy Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under contract No. DE-AC52-07NA27344. A.L. was supported in part by Russian Ministry of Science Mega-grant no.11.G34.31.0068 (PI. Dr Stephen J O’Brien).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
MS, RP, HPK and MO drafted the manuscript. ALL, JH, ST, MH, TR, NM, MH, AP, NNI, VMM, TW and NCK sequenced, assembled, and annotated the genome. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sakamoto, M., Lapidus, A.L., Han, J. et al. High quality draft genome sequence of Bacteroides barnesiae type strain BL2T (DSM 18169T) from chicken caecum. Stand in Genomic Sci 10, 48 (2015). https://doi.org/10.1186/s40793-015-0045-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40793-015-0045-6