
Abstract
Background
Recently, significant interest in 44Sc as a tracer for positron emission tomography (PET) imaging has been observed. Unfortunately, the co-emission by 44Sc of high-energy γ rays (E γ = 1157, 1499 keV) causes a dangerous increase of the radiation dose to the patients and clinical staff. However, it is possible to produce another radionuclide of scandium—43Sc—having properties similar to 44Sc but is characterized by much lower energy of the concurrent gamma emissions. This work presents the production route of 43Sc by α irradiation of natural calcium, its separation and purification processes, and the labeling of [DOTA,Tyr3] octreotate (DOTATATE) bioconjugate.
Methods
Natural CaCO3 and enriched [40Ca]CaCO3 were irradiated with alpha particles for 1 h in an energy range of 14.8–30 MeV at a beam current of 0.5 or 0.25 μA. In order to find the optimum method for the separation of 43Sc from irradiated calcium targets, three processes previously developed for 44Sc were tested. Radiolabeling experiments were performed with DOTATATE radiobioconjugate, and the stability of the obtained 43Sc-DOTATATE was tested in human serum.
Results
Studies of natCaCO3 target irradiation by alpha particles show that the optimum alpha particle energies are in the range of 24–27 MeV, giving 102 MBq/μA/h of 43Sc radioactivity which creates the opportunity to produce several GBq of 43Sc. The separation experiments performed indicate that, as with 44Sc, due to the simplicity of the operations and because of the chemical purity of the 43Sc obtained, the best separation process is when UTEVA resin is used. The DOTATATE conjugate was labeled by the obtained 43Sc with a yield >98 % at elevated temperature.
Conclusions
Tens of GBq activities of 43Sc of high radionuclidic purity can be obtainable for clinical applications by irradiation of natural calcium with an alpha beam.
Similar content being viewed by others

Background
Radionuclide therapy of somatostatin overexpressing tumors is currently being performed with DOTA conjugated to somatostatin analogs (DOTATOC and DOTATATE) labeled with high- and medium-energy β− emitters: 90Y or 177Lu, respectively [1, 2]. Many clinical studies have also shown that 68Ga-labeled somatostatin analogs are relevant positron emission tomography tracers for imaging such tumors and their metastases [3]. DOTATOC labeled with 68Ga showed high binding affinity to the human somatostatin receptor subtype 2, improving tumor imaging capabilities and offering the possibility of low dose imaging followed by higher dose treatment. However, the half-life of 68Ga (T 1/2 = 67.71 min) may limit the spectrum of clinical applications of 68Ga-labeled radiopharmaceuticals. Furthermore, the relatively high cost of the generators and perhaps more importantly the requirement for postelution purification and concentration of 68Ga solution to small volume make this isotope of limited utility in clinical applications [4]. Therefore, the use of radionuclides of extended physical half-life is currently being reconsidered.
An alternative could be cyclotron-produced 64Cu (T 1/2 = 12.7 h) which has been applied in a large number of preclinical and clinical positron emission tomography (PET) studies [5]. The longer half-life offers the possibility to label bigger molecules like mAB fragments and to use 64Cu radiopharmaceuticals in hospitals without cyclotrons and radiopharmaceutical units. However, 64Cu exists in three oxidation states and forms unstable in vivo chelate complexes. In addition, 64Cu presents a relatively low positron branching ratio (17.6 %) and high co-emission of β− particles (39 % branching ratio) which significantly contribute to an additional patient dose.
In 2010, the 44Sc radionuclide was proposed by Roesch as a potential alternative to 68Ga in clinical PET diagnosis [6, 7]. 44Sc decays by the emission of low-energy positrons E β+, with a half-life of T 1/2 = 3.97 h, which is almost four times longer than that of 68Ga. 44Sc can be obtained from the 44Ti/44Sc generator [8] or produced in the 44Ca(p,n)44Sc reaction on small- or medium-sized medical cyclotrons that currently supply 18F to hospitals [9–14]. These properties make it highly attractive for clinical PET applications because they enable transportation of 44Sc-labeled radiopharmaceuticals to hospitals that are located several hundred kilometers away from the radiopharmaceutical production site. Moreover, it was found that Sc3+ likewise Y3+ and Lu3+, forms in aqueous solutions of DOTA complexes with the same coordination sphere (CN = 8) and with similar stability constants [15], whereas the relatively small Ga3+ forms octahedral complexes. As a result, the chemical properties of 44Sc-labeled DOTA conjugates are almost the same as those of the 90Y- and 177Lu-labeled versions; therefore, we can presume that 44Sc-DOTA bioconjugates will demonstrate similar properties in vivo (i.e., receptor affinity, kidney clearance) to the 177Lu- and 90Y-conjugates currently applied in therapy.
It is also important to mention that the other scandium radioisotope, i.e., 47Sc (T 1/2 = 3.4 days, E β−(av) = 162 keV, main E γ = 159.4 keV, I = 68.3 %) is a promising low-energy β− emitter for targeted radiotherapy [16–19]; thus, the β+-emitting 44Sc with the β−-emitting 47Sc represent an ideal theranostic pair as mentioned above regarding 64Cu.
However, the co-emission of high-energy γ rays (E γ = 1157, 1499 keV) has to be taken into consideration with regard to the radiation dose to the patients and clinical staff. Also, co-production of longer lived 44mSc (T 1/2 = 58.6 h) increases the radiation dose.
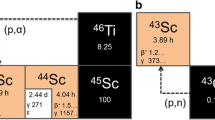
In our work, we propose to use another radionuclide of scandium, i.e., 43Sc, which shows properties similar to 44Sc, but with much lower energy concurrent gamma emissions (Table 1). 43Sc can be produced either by the 43Ca(p,n), or 42Ca(d,n) reactions, but unfortunately, the cost of enriched calcium targets is prohibitive. A more promising method of 43Sc production is alpha irradiation of a natural calcium target via the 40Ca(α,p) and 40Ca(α,n) channels. The possibility of 43Sc production by this route has already been mentioned in four conference communications. The availability of cyclotrons with intense α beams is limited; however, with a near-to-4-h half-life and predicted production cross-section approaching 1 b [20], the potential exists for regional distribution following mass production at a single cyclotron unit. The aim of this study was to investigate the possibility of 43Sc production at an accelerator, allowing its use for preclinical and clinical PET imaging.
Methods
Chemicals and reagents
NaOH micropills and acetic acid were purchased from POCH S.A. Gliwice. Ammonia (ammonium hydroxide solution 25 %), citric acid, and ammonium carbonate were purchased from Sigma Aldrich. N,N,N′,N′-tetra-n-octyldiglycolamide (DGA) 50–100 mesh and UTEVA 100–200 mesh resins were purchased from Eichrom, USA; Chelex 100 resin (Na+ form, mesh size 100–200) was purchased from Bio-Rad, USA; and DOWEX 50 × 8 resin (hydrogen form, 200–400 mesh) was purchased from Fluka Analytical, Germany. [DOTA,Tyr3] octreotate (DOTATATE) 95 % purity (HPLC) was purchased from piChem (Graz, Austria). All chemicals were of analytical grade and were used without further purification.
Natural CaCO3 of chemical purity >99.999 purchased from Sigma Aldrich and enriched [40Ca]CaCO3 (99.99 %) purchased from Isoflex (USA) were used as target materials. The isotopic composition of enriched 40Ca was 99.99 % of 40Ca and 0.01% of 44Ca, while the amounts of other calcium isotopes were below 0.01 %.
Irradiation of natCaCO3 and [40Ca]CaCO3 targets
Irradiations of natural targets were performed using the Scanditronix MC 40 cyclotron at the European Commission’s Joint Research Centre (Ispra, Italy). Irradiations of enriched [40Ca]CaCO3 targets were performed using the Warsaw Heavy Ion Cyclotron operating at the Heavy Ion Laboratory of the University of Warsaw. The Ispra cyclotron is capable of accelerating positive ions such as protons, deuterons, and alphas to variable energies. The Warsaw machine accelerates heavy ions from +He up to Ar with energies from 2 up to about 8 AMeV. For irradiation at the Ispra cyclotron, the target material was wrapped in an aluminum foil of a 25-μm thickness. The samples were irradiated in aluminum capsules with an inner diameter of 10 mm. Each target capsule was inserted in a holder that allowed direct water cooling from both the rear and the front sides. In the Warsaw cyclotron, targets in the form of pellets bundled in thin aluminum foils produced from CaCO3 powder using a hydraulic press were irradiated with an internal α-particle beam. Al energy degraders were used when alpha particle energies lower than maximal were necessary.
In order to optimize the yield of 43Sc production by the 40Ca(α,p)43Sc and natCa(α,n)43Ti→43Sc nuclear reactions, ~100-mg natCaCO3 samples (target thickness ~375 μm) were irradiated for 28–34 min by an alpha beam of 13–25 MeV on the target with an alpha current of 0.5 pμA at the Scanditronix MC 40 cyclotron.
Enriched [40Ca]CaCO3 targets of ~100 mg were irradiated for 30 min by an alpha beam of 20 MeV with an alpha current of 0.25 pμA (He+) at the Warsaw Heavy Ion Cyclotron.
Measurement of radioactivity
The absolute radioactivity of 43Sc and other obtained radionuclides was measured by γ-spectrometry using two high-purity germanium (HPGe) detectors. The detectors were energy and efficiency calibrated in different geometries using certified standard radioactive sources (ENEA Italy, DAMRI and CERCA France). The gamma-ray spectrum analysis software package Genie 2000 (CANBERA, USA) was used to collect the data. The γ-ray peak at 372.8 keV was chosen for 43Sc detection, and the peaks at 1157.00, 271.24, and 159.38 keV were chosen to detect 44Sc, 44mSc, and 47Sc, respectively. Three peaks at 983.52, 1037.52, 1312.10 keV were used to quantify yields of 48Sc [21]. The uncertainty of all the determined activities was below 1 %.
Separation of 43Sc from the target
In order to find the optimal method for the separation of 43Sc from the irradiated calcium targets, three procedures were tested:
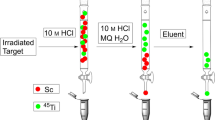
In the first method, described by Valdovinos et al. [22], the irradiated natCaCO3 target was dissolved in 1 ml of 9 M HCl solution. The dissolved target solution was passed through a column containing 50 mg of UTEVA resin, and after adsorption of 43Sc, the column was washed with 5 ml of 9 M HCl. The scandium radionuclides were eluted with a 400-μl portion of H2O.
The second method, reported in the paper by Mueller et al. [10], consists of dissolving the CaCO3 targets in 3 M HCl and adsorption of scandium radionuclides in a column filled with 70 mg of DGA. The adsorbed 43Sc was eluted from the DGA resin with HCl (0.1 M, 2–3 ml). Afterwards, the acidic 43Sc solution was loaded on a second column filled with 100 mg of cation exchange resin DOWEX 50 (hydrogen form, 200–400 mesh). Finally, 43Sc was eluted using 1 M ammonium acetate adjusted to pH = 4 using HCl solution.
The third method, developed by our group [9], consisted of dissolution of the target in 1 M HCl and adsorption of 43Sc on chelating ion exchange resin Chelex 100 of bed size 0.8 × 4.0 cm and conditioned with 5 ml of 1 M HCl. After adsorption of 43Sc and Ca2+, the column was washed with 30 ml of 0.01 M HCl in order to remove Ca2+. The scandium radionuclides were then eluted with 1 M HCl in 0.5-ml fractions.
Radiolabeling and stability studies of DOTATATE conjugate
DOTATATE, octreotate-somatostatin analog conjugated to DOTA chelator, was labeled with the obtained 43Sc using 10, 15, and 25 nmol of the peptide. The most active fraction of 43Sc solution was combined with 0.2 ml of 0.2 M sodium acetate buffer (pH = 6) containing 14, 21, or 36 μl of the peptide (0.7 nmol μl−1) in the buffer. The solution was next heated for 25 min at 95 °C in a water bath. Product formation and reaction yields were estimated by instant thin-layer chromatography (ITLC) using Silica gel 60 TLC plates (Merck). A 0.1 M citric buffer of pH = 5.4 was used as the eluent. Of the solution, 10 μl was dropped on the ITLC strip. Free 43Sc moved with the front boundary of the solution whereas the labeled bioconjugate remained at the starting point. The labeling yield defined as the percentage of 43Sc radioactivity complexed by DOTATATE to the starting activity was calculated as the ratio of the activity of the strip application part to the whole strip activity.
The stability of the labeled DOTATATE in human serum was assessed by adding 20 μl of the radioconjugate solution to 500 μl of the human serum. The mixture was incubated at 37 °C, and the stability was measured by taking aliquots of the human serum solutions at different times and measuring the liberated scandium radionuclide by ITLC analysis.
Results and discussion
Optimization of 43Sc cyclotron production
The first step towards developing a simple, fast, and inexpensive method of 43Sc-DOTATATE production is the optimization of the cyclotron production parameters. For this purpose, we measured the radioactivity yield as a function of alpha particle energy on target. The results are presented in Table 2. The analysis of the results obtained shows that the optimum on-target α particle energies are in the range of 24–26.5 MeV, which is a little higher than that predicted by Levkovskij [23].
The 43Sc activity obtained after irradiation of a ~100 mg natCaCO3 target for 34 min with a 25-MeV alpha particle beam of a 0.5-μA beam current was about 29 MBq. The produced activity can be increased by extending the irradiation time and using a higher beam current, as in the case of 44Sc production by proton irradiation of a [44Ca]CaCO3 target [24]. Extrapolating to an irradiation time of 4 h at 20 μA, the end of bombardment (EOB) yields are expected to approach 5.7 GBq, enough for the preparation of more than 25 patient doses. Of course, the proposed 40-fold scale-up of the current will bring challenging problems with heat dissipation from the CaCO3 target, which can be solved by the use of a metallic Ca target previously tested by Severin et al. [11] for high-current proton irradiation.
Production of 43Sc is accompanied by small co-production of other scandium radionuclides such as 44Sc, 44mSc, 46Sc, and 47Sc (Table 3). The scandium radioisotopes 44gSc and 44mSc were synthesized from 42Ca present in the natural target (0.65 %) via the (α,pn) reaction. 46Sc and 47Sc were produced from the 2 % of 44Ca and the 0.13 % of 43Ca components of the natural calcium in (α,pn) and (α,p) reactions, respectively.
As shown in Table 3, the only significant contaminant is the 44gSc (0.011 %). From the point of view of possible applications of 43Sc in nuclear medicine contamination of the 43Sc product by 44gSc is insignificant due to the similarity of both radioisotopes. Complete elimination of the 43Sc impurities is possible by using an isotopically enriched (and inexpensive) [40Ca]CaCO3 target (1.5 USD/mg). Irradiation of such a target (composed of 99.99 % 40Ca + 0.01 % 44Ca) with 20-MeV alpha particles results in a level of all impurities below 1.5 × 10−5 % of the 43Sc radioactivity, even 20 h after the EOB.
Separation of 43Sc from the target
Three methods previously developed for 44Sc production were tested for the separation of 43Sc from the natural Ca target. One method was based on the application of chelating resin and was developed in our group [9], and in the other two methods, the extraction resins developed by Valdovinos et al. [13] and Mueller et al. [10] were used. These methods were compared with respect to 43Sc recovery, the volume and composition of the 43Sc fraction (Table 4), and the possibility of separation from metallic impurities which could negatively affect the effectiveness of 43Sc bioconjugate labeling.
All separation procedures studied are fast and simple. In the case of Chelex 100 and UTEVA resins, the target dissolution and separation of 43Sc were performed in 30 min and the two-step separation process DGA + Dowex 50 in 45 min. All methods render possible the effective separation of 43Sc from calcium. The efficiency of the separation methods is consistent with the previously reported procedures for separation of 44Sc from enriched and natural targets [9, 10, 13].
High chemical purity of the final 43Sc fraction is important, since the presence of other metals may interact with the DOTA chelator. The most dangerous is Fe3+ for which the log of the stability constant with the DOTA ligand is 29.4 [25] which is greater than that for Sc3+ (log K = 27 [15]). Influence of other possible impurities like Zn2+and Co2+ is negligible due to the much lower stability constants of their DOTA complexes (log K Zn = 19.3, log K Co = 19.3 [25]. Fe concentration, measured with the ICP-MS technique in the dissolved calcium target in HCl solutions, varied between 58 and 87 ppm. After the separation processes, the total level of Fe in the 43Sc samples decreased to 10.50 ppm for separation with Chelex resin, 0.56 ppm for DGA + DOWEX 50 and <0.001 ppm for UTEVA. The amount of Ca2+ in the 43Sc fractions was less than 1 ppm.
Two additional important factors in the labeling processes are the volume of the 43Sc fraction and the composition of the eluate. In the three methods tested here, the volumes of eluates containing more than 80 % of 43Sc which was used for reprocessing varied between 0.4 and 0.65 ml. The best composition of the eluate was obtained using the tandem of DGA and Dowex 50 resins where the eluate containing ammonium acetate buffer can be used directly to label DOTA or DTPA bioconjugates. The acidic eluates from Chelex 100 and UTEVA resins need neutralization. From the experiments performed, it can be concluded that, as with 44Sc [13], due to the simplicity of the operations, the best methods for isolation of 43Sc from the target material are procedures in which Chelex 100 or UTEVA resins are used. In respect to the chemical purity of the obtained 43Sc solutions, the best separation is obtained using UTEVA resin. Therefore, for further experiments related to the labeling of DOTATATE, we chose this process.
Radiolabeling and stability studies of DOTATATE conjugate
The DOTATATE was used as a model system for radiolabeling with the 43Sc radionuclide. High efficiency of labeling the DOTATATE with 43Sc was achieved as shown by the labeling yield exceeding 98 % for an amount of bioconjugate equal to or higher than 15 nmol (Table 5). The high yield showed that highly pure 43Sc was obtained after the separation process with UTEVA resin and is suitable for labeling biomolecules. When the reaction yield is not high enough, the labeled peptide can be easily purified using the Sep-Pak® C-18 column.
The labeled DOTATATE radioconjugate exhibited high stability in human serum at 37 °C. After 14 h of incubation in the serum, more than 98 % of 43Sc remained in the radioconjugate.
In the present study, we found that synthesis of 43Sc-DOTATATE using cyclotron produced 43Sc could be adequate for nuclear medicine applications. Therefore, we believe that our method could be suitable for labeling different bioconjugates, regardless of whether they are other somatostatin analogs or a useful diagnostic peptide such as bombesin, substance P, or an oxytocin analog.
The procedure for labeling with 43Sc is as easy as that in the case of 68Ga and 44Sc which makes it possible to use commercially available kits. The 4 h half-life and obtainable GBq activities of 43Sc make possible the production and transport of the labeled bioconjugates to satellite PET centers, in analogy to 18F-FDG.
Conclusions
The production of 43Sc in (α,n) and (α,p) nuclear reactions on a natural CaCO3 target was successfully performed, and extrapolation of the results obtained creates the opportunity to produce activity levels of 43Sc sufficient for medical applications. The 43Sc radionuclide has several advantages in comparison to the 44Sc recently proposed for PET imaging. Firstly, in contrast to 44Sc, it does not emit high-energy gamma rays that should be taken into consideration with regard to the radiation dose delivered to the patients and clinical staff. Emission of high-energy gamma rays also generates radiolytic decomposition of biomolecules, which is thought to be mediated by the formation of free radicals. This becomes an important issue when high quantities of radioactivity are used for labeling, as is necessary for clinical applications [26]. Also, co-production of the longer lived 44mSc (T 1/2 = 58.6 h) increases the radiation dose. Furthermore, in contrast to 44Sc, the production of 43Sc does not require an expensive highly enriched 44CaCO3 target, the price of which currently exceeds 14 USD/mg.
The proposed separation process of 43Sc from the calcium target is simple, reliable, efficient, and fast. Therefore, it is possible to use the same commercially available modular entity as that commonly used for preparation of 68Ga-radiopharmaceuticals. Unfortunately, availability of cyclotrons with high-current alpha beams is limited. Despite this, regional distribution following massive production at a single alpha facility is possible. We believe that the 43Sc obtained could be used instead of 68Ga in PET imaging and in planning peptide receptor radionuclide therapy with 177Lu- and 90Y-DOTA radiobioconjugates.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
References
Maecke HR, Reubi JC. Somatostatin receptors as targets for nuclear medicine imaging and radionuclide treatment. J Nucl Med. 2011;52:841–4.
Pool SE, Krenning EP, Eric P, Koning GA, Van Eijck CHJ, Casper HJ, et al. Preclinical and clinical studies of peptide receptor radionuclide therapy. Sem Nucl Med. 2010;40:209–18.
Antunes P, Ginj M, Zhang H, Waser B, Baum RP, Reubi JC, et al. Are radiogallium-labelled DOTA-conjugated somatostatin analogues superior to those labelled with other radiometals? Eur J Nucl Med Mol Imag. 2007;34:982–93.
Roesch F, Riss PJ. The renaissance of the 68Ge/68Ga radionuclide generator initiates new developments in 68Ga radiopharmaceutical chemistry. Curr Top Med Chem. 2010;10:1633–8.
Anderson CJ, Ferdani R. Copper-64 radiopharmaceuticals for PET imaging of cancer: advances in preclinical and clinical research. Cancer Biother Radiopharm. 2009;24:379–93.
Pruszyski M, Loktionova N, Filosofov D, Roesch F. Post-elution processing of 44Ti/44Scgenerator derived 44Sc for clinical application. Appl Radiat Isot. 2010;68:1636–41.
Rösch F. Scandium-44: benefits of a long-lived PET radionuclide available from the 44Ti/44Sc generator system. Curr Radiopharm. 2012;5:187–201.
Filosofov DV, Loktionova NS, Roesch F. A 44Ti/44Sc radionuclide generator for potential nuclear-medical application of 44Sc-based PET-radiopharmaceuticals. Radiochim Acta. 2010;98:149–56.
Krajewski S, Cydzik I, Abbas K, Bulgheroni A, Simonelli F, Holzwarth U, et al. Cyclotron production of 44Sc for clinical application. Radiochim Acta. 2013;101:333–8.
Müller C, Bunka M, Reber J, Fischer C, Zhernosekov K, Türler A, et al. Promises of cyclotron-produced 44Sc as a diagnostic match for trivalent β− emitters: in-vitro and in-vivo study of a 44Sc-DOTA-folate conjugate. J Nucl Med. 2013;54:2168–74.
Severin G, Engle J, Valdovinos H, Barnhart T, Nickles R. Cyclotron produced 44gSc from natural calcium. Appl Radiat Isot. 2012;70:1526–30.
Duchemin C, Guertin A, Haddad F, Michel N, Metivier V. Production of scandium-44m and scandium-44g with deuterons on calcium-44: cross section measurements and production yield calculations. Phys Med Biol. 2015;60:6847-6864.
Haddad F, Ferrer L, Guertin A, Carlier T, Michel N, Barbet J, et al. ARRONAX, a high-energy and high-intensity cyclotron for nuclear medicine. Eur J Nucl Med Mol Imaging 2008;35:1377–87.
Huclier-Markai S, Kerdjoudj R, Alliot C, Bonraisin AC, Michel N, Haddad F, et al. Optimization of reaction conditions for the radiolabeling of DOTA and DOTA-peptide with 44m/44Sc and experimental evidence of the feasibility of an in vivo PET generator. Nucl Med Biol. 2014;41:e36–43 [Suppl.].
Majkowska-Pilip A, Bilewicz A. Macrocyclic complexes of scandium radionuclides as precursors for diagnostic and therapeutic radiopharmaceuticals. J Inorg Biochem. 2011;105:313–20.
Bartoś B, Majkowska A, Kasperek A, Krajewski S, Bilewicz A. New separation method of no-carrier-added 47Sc from titanium targets. Radiochim Acta. 2012;100:457–62.
Mausner LF, Kolsky KL, Joshi V, Srivastava SC. Radionuclide development at BNL for nuclear medicine therapy. Appl Radiat Isot. 1998;49:285–94.
Kolsky KL, Joshi V, Mausner LF, Srivastava SC. Radiochemical purification of no-carrier-added scandium-47 for radioimmunotherapy. Appl Radiat Isot. 1998;49:1541–9.
Majkowska A, Neves M, Antunes I, Bilewicz A. Complexes of low energy beta emitters 47Sc and 177Lu with zoledronic acid for bone pain therapy. Appl Radiat Isot. 2009;67:11–3.
Howard AJ, Jensen HB, Rios M, Fowler WA, Zimmerman BA. Measurement and theoretical analysis of some reaction rates of interest in silicon burning. Astroph J. 1974;188:131.
Browne E, Firestone RB. In: Shirley VS, editor. Table of radioactive isotopes. USA: John Wiley & Son; 1986.
Valdovinos HF, Hernandez R, Barnhart TE, Graves S, Cai W, Nickles RJ. Separation of cyclotron-produced 44Sc from a natural calcium target using a dipentyl pentylphosphonate functionalize dextraction resin. Appl Radiat Isot. 2015;95:23–9.
Levkovskij VN. Activation cross section for medium mass nuclei (A = 40–100) by medium energy protons and alpha particles (E = 10–50 MeV). Moscow: USSR; 1991.
van der Meulen NP, Bunka M, Domnanich KA, Müller C, Haller S, Vermeulen C, et al. Cyclotron production of 44Sc: from bench to bedside. Nucl Med Biol. 2015;42:745–51.
Martell A, Smith R, Motekaitis R. NIST critically selected stability constants of metal complexes database. 2004. http://www.nist.gov/srd/nist46.cfm.
Scott PJ, Hockley BG, Kung HF, Manchanda R, Zhang W, Kilbourn MR. Studies into radiolytic decomposition of fluorine-18 labeled radiopharmaceuticals for positron emission tomography. Appl Radiat Isot. 2009;67:88–94.
Acknowledgements
The authors thank Prof. Sławomir Siekierski for helpful discussions. This work was carried out as part of the National Center for Applied Research of Poland project Nr PBS3/A9/28/2015.
Funding
This study was funded by a grant from the National Center for Applied Research of Poland (grant no.: PBS3/A9/28/2015). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author declares that they have no competing interests.
Authors’ contributions
RW carried out separation of 43Sc from irradiated natural calcium targets. SK performed irradiation of the natural targets and chemical separation in JRC Ispra. KS, MS, and AJ participated in the design and performed irradiations on [40Ca]CaCO3-enriched targets at the Heavy Ion Laboratory of the University of Warsaw. KA carried out the calculations for targets and optimal proton beams. JCh participated in the design of the studies and helped to write the manuscript. JJ was involved in planning the experiments and writing the manuscript. AM carried out radiolabeling studies. FS prepared targets from natural calcium in JRC Ispra. AS designed and prepared targets from natural and enriched [40Ca]CaCO3. AT and WZ performed the analysis of gamma spectra of irradiated enriched targets and performed the statistical analysis. AB presented the idea of this work and wrote the publication. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Walczak, R., Krajewski, S., Szkliniarz, K. et al. Cyclotron production of 43Sc for PET imaging. EJNMMI Phys 2, 33 (2015). https://doi.org/10.1186/s40658-015-0136-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40658-015-0136-x