
Abstract
Background
As an important player during food digestion, gut microbiota has attracted much attention in diet adaptation studies in birds. Microbiota extracted from feces has been widely used as a proxy for gut microbiota. Although several methods have been developed for microbial DNA extraction, their performances in the bird feces have not been systematacially evaluated yet.
Methods
In this study, we applied three DNA extraction methods (Qiagen, MoBio and Bead) to extract DNA from feces of three avian dietary guilds (granivore, omnivore and carnivore), sequenced V4 region of 16S rRNA gene for each extract and evaluated the performances of DNA yield, DNA integrity, microbial composition, cell lysis capacity and alpha diversity for the three methods on each dietary guild.
Results
Bead method was the best on the performance of both DNA yield and DNA integrity regardless of dietary guild. In granivore, microbial relative abundance at both species and phylum levels, alpha diversity and cell lysis capacity were comparable among all methods. In omnivore, Qiagen had the best performance on alpha diversity, followed by Bead and MoBio. There were small variations on microbial relative abundance at both species and phylum levels among different extraction methods. MoBio exhibited the best performance on cell lysis capacity. In carnivore, considerable variations were found on microbial relative abundance at both species and phylum levels. Qiagen had the best performance on alpha diversity, followed by MoBio and Bead. MoBio had the highest cell lysis capacity.
Conclusions
DNA yield and integrity have no obvious impact on microbial composition, alpha diversity or cell lysis capacity. The microbiota results (e.g., microbial composition, cell lysis capacity, alpha diversity) obtained from different methods are comparable in granivorous avian species but not in omnivorous or carnivorous birds. Either method could be used in granivore microbiota studies. For omnivores and carnivores, we recommend Qiagen method when the research purpose is microbial diversity and MoBio when gram-positive bacteria is the research target.
Similar content being viewed by others


Background
Birds, a diverse and evolutionarily successful lineage, are composed of over 10,000 extant species (Gill and Donsker 2016), with different species inhabiting diversified ecological niches, which results in varied dietary guilds. Based on its main diet, each bird species can be grouped into one of the following nine guilds: carnivores, frugivores, granivores, herbivores, insectivores, nectarivores, omnivores, piscivores and scavengers (Burin et al. 2016). Since dietary habits can affect the evolution of avian species (e.g., speciation) (Darwin 1859), elucidating the diet adaptation for different bird species has become an important step towards the full understanding of bird evolution.
Gut microbiota has become an interesting and hot topic in diet adaptation (e.g., Di Rienzi and Britton 2020; Fuertes et al. 2019), for which microbiota extracted from feces have been widely used as a proxy (Yang et al. 2019; Berlow et al. 2020). Unlike other species (e.g., human), microbial DNA extraction from avian feces has been proved challenging due to the high urinary excretions (Eriksson et al. 2017). Currently, it is suggested that only the methods based on physical disruption (e.g., bead beating) could yield acceptable amounts of DNA (Eriksson et al. 2017) from bird fecal samples. However, further performances, such as relative abundance and bacterial diversity, two basic parameters depicting microbiota composition, have not been reported yet. Furthermore, how these DNA extraction methods behave among different avian dietary guilds also remains unknown.
In this study, we evaluated the performance of three methods (two commercial and one non-commercial methods) with different cell lysis strategies on fecal samples from the birds with plant dominant (granivore as a representative), meat dominant (carnivore) and omnivorous (omnivore) diets, respectively, aiming to explore suitable feces DNA extraction method for birds with specific dietary guilds (i.e., granivore, carnivore or omnivore). This study would provide a good reference for researchers who study microbiota in birds.
Methods
Ethics statement
All lab experiment procedures were under the guidance of the Ethics Committee of the Institute of Zoology, Chinese Academy of Sciences (IoZ, CAS) and were in full compliance with the Institutional Animal Care and Use Committee at the IoZ, CAS.
Study design and sample collection
The design of this study was shown in Fig. 1. Briefly, for the granivorous group, we chose domesticated budgerigars (Melopsittacus undulatus) (N = 16) and fed them with millets in cages for a week before feces collection. During collection, we placed a silver paper under the cages in advance. After defecation, the feces were immediately transferred from the silver paper to 1.5 mL sterile microcentrifuge tubes using a sterile forceps and stored in dry ice promptly. We collected fecal samples from a total of 16 individuals and pooled them before DNA extraction.

The experiment design in this study
For the omnivorous group, the domesticated crested mynas (Acridotheres cristatellus) (N = 1) was chosen and fed with millets and mealworms (the larval form of the Mealworm Beetle, Tenebrio molitor). Feces were collected following the approach mentioned above. For the group of carnivores, we chose a common kestrel (Falco tinnunculus) (N = 1) fed by beef and mice at IFAW Beijing Raptor Rescue Center. The feces were collected immediately and put into 1.5 mL sterile microcentrifuge tubes after defecation, and stored in dry ice. All samples were sent to the lab and stored in a − 80 °C freezer before use.
DNA extraction
In this study, we focused on three commonly used DNA extraction methods, Qiagen (QIAamp Fast DNA Stool Mini Kit, Qiagen, Germany), Bead (non-commercial protocol published in Li et al. 2014) and MoBio (MoBio’s PowerSoil DNA Isolation Kit, Qiagen, Germany). These methods are different in cell lysis strategies. Briefly, Qiagen method uses a chemical strategy (buffer ASL mainly composed of sodium dodecyl sulfate) to lyse the cell at high temperature (95 °C). Bead method from Li et al. (2014) adopts a chemical lysing (guanidine thiocyanate) with high temperature incubation (70 °C), followed by a mechanical strategy (ceramic beads of 0.1 mm diameter) to lyse cells. MoBio approach lyses the cells using the combination of both mechanical (garnet beads of 0.7 mm diameter) and chemical (sodium dodecyl sulfate) strategies but without high temperature incubation.
To minimize the potential effects of heterogeneity of feces from different individuals or different collection time, the samples were thoroughly mixed with phosphate buffered saline (PBS) and divided into 9 aliquots in granivore and omnivore groups, 15 aliquots in carnivore group with each weighing 200 mg. Subsequently, 3 (for granivore and omnivore) and 5 (for carnivore) aliquots were randomly allocated for each DNA extraction method (for details, please see Additional file 1: Supporting information).
Determination of DNA yield and integrity
DNA yield in each extract was estimated by using Qubit 2.0 Fluorometer (Thermo Fisher Scientific, United States) and Qubit dsDNA HS Assay kit (Thermo Fisher Scientific, United States). To evaluate the DNA integrity, we used the degree of DNA degradation as the indicator (i.e., the higher degree of DNA degradation, the less complete the DNA integrity). Three grades of DNA degradation were classified based on the electrophoresis gel band, according to a previous study (Fiedorová et al. 2019): (1) normal-intensity band (++); (2) faint band (+) and (3) very faint band (±), representing the most, moderately and least complete DNA integrity, respectively. DNA degradation was assessed by electrophoresis (100 V for 25 min) on a 1% agarose gel followed by staining with ethidium bromide and visualization under UV light.
Library construction and 16S rRNA gene sequencing
Each DNA sample was firstly normalized to 1 ng/μL. Subsequently, primers (U515F: 5′-GTGCCAGCMGCCGCGGTAA-3′/806R: 5′-GGACTACHVHHHTWTCTAAT-3′) were used to amplify the V4 hypervariable region of the 16S rRNA gene using the high-fidelity thermostable DNA polymerase and Phusion High-Fidelity PCR Master Mix with GC Buffer (New England Biolabs, United States). Thermal cycling reactions were set as: 1 min of initial denaturation at 98 °C, followed by 30 cycles of denaturation at 98 °C for 10 s, annealing at 50 °C for 30 s, elongation at 72 °C for 30 s, and with a final extension at 72 °C for 5 min. Then, adapters were added to the PCR products with the TruSeq DNA PCR-Free Sample Preparation Kit (Illumina, United States) to finish the library construction. The constructed library was subjected for sequencing on an Illumina NovaSeq 6000 sequencer.
Taxonomic assignment and bacterial diversity analyses
Sequencing data analysis was carried out using QIIME 2 (Bolyen et al. 2019). Briefly, adapters and 16S rRNA gene primer sequences were removed. Next, paired bacterial 16S rRNA reads were merged with a minimum merge length of 250 bp. Subsequently, the chimeras due to PCR artifacts and singletons (unmerged paired reads) were removed. Sequences were clustered into operational taxonomic units (OTU) at a threshold of 99% identity and taxonomy was assigned to each sequence using the Greengenes database version 13_8 (DeSantis et al. 2006). Alpha diversity metrics were calculated based on the observed OTU, Shannon and phylogenetic diversity.
Cell lysis capacity analysis
Stronger cell lysis capacity leads to a higher probability of breaking the hard cell wall of gram-positive bacteria and results in a higher relative abundance of gram-positive bacteria (Costea et al. 2017). So we estimated cell lysis capacity of each method according to the relative abundance of a dominant gram-positive bacteria (Firmicutes here).
Statistical analyses
Differences in DNA quantity, OTU numbers, relative abundance of bacterial taxa, cell lysis capacity and alpha diversity were compared pairwisely between methods using the Student’s t-test followed by a Benjamini–Hochberg correction. A false discovery rate (FDR) < 0.05 was considered to be statistically significant.
Result
Assessments on DNA yield and integrity
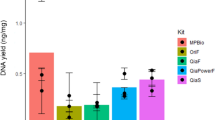
DNA yield comparisons between different methods in each dietary guild were shown in Fig. 2 and Additional file 2: Table S1. Bead method generated significantly higher DNA yield than Qiagen and MoBio regardless of dietary guilds (omnivore: FDR = 0.03 for Qiagen vs. Bead, FDR < 0.0001 for MoBio vs. Bead; granivore and carnivore: FDR < 0.0001 for both comparisons). The latter two methods showed no significant difference for both granivore and omnivore. MoBio generated a significantly higher DNA yield than Qiagen for carnivore (FDR < 0.0001).

DNA quality comparisons between different DNA extraction methods in granivore, omnivore and carnivore. The median, first and third quartiles were shown in the box (middle bar represents median, upper bound the third quartile, lower bound the first quartile). Statistical significance was determined using a Student’s t-test with a Benjamini–Hochberg correction. ns: no significant difference; *: FDR < 0.05; **: FDR < 0.01; ***: FDR < 0.001
For DNA integrity evaluation, both Bead and MoBio showed faint bands but Qiagen showed very faint bands for granivore (Additional file 3: Fig. S1). In omnivore, normal-intensity bands were observed in both Bead and Qiagen but faint bands in MoBio. In carnivore, normal-intensity bands were observed only for Bead method and very faint bands in others.
16S rDNA data generation
On average, 48,147, 48,622, 50,183 reads per sample were generated after filtering and chimera removal, for Bead, MoBio and Qiagen, respectively, in granivore. Comparable read numbers were also generated for omnivore (58,841, 48,622, 52,457 reads per sample) and carnivore (52,686, 60,572, 50,314 reads per sample). None of any two methods showed a significant difference in read number (FDRs > 0.05 for all comparisons) for any dietary guild.
Evaluation on microbial communities
In granivore, an average of 330 (± 49), 483 (± 202), 465 (± 150) OTUs were identified for Bead, MoBio and Qiagen methods, respectively. Our pairwise comparison on OTU numbers found no significant difference between the methods (all FDRs > 0.05). The comparison of taxa abundance, at the species level, found eight, seven and five OTUs, exhibiting significant differences between Qiagen and Bead, MoBio and Qiagen, as well as MoBio and Bead, respectively. However, at the phylum level the relative abundance of the ten dominant phyla (> 99% of total abundance; Table 1, Fig. 3a) were similar between the methods. The relative abundance of Firmicutes, the dominant gram-positive bacteria (77.89%), was similar between the methods (Fig. 3a).

Relative abundance comparisons between different methods at the phylum level in granivore (a), omnivore (b) and carnivore (c)
In omnivore, Bead, MoBio and Qiagen methods detected 368 ± 43, 359 ± 53, 484 ± 139 OTUs, respectively. OTU numbers were similar between the tested methods (all FDRs > 0.05). Similar to that in granivore, significant differences in relative abundance were detected for only a small number of species in the three comparison pairs (12 for Qiagen vs. Bead, 16 for MoBio vs. Qiagen, 6 for MoBio vs. Bead). Among the five dominant taxa at the phyla level (Firmicutes, Proteobacteria, Cyanobacteria, Acidobacteria, Gemmatimonadetes), significant differences were found in Proteobacteria between Qiagen and Bead (FDR < 0.01), Qiagen and MoBio (FDR < 0.001) with Qiagen method showing significantly higher relative abundance. A significant difference was also detected for Firmicutes but only in the comparison between Qiagen and MoBio (FDR < 0.001, Table 1) with the latter giving a higher relative abundance.
In carnivore, Qiagen identified the largest number of OTUs (1456 ± 632), followed by MoBio (964 ± 296) and Bead (221 ± 63) (Qiagen vs. MoBio, FDR > 0.05; Qiagen vs. Bead, FDR < 0.05; MoBio vs. Bead, FDR < 0.01). A total of 110, 94, 114 taxa at the species level exhibited significant differences in the relative abundance (Qiagen vs. Bead, FDR < 0.05; MoBio vs. Qiagen, FDR < 0.05; MoBio vs. Bead, FDR < 0.05). At the phyla level, a significant difference between Bead and MoBio was found for the most dominant taxa (Table 1). For Firmicutes, MoBio detected a significantly higher relative abundance compared with the other two methods (MoBio vs. Bead, FDR < 0.001; MoBio vs. Qiagen, FDR = 0.001).
Evaluation on microbial diversity
The results of microbial diversity comparison between these methods in each dietary guild were shown in Fig. 4 (species list: Additional file 4). For granivore, the alpha diversity was similar among different methods (FDR > 0.05, Fig. 4a). For omnivore, Shannon diversity was the highest in Qiagen (Qiagen vs. MoBio, FDR < 0.05; Qiagen vs. Bead, FDR < 0.05, Fig. 4b), followed by Bead and then MoBio (FDR = 0.001). The other two indices were similar among these methods. For carnivore, a significantly higher alpha diversity was found in Qiagen or MoBio than that of Bead (Qiagen vs. Bead, FDR < 0.05, MoBio vs. Bead, FDR < 0.001). Qiagen has a higher alpha diversity than MoBio though not significant, possibly due to the small number of simple size (mean: 1456 vs. 964 in observed OTU, 4.77 vs. 3.52 in Shannon diversity, 63 vs. 52 in phylogenetic diversity, Fig. 4c).

Alpha diversity comparisons between different methods in granivore (a), omnivore (b) and carnivore (c). Observed OTU, Shannon diversity and phylogenetic diversity were used to estimate alpha diversity. The median, first and third quartiles were shown in the box (middle bar represents median, upper bound the third quartile, lower bound the first quartile). The statistical significance was determined using a Student’s t-test with a Benjamini–Hochberg correction. ns: no significant difference; *: FDR < 0.05; **: FDR < 0.01; ***: FDR < 0.001
Discussion
In this study, we evaluated the performances of DNA yield and integrity, microbial community composition, cell lysis capacity and alpha diversity of three different DNA extraction methods used in bird fecal microbiota research. We mainly focused on their performances on three avian dietary guilds (i.e., granivore, carnivore and omnivore).
For all dietary guilds examined, Bead method had the best performance on both DNA yield and integrity, consistent with a previous study (Eriksson et al. 2017). This is likely because: (1) Bead method adopts a mechanical lysis approach, which is more powerful in cell lysis than the chemical one applied in Qiagen; (2) the diameter of beads used is much smaller than those in MoBio (0.1 vs. 0.7 mm), leading to lysis of more cells and subsequently more DNA release (Fujimoto et al. 2004).
The measurements of other indices (microbial relative abundance, cell lysis capacity and alpha diversity) varied for each method in different dietary guilds. In granivore, we found little difference regardless of indices above among all methods, indicating that these indices were not conditioned on the DNA yield and integrity, which coincided with the studies in humans (Knudsen et al. 2016; Huseyin et al. 2017; Rintala et al. 2017; Lim et al. 2018; Fiedorová et al. 2019). Our findings also indicated that the results were comparable when employing these methods in the microbiota research of granivorous avian species.
In omnivore, the highest relative abundance of Firmicutes observed in MoBio indicated that this method was the most powerful on cell lysis. For alpha diversity, Qiagen showed the best performance (highest Shannon diversity). This result is different from a previous human study (Costea et al. 2017), which showed a higher Shannon diversity in the method using mechanical strategy than that using chemical ones (e.g., Qiagen). To figure out the possible reasons for this discrepancy, we assessed the main factors leading to the observed higher Shannon diversity in Qiagen in our study using species richness and evenness, two key components for Shannon diversity. Although the species richness was comparable between the methods, the evenness was the highest in Qiagen (Qiagen vs. MoBio, FDR < 0.05; Qiagen vs. Bead, FDR < 0.05). Such high evenness was caused by the relative high abundance of some gram-negative bacteria groups (e.g., Nitrospirae and Chlamydiae), rather than the high abundance of gram-positive bacteria reported in Costea et al. (2017). In summary, different methods were advantageous in different aspects for omnivorous fecal DNA extraction. For instance, Qiagen had the best performance on alpha diversity but not DNA yield and integrity and cell lysis capacity.
In carnivore, Qiagen had better performance on alpha diversity in contrast with Bead and MoBio. The lower alpha diversity observed in Bead and MoBio may be because: (1) the fecal DNA in carnivore was not exclusively microbial, but also originated from the food (i.e., mice and beef in our case); (2) Due to the presence of cell wall, bacteria were more difficult to be lysed than food cells (as well as mammalian cells in our study) when use mechanical strategy, hence decreasing the efficiency of bacteria lysis and leading to a relative lower alpha diversity.
We noted that one limitation in our study was the small sample size (i.e., N = 3 and 5) per group, which might not have enough statistical power to detect the significance. For example, using a small sample size may underestimate the significance level. Hence, the insignificant results need to be further confirmed by enlarging the sample size in the future. We also noted that a mock microbial community, a defined mixture of microbial cells created in vitro to simulate the composition of a microbial sample (Highlander 2014), is required to quantify the bias for microbial composition associated with different extraction methods.
Conclusions
Our results suggested that DNA yield and integrity have no obvious impact on the evaluation of microbial composition and alpha diversity. We think any of the three methods could be used for microbiota studies in granivorous birds. For omnivores and carnivores, we recommend Qiagen method if the research target is microbial diversity, but MoBio if the research target is gram-positive bacteria.
Availability of data and materials
The datasets used in the present study are available from the corresponding author on reasonable request.
Change history
27 May 2021
A Correction to this paper has been published: https://doi.org/10.1186/s40657-021-00262-9
References
Berlow M, Kohl KD, Derryberry EP. Evaluation of non-lethal gut microbiome sampling methods in a passerine bird. Ibis. 2020;162:911–23.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;8:852–7.
Burin G, Kissling WD, Guimarães PR, Şekercioğlu ÇH, Quental TB. Omnivory in birds is a macroevolutionary sink. Nat Commun. 2016;7:1–10.
Costea PI, Zeller G, Sunagawa S, Pelletier E, Alberti A, Levenez F, et al. Towards standards for human fecal sample processing in metagenomic studies. Nat Biotechnol. 2017;35:1069–76.
Darwin C. The origin of species. 6th ed. London: John Murray Press; 1859.
DeJong TM. A comparison of three diversity indices based on their components of richness and evenness. Oikos. 1975;26:222–7.
DeSantis TZ, Hugenholtz P, Keller K, Brodie EL, Larsen N, Piceno YM, et al. NAST: a multiple sequence alignment server for comparative analysis of 16SrRNA genes. Nucleic Acids Res. 2006;34:394–9.
Di Rienzi SC, Britton RA. Adaptation of the gut microbiota to modern dietary sugars and sweeteners. Adv Nutr. 2020;11:616–29.
Eriksson P, Mourkas E, González-Acuna D, Olsen B, Ellström P. Evaluation and optimization of microbial DNA extraction from fecal samples of wild Antarctic bird species. Infect Ecol Epidemiol. 2017;7:1386536.
Fiedorová K, Radvanský M, Němcová E, Grombiříková H, Bosák J, Černochová M, et al. The impact of DNA extraction methods on stool bacterial and fungal microbiota community recovery. Front Microbiol. 2019;10:821.
Fuertes A, Pérez-Burillo S, Apaolaza I, Vallès Y, Francino MP, Rufián-Henares JÁ, et al. Adaptation of the human gut microbiota metabolic network during the first year after birth. Front Microbiol. 2019;10:848.
Fujimoto S, Nakagami Y, Kojima F. Optimal bacterial DNA isolation method using bead-beating technique. Memoirs Kyushu Univ Dep Of Health Scis Of Medical Sch. 2004;3:33–8.
Gill F, Donsker D. IOC World Bird List (v 6.4). 2016. http://www.worldbirdnames.org. Accessed 31 Dec 2016.
Highlander S. Mock community analysis. In: Nelson K, editor. Encyclopedia of metagenomics. New York: Springer Press; 2014. p. 1–7.
Huseyin CE, Rubio RC, O’Sullivan O, Cotter PD, Scanlan PD. The fungal frontier: a comparative analysis of methods used in the study of the human gut mycobiome. Front Microbiol. 2017;8:1432.
Knudsen BE, Bergmark L, Munk P, Lukjancenko O, Prieme A, Aarestrup FM, et al. Impact of sample type and DNA isolation procedure on genomics inference of microbiome composition. mSystems. 2016;5:e00095-16.
Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol. 2014;32:834–41.
Lim MY, Song EJ, Kim SH, Lee J, Nam YD. Comparison of DNA extraction methods for human gut microbial community profiling. Syst Appl Microbiol. 2018;41:151–7.
Rintala A, Pietilä S, Munukka E, Eerola E, Pursiheimo JP, Laiho A, et al. Gut microbiota analysis results are highly dependent on the 16SrRNA gene target region, whereas the impact of DNA extraction is minor. JBT. 2017;28:19–30.
Yang N, Yan W, Sun C, Zheng J, Wen C, Ji C, et al. Efficacy of fecal sampling as a gut proxy in the study of chicken gut microbiota. Front Microbiol. 2019;10:2126.
Acknowledgements
We thank IFAW Beijing Raptor Rescue Center for their assistance in sample collection. We also thank Mr. Xuemin Dong for his help with data analysis.
Funding
This study was supported by National Natural Science Foundation of China (No. 31930013, 31872240), the National Key Program of Research and Development, Ministry of Science and Technology (2016YFC0503200), Youth Innovation Promotion Association of Chinese Academy of Sciences (2020086) to SP.
Author information
Authors and Affiliations
Contributions
XZ and JX designed the project. XH and ZL collected samples and XH performed experiments. XH and SP analyzed the data. XH wrote the manuscript. XZ, JX and SP revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Non-invasive sample collection was used in this study. Permission was obtained from the Institutional Animal Care and Use Committee at the IoZ, CAS (IOZ20180062).
Consent for publication
Not applicable.
Competing interests
All the authors declare that they have no competing interests.
Additional information
The original online version of this article was revised: The wrong Figure 1 had been published. Now, this has been corrected in the original publication.
Supplementary Information
Additional file 1.
Supporting Information.
Additional file 2: Table S1.
DNA yield and integrity obtained from different methods in the examined dietary guilds.
Additional file 3: Fig. S1.
DNA integrity assessments using three extraction methods for granivore, omnivore and carnivore.
Additional file 4.
The microbial species list and relative abundance of each dietary guilds.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hou, X., Pan, S., Lin, Z. et al. Performance comparison of different microbial DNA extraction methods on bird feces. Avian Res 12, 19 (2021). https://doi.org/10.1186/s40657-021-00254-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40657-021-00254-9