
Abstract
The Earth's fluid budget is dominated by species in the system C–O–H–N–S together with halogens such as F and Cl. H2O is by far the most abundant. Such fluids are one of the two main mass transport agents (fluid and magma) in the Earth. Among those, in particular aqueous fluids are efficient solvents of geochemically important components at high temperature and pressure. The solution capacity of aqueous fluids can be enhanced further by dissolved halogens and sulfur. CO2 or nitrogen species has the opposite effect.
Fluid-mediated transport in the Earth is by fluids passing through cracks at shallow depth and via percolation channels along grain boundaries at greater depth. Percolation velocity is linked to permeability, which, in turn is governed by rock porosity. Porosity is controlled by wetting angles, θ, at the interface between fluid and mineral surfaces. When θ < 60°, fluid will wet all grain boundaries of an isotropic crystalline material, whereas when greater than 60°, grain boundary wetting does not occur as readily, and fluid-mediated transport efficiency can be greatly reduced. The size of the wetting angle is negatively correlated with the solubility of silicate components in the fluids, which means that fluid composition, temperature, and pressure affect the wetting angles and, therefore, fluid-mediated mass transport efficiency in the interior of the Earth.
Geophysical and geochemical anomalies in the Earth's interior have been linked to the presence of fluids. Fluid infiltration in crustal and mantle rocks will enhance electrical conductivity and seismic wave attenuation. For example, 5–10% H2O-rich fluids in the mantle wedge above subducting plates have been suggested from enhanced electrical conductivity. Similar fluid fractions have been suggested to be consistent with seismic velocities in these regions. The geochemistry of the crust and the mantle can be affected by fluid-mediated transport of major, minor, and trace elements. When such altered materials serve as source rocks of partial melts, those geochemical alterations also lead to changes in partial melt compositions. As an example, the presence of such aqueous fluid in the mantle wedge above subducting and dehydrating subducting slabs is consistent with partial melting of an H2O-bearing mantle wedge above subducted oceanic crust.

Similar content being viewed by others


1 Introduction
Fluids are one of the two main mass transport agents in the Earth. Magma is the other transport agent. Fluids can be comprised of oxidized species such as H2O, CO2, SO3, and N2 and reduced species such as H2, CH4, H2S, and NH3 depending on oxygen fugacity, fO2, conditions. H2O is by far the most abundant of these fluid species (Jambon 1994).
The two main fO2-dependent carbon species are CO2 or CH4 (Eggler and Baker 1982; Taylor and Green 1989). Redox-dependent sulfur and nitrogen species can be found under specific circumstances such as during subduction zone melting, for example (Busigny et al. 2011; Wallace and Edmonds 2011).
The impact of fluids as mass transport agents on properties and processes of rock-forming materials depends on temperature, pressure, composition of the fluid, bulk rock composition, and redox conditions. The composition of fluids, in turn, reflects the conditions of fluid of formation, including the composition of their source rock. Conditions of fluid formation in metamorphic processes depend primarily on stability and phase boundaries of volatile-bearing crystalline materials (e.g., Winkler 1965; Connolly 2005; Evans and Tomkins 2020). The conditions also can include those that govern solubility of volatiles in magma, and, therefore, the circumstances under which one or more fluid species may exsolve during cooling and crystallization of fluid-rich magma (Eggler and Kadik 1979; Aubaud et al. 2005; Papale et al. 2006; Moretti et al. 2018; Audetat and Edmonds 2020). Oxygen fugacity also can be important for the solubility in magma of elements that can exist in multiple oxidation states (Peiffert et al. 1996; Klein-BenDavid et al. 2011).
The properties and composition of the rock matrix through which fluid migration takes place also are important for penetration of fluids into rocks (see Holness 1997; for review). The stress field can also influence fluid migration (Riley and Kohlstedt 1991; Hustoft and Kohlstedt 2006). Fluid density and viscosity are additional variables that can influence fluid migration although in general the density and viscosity contrasts between fluids, regardless of their composition, and rock matrix are so great that these often can be ignored. An exception to this suggestion is that where the temperature/pressure/compositions are such that fluids are supercritical and cannot be distinguished from fluid-rich magma. Under such conditions, density and viscosity of fluid can resemble those of volatile-rich magmatic liquids. This situation is not uncommon for H2O-rich systems in the upper mantle, for example (Shen and Keppler 1995; Bureau and Keppler 1999; Kessel et al. 2005; Mibe et al. 2007).
Carbon in its oxidized form, CO2, is the second-most abundant fluid species in the Earth (Jambon 1994). In the modern Earth, which likely becomes increasingly reducing with depth (Frost and McCammon 2008), methane (CH4) may be the dominant C-species in the lower mantle. Methane may also have been the dominant C-species in the Early Earth (O'Neill 1991; O'Neill et al. 1998). Reduced carbon as CH4 also has been reported from portions of descending slabs in modern subduction zones, for example (Tao et al. 2018). Absent hydrogen, carbides are possible. Carbide minerals are found as inclusions of deep-seated diamonds, for example (Kaminsky and Wirth 2017).
H2O is the most important and abundant fluid component in the Earth (Jambon 1994). H2O also is the most effective solvent of major, minor, and trace elements at high temperature and pressure (Manning 1994; Zhang and Frantz 2000; Newton and Manning 2007, 2008). The transport properties of H2O-rich fluids and their role in mass transport processes are, therefore, a central theme of this review. The impact of other components such as CO2, halogen, and sulfur species, on mass transport and rock-forming processes, will be incorporated as appropriate.
2 Review
In order to characterize the behavior of fluids in the Earth's interior, we will first discuss fluid sources. This will be followed by fluid properties including solubility of geochemically important elements and partitioning of elements between fluid and magma. The presentation will conclude with a discussion of how fluids migrate through a crystalline matrix and consequences of fluid distribution for geochemical and geophysical properties of the Earth's interior.
2.1 Sources of fluid
Except for the Earth's primordial volatiles, fluids are recycled usually with sediments at the beginning of a cycle. These sediments typically were deposited on the ocean floor and are comprised of both inorganic and organic components. Early stages of fluid cycles also can include metamorphic rocks formed in the hydrothermal environment existing during cooling of mid-ocean ridge volcanics that interact with H2O and its dissolved salts (see, for example, Evans and Tomkins 2020; for recent review).
During metamorphism of sediments, fluid components become part of hydrous, carbonate, and sulfide minerals and, sometimes, halogens such as F and Cl. These fluids, in turn, for the most part are gradually released with increasing metamorphic grade such as seen with increasing depth in subduction zones, for example. Under some circumstances, fluids might be transported through the transition zone and into the lower mantle. The extent to which this may take place, depends on the bulk composition, redox conditions, and thermal environment of the descending slab (van Keken et al. 2011; Bebout et al. 2013; Ohtani 2019).
Fluids derived from dehydration reactions during metamorphism also can trigger partial melting followed by fluid incorporation in the magmatic liquids thus formed (Wyllie 1982; Ulmer 2001). During crystallization and decompression of such fluid-bearing magma, some or all of the fluid will be exsolved to form a separate fluid phase. The composition of those fluids will depend on the fluids in the source region of melting and temperature and pressure conditions during cooling and crystallization (Audetat and Edmonds 2020).
Under certain circumstances fluids in the crust and upper mantle can migrate toward the surface along grain boundaries (Mysen et al. 1978; Watson et al. 1993) or, under some circumstances, through cracks, such as may be found in shear zones below some island arcs (White et al. 2019), without causing partial melting. Such movement is likely for fluids with low solubility in magmatic liquids and has limited impact solidus temperatures of rocks.
2.1.1 Fluids and devolatilization during metamorphism
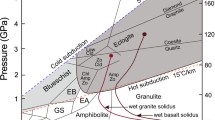
Fluids released in metamorphic processes by exceeding stability fields of minerals that contain volatiles, are dominated by H2O because most major volatile-bearing metamorphic minerals are H2O-bearing (clay minerals, serpentine, mica, and amphiboles). Their H2O contents typically are greater the lower their upper temperature stability (Evans and Tomkins 2020; see also Fig. 1A).

Modified from Evans and Tomkins (2020)
H2 and CO2 generation during metamorphic processes. A. Cumulative H2O loss as a function of increasing metamorphic grade. B. CO2 loss from decarbonation reactions at 0.5 GPa as a function of CO2–H2O fluid composition.
Carbon dioxide is the second-most important fluid component in metamorphic rocks such as in the subducting plates although under some circumstances, reduced carbon in the form of CH4 as well as more complex hydrocarbons may form (Chu and Ague 2013; Yardley and Bodnar 2014; see also Manning et al. 2013; for review). The CO2 is primarily found in carbonate minerals such as calcite, aragonite, dolomite, and magnesite, but can also occur in smaller concentrations as parts of solid solutions in apatite and scapolite (Moecher and Essene 1990; Harlov 2015). In metamorphic systems with mixed CO2–H2O fluid, the stability of the CO2 and H2O-bearing minerals depends not only on temperature, pressure, and bulk chemical composition, but also varies with the proportion of CO2 and H2O (Connolly 2005; Evans and Tomkins 2020; see also Fig. 1B). For example, during subduction, fluid is predominantly H2O + CO2 with its CO2/H2O ratio increasing with depth of fluid release (Poli and Schmidt 2002; Connolly 2005). This CO2/H2O increase is because carbonate minerals (calcite, aragonite etc.) generally are stable to higher pressure and temperatures (greater depth) than many of the OH-bearing minerals in metamorphosed subducting slabs. These stability features lead to increased proportion of CO2 (Connolly 2005) relative to that of H2O from hydrous minerals (Poli and Schmidt 2002), and, therefore, the increased CO2/H2O ratio of released fluid with increasing depth in subduction zones.
This released fluid provides a means of mass transfer to the overlying mantle wedge that ultimately undergoes partial melting. This changing CO2/H2O ratio depending on depth (pressure) affects the solubility of geochemically important elements, the migration behavior of the fluid through crystalline subduction zone rocks, and the bulk composition of partial melts from the metasomatically altered mantle wedge (see, for example, Mysen and Boettcher 1975; Watson 1990; Manning 2004; Manning and Frezzotti 2020).
Chlorides can be important fluid components in particular during release of volatiles in subduction zones (Scambelluri and Philippot 2001; Kawamoto et al. 2014). The origin of such salts typically is ocean water and/or hydrothermal fluids trapped in pore space during sedimentation, diagenesis, and hydrothermal action near active mid-ocean ridges. Halite (NaCl) sometimes could have formed and been transported into the mantle with other sediments during subduction (e.g., Yardley and Graham 2002). Chlorine also can form solid solutions in minerals such as biotite, amphibole, and scapolite in addition to its entrapment in fluid inclusions (Goldschmidt and Newton 1977; Pillippot et al. 1998; Chevychelov et al. 2008; Henry and Daigle 2018).
Sulfur is a minor component of metamorphic fluids in most settings. It can exist, however, as pyrite and as a minor component in scapolite solid solutions together with components such as CO32− and Cl− (Orville 1975; Goldsmith and Newton 1977; Morrissey and Tomkins 2020). Nitrogen when in reduced form (e.g., NH+4) can be exchanged in K+ in feldspar, mica, and dense magnesian phases. Under such conditions, nitrogen can be transported deep into the mantle near subducting plates (Hallam and Eugster 1976; Plessen et al. 2010) because such phases are stable to pressures sometimes in excess of 20 GPa (Konzett and Fei 2000; Trønnes 2002).
2.1.2 Fluids exsolved from magma
The composition of fluids released from cooling magma depends on magma composition, and on its partial melting source. The composition of fluids exsolved from cooling magma also varies with the temperature and pressure at which the fluids are released. Such variables result in varying partition coefficients between fluid and magmatic liquid. Therefore, partition coefficients of fluid species between magma and a coexisting fluid phase govern the composition of the fluid.
2.1.2.1 Partitioning of H2O between fluid and melt
The temperature–pressure coordinates of the H2O (fluid)/hydrous melt equilibria as well as the coordinates of the critical point, above which fluids and melts are completely miscible, are significantly dependent on silicate composition (e.g., Shen and Keppler 1997; Bureau and Keppler 1999; Kessel et al. 2005). The fluid/melt partition coefficient of H2O in silicate–H2O systems at temperatures above their liquidii also varies significantly with temperature and pressure at conditions less than those of the critical point of rock–H2O systems. The exact temperature–pressure trends of fluid/melt partition coefficient curve toward the critical endpoint in the example in Fig. 2 in differ ways because the composition of the two systems shown in that figure differs. The temperature and pressure ranges of the two experimental data sets also are significantly different. Such differences are even more obvious when comparing compositionally different hydrous magmatic systems because temperatures and pressures of the critical endpoints increase the more mafic the silicate composition. For example, the critical point of the system MgO–SiO2–H2O may be at pressures in excess of 10 GPa although this pressure is the subject of considerable discussion with suggested pressures for the critical point in peridotite–H2O varying between 3.4 and about 11–13.5 GPa (Stalder et al. 2001; Mibe et al. 2002, 2007; Melekhova et al. 2007). In basalt–H2O systems, the critical point has been suggested to be near 3.4–5 GPa (Kessel et al. 2005; Mibe et al. 2011), and in granite–H2O systems near 1 GPa (Shen and Keppler 1997; Sowerby and Keppler 1998).

In the case of multicomponent fluids, the fluid/melt partition coefficient of H2O becomes a function of fluid composition (Botcharnikov et al. 2015; Webster et al. 2009). For example, Webster et al. (2009) reported that the H2O content of fluid increases with increased salinity (NaCl and KCl). In melt-H2O–CO2 systems, the partition coefficient is particularly sensitive to pressure and melt composition because the much greater solubility of H2O in silicate melts compared with the solubility of CO2 (Eggler and Kadik 1979; Iacono-Marziano et al. 2012).
2.1.2.2 Partitioning of carbon-bearing species between fluid and melt
Oxidized carbon in magmatic systems, absent other components such as H2O, exists as CO2 in a C–O fluid phase and as CO2 and CO32− complexes in silicate melts. In melt-C–O–H systems, additional speciation is possible. Here, oxidized carbon can occur as CO2, CO32−, and HCO3− in both melts and fluids (Mysen 2015a, 2018). Under reducing conditions, CH4 and CH3− groups can be stabilized in both silicate melts and silicate-bearing fluids (Mysen et al. 2009, 2011). The proportion of these species and their partitioning behavior between fluid and melt vary with fluid and silicate composition, temperature and, likely, with pressure.
The exchange equilibrium of carbon-bearing species between melt and fluid under oxidizing conditions is (Fig. 3A):
whereas reducing under conditions the exchange equilibrium is (Fig. 3B):

Under oxidizing conditions and with increasing temperature, the CO3/HCO3 abundance ratio in melts increases faster than in coexisting fluid. The enthalpy change, ∆H, for reaction (1) is − 44 ± 9 kJ/mol with the assumption of ideal mixing (Mysen 2015a). In comparison, under sufficiently reducing conditions, the temperature dependence of the CH4/CH3 abundance ratio in fluid is greater than in coexisting melt with a ∆H for reaction (2) of 34 ± 3 kJ/mol (Mysen 2015a). It must be kept in mind, however, that because the experiments used to extract the data in Fig. 3 were carried out in a fixed volume hydrothermal diamond anvil cell (Bassett et al. 1994), the pressure also increased when the temperature increased. It was assumed, therefore, that the ∆V of the exchange equilibria (1) and (2) is negligible and that pressure did not, therefore, impact on the calculated ∆H-values.
2.1.2.3 Partitioning of halogens between fluid and melt
Chlorine has attracted the most attention among experimental studies of the partitioning of halogens between fluid and magma. This attention likely at least in part is because Cl-complexes are often considered responsible for enrichments of economically important metals such as, for example, Mo, Cu, and Au in fluids and melts (Frank et al. 2011; Zajacz et al. 2013).
At the pressures of the subcritical region in melt-H2O–Cl systems such as, for example, the phonolite magma + H2O + Cl (below about 180 MPa; see also Fig. 4A), the Cl concentration in melt changes little as a function of the Cl in the coexisting fluid phase. However, at higher pressures the concentrations in coexisting melts and fluids are correlated albeit in a nonlinear way (Fig. 4B). It is also notable that the Cl concentration in the melt decreases with increasing pressure because of the partial molar volume difference of NaCl in aqueous fluid and H2O-rich melt is negative (Shinohara et al. 1989; Signorelli and Carroll 2002). Under anhydrous conditions, on the other hand, the Cl solubility in silicate melts is a positive function of pressure (Webster et al. 1999; Dalou and Mysen 2015).

Modified from Signorelli and Carroll (2000)
Chlorine distribution between saline fluid and phonolitic magma, A. Fluid–melt equilibria with immiscibility gap as a function of NaCl concentration in H2O–NaCl fluid and pressure. B. Evolution of Cl concentration in coexisting melt and saline fluid as a function of chlorine concentration in fluid and melt at pressures indicated.
The chlorine partition coefficient between brine and hydrous magma, DClfluid/melt, decreases with increasing pressure and decreasing temperature (Kilinc and Burnham 1972; Shinohara et al. 1989; Signorelli and Carroll 2000; Webster 1992; Hsu et al. 2019). Moreover, the DClfluid/melt is a strong function of Cl concentration and also changes with SiO2 content of the magma (Webster et al. 2009; Beerman et al. 2015; see also Fig. 5A–C). The more silica-rich, and, therefore, more felsic, the greater the fluid/melt partition coefficient (Botcharnikov et al. 2015). This relationship would likely be even more pronounced if the NBO/T parameter of the melt was used in replacement of the SiO2 (Signorelli and Carroll 2002; Metrich and Rutherford 1992). The SiO2 of magmatic liquids typically is negatively correlated with the melt NBO/T so that the lower the SiO2 concentration, generally the greater is the NBO/T of the melt (Mysen and Richet 2019; Chapter 18). Other compositional variables affecting the fluid/melt partition coefficient of Cl include Al2O3/(CaO + Na2O + K2O) (Iveson et al. 2017; Signorelli and Carroll 2002).

Fluid/melt partition coefficient of Cl A. as a function of pressure for two different magma compositions as indicated. B. As a function of Cl concentration in andesitic melt at two different temperatures as indicated, and C. as a function of SiO2 concentration in melt.
The chlorine concentration also affects DClfluid/melt (Beermann et al. 2015; Hsu et al. 2019). Increasing Cl concentration such as from NaCl dissolved in aqueous fluid, for example, results in increasing fluid/melt partition coefficient (Fig. 6A). Increasing CO2 concentration in an H2O–CO2–NaCl environment, on the other hand, results in decreasing DClfluid/melt (Hsu et al. 2019; See also Fig. 6B).

Fluid/melt partition coefficient of chlorine, \({D}_{\mathrm{Cl}}^{\mathrm{fluid}/\mathrm{melt}}\) A. as a function of mol fraction of NaCl in fluid at two different temperatures as indicated. B. \({D}_{\mathrm{Cl}}^{\mathrm{fluid}/\mathrm{melt}}\) as a function of mol fraction of CO2 in H2O–CO2 fluid with NaCl added. The melt composition is that of a composition along the join SiO2–NaAlO2 with a slight excess of Al2O3 (Composition (wt%) on an anhydrous basis, SiO2: 80., Al2O3: 12.6, Na2O: 7.3) Modified after Hsu et al. (2019)
Relatively few experiments have been carried out to determine fluid/melt partitioning of F (Xiong et al. 1998; Kravchuk et al. 2004; Chevychelov et al. 2008; Webster et al. 2009). For those for which experimentally determined partition coefficients, DFfluid/melt, exist, these partition coefficients typically are less than 1 (Fig. 7). The partitioning behavior of fluorine differs, therefore, from all other halogens for which the fluid/melt partition coefficients are greater than 1 (Dolejs and Zajacz 2018). This difference between F and Cl partition coefficients reflects the different solubility behavior of Cl and F in magmatic liquids (Dalou et al. 2015; Dalou and Mysen 2015). For example, whereas Cl solubility decreases as silicate melts become more aluminous, the opposite trend was observed for F (Dalou et al. 2015). The fluorine solubility in silicate melts also increases with increasing H2O content, a behavior that contrasts with that of Cl, the solubility of which decreases with increasing H2O content of silicate melts (Dalou and Mysen 2015).

Fluorine concentration in coexisting aqueous fluid and H2O-rich melt for various granitic melt compositions. Modified after Dolejs and Zajacs (2018)
Fluid/melt partition coefficients of F, Cl, Br, and I follow a simple relationship of the form (Bureau et al. 2000);
where r is the radius of the halogen (Fig. 8). This relationship likely is because the solubility of halogens in silicate melts is less the greater their ionic radius.

Fluid/melt partitioning at 200 MPa and 900° as a function of the ionic radius of the halogen. (The melt composition is (wt%): SiO2: 67.09, Al2O3: 18.08, Na2O: 11.06.) (Modified after Bureau et al. 2000)
2.1.2.4 Partitioning of sulfur between fluid and melt
Sulfur is the third-most abundant volatile component in the Earth (Jambon 1994). It is of particular interest because S-rich fluids can be important transport media of metals to form economically viable ore deposits because transition metals such as, for example, Zn, Cu, Mo, Pb, and Ag can form sulfide complexes when dissolved in fluids and magma (Pokrovski et al. 2008; Botcharnikov et al. 2011). Oxidized sulfur, whether in fluid or magma, does not enhance the solubility of such elements significantly. Oxidized sulfur also can govern degassing processes of magma during their ascent and cooling (Oppenheimer 2003).
Oxygen fugacity, fO2, is an important variable governing the behavior of sulfur in fluids (and magmatic liquids) because the oxygen fugacity can govern the redox state of sulfur (e.g., Nagashima and Katsura 1973; O'Neill and Mavrogenes 2002; Jugo et al. 2010). It has proposed, for example, that fluid/melt partition coefficients can be described with an expression of the type (Gennaro et al. 2020):
In this equation, T is temperature (°C), P is pressure (MPa) and ∆NNO is the oxygen fugacity difference from that of the nickel–nickel oxide (NNO) oxygen buffer (log units).
Under oxidizing conditions, where sulfur exists predominantly as S6+, its solution behavior in fluids appears to follow Henry's law (Fig. 9). However, the slope of the curves in Fig. 9, and, therefore, the Henry's Law constant, depends on the bulk composition of the system. It also depends on H2O content (Webster and Botcharnikov 2011), SiO2 content (Scaillet et al. 1998), the proportion of the sum of alkali metals and alkaline earths versus Si + Al + Fe3+ (Webster and Botcharnikov 2011) and, therefore, the NBO/T of the melt (Zajacz 2015). Increased peralkalinity also leads to increased DSfluid/melt. Finally, the fluid/melt partition coefficient of reduced sulfur decreases rapidly with increasing FeO concentration, which is not surprising given the particularly strong affinity of S2− for Fe2+ (Richardson and Fincham 1954; O'Neill and Mavrogenes 2002).

Sulfur content coexisting aqueous fluid and hydrous melt as a function of their sulfur content at 500 MPa and 1240 °C with the oxygen fugacity controlled 1.4 log units above that of the NNO buffer for systems as indicated on diagrams. (Modified after Zajacs 2015)
2.2 Solubility of major elements in fluids
Fluids in the Earth are important transport agents because of significant solubility in fluids of geochemically and geophysically important components. This solubility depends on the fluid composition, the element of interest, temperature, and pressure. For aliovalent elements, the oxygen fugacity also can be important.
2.2.1 SiO2 in aqueous fluid
The concentration of SiO2 in most terrestrial rocks exceeds 40 wt% (Allegre et al. 2001), which typically is more than twice the terrestrial abundance of any other major oxide component. Characterization of the interaction between H2O fluid and SiO2 to high temperatures and pressures is, therefore, fundamental to our understanding of the role of H2O as a transport agent of rock-forming components in the Earth.
Solubility determination of SiO2 in H2O fluid in the SiO2–H2O system at pressures and temperatures below the second critical end point (near 1 GPa and 1080 °C; see Kennedy et al. 1962) have, therefore, been the subject of extensive experimental work (Kennedy 1950; Morey and Hesselgesser 1951; Weill and Fyfe 1964; Anderson and Burnham 1965; Fournier and Potter 1982; Manning 1994; Newton and Manning 2000). The SiO2 solubility increases with both temperature and pressure (Fig. 10). It varies particularly rapidly with temperature near the critical temperature of H2O resulting in an inflection of the solubility curve (Fig. 10A). As the pressure is increased, the extent of the inflection of those solubility curves diminishes so that above about 100 MPa, the inflection point is barely discernible. Moreover, the pressure at which the inflection of the solubility curve occurs, shifts to high temperature higher the total pressure.

Solubility of SiO2 in aqueous fluid. A. As a function of temperature across the critical endpoint at pressures as indicated on individual curves. B. As a function of temperature at pressures from 100 to 700 MPa as indicated on individual curves. C. As a function of pressure at temperatures indicated. Data from Weill and Fyfe (1964) (A), Anderson and Burnham (1965) (B), and Manning (1994) (C)
Aqueous fluids are important in rock-forming processes to much greater depth than the approximately 5 km (equivalent to about 150 MPa) of the early experimental data from Kennedy (1950). Weill and Fyfe (1964) extended the pressure and temperature ranges to 400 MPa in the 400°-550 °C respectively (Fig. 10B). More recent experimental SiO2 solubility data have been dominated by the experiments of Craig Manning and coworkers. They have reported SiO2 solubility in aqueous fluids to pressures near 2 GPa (see, for example, Manning 1994; Newton and Manning 2000, 2008; Hunt and Manning 2012). They found that the rate of SiO2 solubility increases with pressure is greater the higher the temperature (Fig. 10C), an observation that also is similar to earlier experimental studies of SiO2 solubility in the SiO2–H2O system (Weill and Fyfe 1964; Anderson and Burnham 1965). Moreover, the isothermal SiO2 solubility becomes linear when expressed as a function of the log ρH2O (density of pure H2O). From regression analysis of their own data together with other published experimental data over a range of temperatures, Manning (1994) arrived at an empirical expression that may be used to calculate the solubility in SiO2 in H2O to perhaps 2 GPa total pressure;
where mSiO2 is molality of SiO2 in the aqueous solution (kg/mol), \({\rho }_{\mathrm{H}2\mathrm{O}}\) is density (g/cm3) of pure H2O and T is temperature (kelvin). In Eq. (5), pressure effects are built into the relationship between solubility and density of H2O. Of course, \({\rho }_{\mathrm{H}2\mathrm{O}}\) also depends on temperature even though in Eq. (5), temperature also is one of the explicit variables in the regression of SiO2 solubility.
Most of the proposed solution mechanisms for SiO2 in H2O fluid refer to OH-bearing silicate monomers and dimers and perhaps even trimers as the structural entities of dissolved silica (Wendlandt and Glemser 1964; Newton and Manning 2003; Zotov and Keppler 2002; Mysen 2010; Mysen et al. 2013). For example, Manning and coworkers (Newton and Manning 2003, 2008; Hunt and Manning 2012) modeled the SiO2 solubility mechanisms in aqueous fluids in terms of degree of polymerization of SiO2 species as a function of total SiO2 content of the fluid. As an example, near the second critical endpoint of the SiO2–H2O system (1080 °C and 1 GPa; see Kennedy et al. 1962), speciation in SiO2–H2O fluid as a function of SiO2 concentration such as illustrated in Fig. 11 was proposed (Newton and Manning 2008). In this model, the degree of polymerization of the silicate species in aqueous fluid is correlated positively with the total SiO2 concentration, a structural feature apparently originally proposed by Wendlandt and Glemser (1964) on the basis of their silicate solubility data.

Modified from Newton and Manning (2008)
Speciation of dissolved SiO2 in aqueous fluid as a function of SiO2 concentration in the fluid.
Direct experimental determination of the structure of SiO2–H2O fluids at high temperature and pressure initially was reported by Zotov and Keppler (2002) and subsequently expanded upon by Mysen (2010) and Mysen et al. (2013). At pressures and temperatures below 0.6 GPa and 500 °C, only monomers [Si(OH)4] were detected by Zotov and Keppler (2002). With an additional temperature and pressure increase, the latter authors also found silicate dimers in SiO2–H2O fluid and proposed a dimerization reaction such as
for which the equilibrium constant as a function of temperature and pressure was reported as;
where ∆Veqn.(6) denotes the volume change for reaction shown as Eq. (6), the VH2O is molar volume of pure H2O, P is pressure, and T is temperature (kelvin).
Zotov and Keppler (2002) reported an enthalpy for Eq. (6) of 12.6 ± 1.3 kJ/mol. This enthalpy value is considerably greater than that which Sverjensky et al. (2014) from thermodynamic modeling and Mysen (2010) from Raman spectroscopy reported for reaction (6). The difference between the results of Zotov and Keppler (2002) and Mysen (2010) reflects different structural assignments of the Raman intensities used to deduce silicate species abundance in SiO2–H2O fluids. The enthalpy values in those two studies are in accord, however, when using the same assignments of the Raman bands reported in those two experimental studies.
Q0, Q1, and Q2 speciesFootnote 1 of silica were detected in aqueous fluid from vibrational spectra of the SiO2–H2O system when temperatures and pressures were extended to 900 °C and 5.4 GPa, respectively (Mysen et al. 2013). Here, the abundance of the variously polymerized Qn-species (n > 0) is positively correlated with the concentration of SiO2 in the aqueous fluid, which, of course, is the same relationship as proposed from the SiO2 solubility in the SiO2–H2O system (Wendlandt and Glemser 1964; Newton and Manning 2008) and from the thermodynamic modeling of solubility in this system (Sverjensky et al. 2014).
For the mol fraction of Q0, Q1, and Q2 species, for example, the following relationship holds (Mysen et al. 2013);
where the X-values are mol fractions and mSiO2 is molality (Fig. 12). It is clear, therefore, the concentration of SiO2 is a critical factor in determining the degree of polymerization of dissolved SiO2. This relationship between polymerization of Qn-species and SiO2 concentration in fluids resembles qualitatively the relationship between SiO2 content and the degree of polymerization Qn-species of silicate melts (Mysen et al. 1982; McMillan 1984; Buckermann et al. 1992; Cody et al. 2005).

Modified from Mysen et al. (2013)
Silicate species (Q.n-species) in SiO2–H2O as a function of total SiO2 concentration at temperatures and pressures indicated.
2.2.2 SiO2 in saline fluids
Aqueous fluids, in particular in subduction zone settings, can be saline with NaCl the dominant salt (Keppler 1996; Scambelluri and Philippot 2001; Manning and Aranovich 2014; Kawamoto et al. 2013). There are, therefore, numerous reports on experimental determination of SiO2 solubility in H2O–NaCl fluids at high temperature and pressure (Anderson and Burnham 1967; Xie and Walther 1993; Newton and Manning 2000, 2006; Shmulovich et al. 2001; Cruz and Manning 2015; Scheuermann et al. 2018).
The SiO2 solubility in H2O–NaCl fluids decreases with increasing NaCl concentration at pressures at and above about 0.5 GPa. At such pressures, the log mSiO2 is a linear or near linear function of NaCl mol fraction in the fluid, XNaCl (Fig. 13). Notably, the slope of this relationship is nearly independent of temperature in the temperature range examined experimentally (500°–900 °C), while the solubility itself increases with increasing temperature (Fig. 13). However, at pressures below 0.5 GPa, in H2O–NaCl fluids, there is an initial SiO2 solubility increase with increased mol fraction of NaCl (XNaCl) equal to or less than about 0.1 before a further XNaCl increase results in lowered SiO2 solubility (Xie and Walther 1993; Newton and Manning 2000).

Modified from Newton and Manning (2000)
Solubility of SiO2 in H2O–NaCl fluid as a function of salinity at different temperatures as indicated and 0.5 GPa total pressure.
The reason for the changing SiO2 solubility dependence on NaCl concentration below and above about 0.5 GPa is not well known. One might surmise, however, that this solubility behavior is because two different solution mechanisms are active in the SiO2–H2O–NaCl system. One is dilution of H2O by NaCl in the fluid, which is likely to shift to the left a solubility reaction such as: Footnote 2
This shift would decrease the silica solubility in the fluid. The extent to which this shift affects the SiO2 solubility depends on the fugacity of H2O, fH2O, which by itself decreases with decreasing pressure (Burnham et al. 1969). Therefore, one would expect the influence on SiO2 by the dilution of the fluid with NaCl, would be less the lower the pressure, the second mechanism involves chemical interaction between dissolved silica and Na+ from the NaCl. Such interaction results in formation of depolymerized Qn-species in the fluid where Na+ forms bonding with nonbridging oxygen in those Qn-species. Based on the analogy between structural behavior of Na2O–SiO2 melts as a function of their Na/Si ratio (e.g., Maekawa et al. 1991; Buckermann et al. 1992) and with H2O in solution (Cody et al. 2005), from steric considerations of the local charge environment surrounding nonbridging oxygens, Na–O bonding is favored over H–O bonding in these structures (Cody et al. 2005) because of the much smaller ionic radius of H+.Footnote 3 It is likely that the solution mechanism of silicate components in silicate-rich H2O–NaCl fluids resembles those documented for silicate melts and that this mechanism would enhance the solubility of SiO2 in saline fluids.
Whether the first or the second mechanism dominates would depend significantly on pressure, which, in turn, governs the fH2O. The lower the pressure, the smaller the effect of fH2O in reaction (9) and the more important is the second process. It is suggested that those relationships would explain the pressure-dependent effect of NaCl on the solubility of SiO2 in H2O–NaCl fluids. This explanation also implies that the pressure at which the solubility crossover takes place will depend on both temperature, which affects fH2O and the concentration of chloride in the H2O–NaCl fluid. It also means that different chlorides will have different effects on the solubility of SiO2 in the fluid.
A number of additional models for solution of SiO2 in H2O–NaCl fluids has been proposed (Franck 1973; Walther and Schott 1988; Newton and Manning 2000, 2016; Cruz and Manning 2015; Shi et al. 2019). Among those models, that of Shi et al. (2019) seems to reproduce the SiO2 solubility in H2O–NaCl fluids over the widest range of temperature, pressure and NaCl concentration.
Shi et al. (2019) considered the simple solubility reaction expressed with Eq. (9), with the equilibrium constant for this reaction
where
and
In Eqns. (9–12), a is activity, γ is activity coefficient and dH2O is the concentration of H2O.
These equations can be combined to yield (Shi et al. 2019);
where γSiO2 and γH2O are the activity coefficients of SiO2 and H2O in H2O–NaCl fluid, respectively. F is the mass fraction of H2O, and ρsoln is the density of the solution. This model describes the experimental data for SiO2–H2O systems quite accurately (Fig. 14).

2.2.3 MgO-SiO2 in aqueous fluid
Characterization of SiO2-bearing aqueous solutions is a critical first step toward understanding the behavior of aqueous solutions in natural processes. However, determination of only SiO2 solubility and only in the SiO2–H2O system is an obvious oversimplification of conditions in nature.
As a next step toward characterization of the solution behavior of chemically more complex silicates in fluids in the Earth's mantle, the system SiO2–MgO–H2O often has been employed as model peridotite system because the abundance of SiO2 + MgO comprises 70–80% of mantle peridotite (McDonough et al. 1995; Nakamura and Kushiro 1974; Konzett and Ulmer 1999; Zhang and Frantz 2000; Newton and Manning 2002; Mibe et al. 2002; Stalder et al. 2001; Kawamoto et al. 2004).
In the MgO–SiO2–H2O system at pressures near 1.5 GPa, there is a continuous solubility from melt near the SiO2 corner to the H2O corner where aqueous fluid contains only SiO2 (Fig. 15A). In other words, at least at this pressure, the solute in aqueous fluids is essentially pure SiO2 in equilibrium with Mg-rich crystalline phases such as Mg2SiO4 (forsterite) or MgSiO3 (enstatite). This finding is in accord with more recent experimental data in the same system both near this as well as at higher pressure (Zhang and Frantz 2000; Stalder et al. 2001; Mibe et al. 2002; Newton and Manning 2002; Kawamoto et al. 2004). In fact, the Mg/Si ratio of the silicate solute is near 0 at pressures at or below about 2 GPa before this ratio begins to increase as pressure is increased beyond 2 GPa (Kawamoto et al. 2004; Mibe et al. 2002; Zhang and Frantz 2000; see also Fig. 15B).

By extending the solubility data in the MgO–SiO2–H2O system from the 1.5 GPa in the Nakamura and Kushiro (1974) study to higher pressures, a second critical endpoint may be approached (Mibe et al. 2007; Melekhova et al. 2007). For example, at and above the 10 GPa pressure, Melekhova et al. (2007) reported that the MgO content of the fluid increased rapidly with increasing temperature, until near 13.5 GPa where the temperature effect on MgO solubility had disappeared. This evolution led Melekhova et al. (2007) to suggest that the critical endpoint in the MgO–SiO2–H2O system is somewhere between 11 and 13.5 GPa in the 1000–1350 °C temperature range of their study.
The estimated pressure–temperature coordinates of the proposed critical point from the Melekhova et al. (2007) study from the simple MgO–SiO2–H2O system (11–13.5 GPa/1000–1350 °C) differ significantly, however, from the pressure/temperature coordinates of a synthetic peridotite with typical peridotite composition (3.8 GPa/1000 °C) reported by Mibe et al. (2007). There are, of course, some important compositional differences that could affect the different critical point coordinates. The MgO–SiO2–H2O system examined by Melekhova et al. (2007) did not contain FeO, Al2O3, and alkali oxides, whereas the peridotite composition employed by Mibe et al. (2007) did. Addition of any and all of those latter components enhance the solubility in aqueous fluids as discussed in more detail later in this presentation. Enhanced solubility in aqueous fluid typically correlates with lowered pressure (and temperature) of the critical point. This latter observation is, therefore, consistent with expecting the pressure–temperature coordinates of critical point in a peridotite–H2O system (Mibe et al. 2007) to be lower than in the simpler MgO–SiO2–H2O system (Melekhova et al. 2007). However, existing information is insufficient to quantify those difference and, therefore, whether this explains the different pressures and temperatures reported on those two experimental studies.
There is, however, an additional difference between the two sets of experiments, a difference that also aid in explaining why the pressure/temperature coordinates of the critical points reported for the MgO–SiO2–H2O (Melekhova et al. 2007) and peridotite–H2O (Mibe et al. 2007) differ. In the MgO–SiO2–H2O system, the critical point was estimated from the discontinuous evolution of MgO concentration of quenched fluid (analyzed at ambient temperature and pressure after extraction of the sample) as a function of temperature at 11 and 13.5 GPa. This evolution led Melekhova et al. (2007) to bracket the critical point in the MgO–SiO2–H2O system between 11 and 13.5 GPa and between 1000 and 1350 °C. Notably, though, the temperature evolution of the SiO2 concentration in fluid did not show any discontinuity as a function of temperature in the same pressures and the same temperature ranges. It is not clear, therefore, how reliable the estimated pressure–temperature coordinates of the critical point determined solely from the discontinuous MgO concentration of fluid actually are.
The critical point reported for the peridotite–H2O system (Mibe et al. 2007) was determined by using X-ray imaging of the sample in situ, while it was at any pressure–temperature condition. A sample consisting of melt + fluid transformed to a single supercritical fluid phase going up temperature near 3.8 GPa and 1000 °C. There was exsolution of fluid from this fluid to form a melt + fluid during cooling. This method is closely similar to that used in the original studies of critical points in granite–H2O systems (Nowak and Behrens 1995; Shen and Keppler 1997; Bureau and Keppler 1999). In light of the discussion above, it is concluded that most likely, the pressure–temperature coordinates of the critical endpoint in the peridotite–H2O system from the Mibe et al. (2007) experiments should be considered more reliable and those from the Melekhova et al. (2007) study.
This conclusion also means that the reported pressure-temperature coordinations of the critical point of basalt–H2O and eclogite–H2O by Kessel et al. (2005), using the same method as that of Melekhova et al. (2007), probably also are not accurate.
The SiO2 concentration in aqueous fluids in equilibrium with enstatite in the MgO–SiO2–H2O system is less polymerized than the SiO2 solute in fluid in the SiO2–H2O system at the same temperature and pressure (Zhang and Frantz 2000; Mysen et al. 2013). This difference happens because the silica activity defined by crystalline phases coexisting with fluid (forsterite and enstatite) in MgO–SiO2–H2O system is lower than in the SiO2–H2O system where at silica saturation, quartz coexists with fluid. The lower SiO2 concentration in MgO–SiO2–H2O fluid leads to less polymerization of silicate species in aqueous solution. A comparison of the Qn-species evolution in fluids with temperature and pressure in SiO2–H2O and MgO–SiO2–H2O system illustrates this difference (Fig. 16).

Q.n speciation in SiO2–H2O and MgO–SiO2–H2O fluid as a function of temperature at the pressures indicated on individual curves. Modified after Mysen et al. 2013
The equilibrium among the Qn-species in the MgO–SiO2–H2O fluid at any pressure and temperature is, therefore, simpler than in the SiO2–H2O [Eqns. (6), (8), and (9)]:
A striking difference between the results for MgO–SiO2–H2O fluids and those of SiO2–H2O fluids is that whereas the ∆H and ∆V for the polymerization reaction in SiO2–H2O fluids depend on pressure and temperature, there are no such effects for the structurally simpler equilibrium relations in MgO–SiO2–H2O fluids (Mysen et al. 2013). This difference may also reflect the lesser extent of silicate polymerization in the MgO–SiO2–H2O fluids. The less polymerized silicate species in the latter fluids might lead to lesser excess volume of mixing in these latter MgO–SiO2–H2O fluids.
2.2.4 MgO–SiO2 in saline fluids
The solubility of Mg2SiO4 (forsterite) and MgSiO3 (enstatite) in H2O–NaCl fluid has been determined at 1 GPa (Macris et al. 2020) who reported incongruent solution of enstatite in H2O–NaCl fluids, whereas forsterite dissolved congruently. Both solubility and the Mg/Si ratio in the fluid increase with increasing NaCl concentration in the fluid (Fig. 17). This solubility behavior differs from that of SiO2 in H2O–NaCl fluids where the silicate solubility as a function of NaCl concentration varies with both NaCl concentration in fluid and with pressure (Xie and Walther 1993; Newton and Manning 2000; see also Sect. 2.2.2 and Fig. 13) This different solution behavior in fluids in the SiO2–H2O–NaCl and –NaCl systems probably results from additional solution mechanisms in MgO–SiO2–H2O–NaCl fluids. First, the decreasing SiO2 concentration (increasing Mg/Si ratio) with increasing NaCl of the fluid such as seen in the Mg2SiO4–H2O–NaCl system (Fig. 17B) in principle is the same trend as the solubility behavior of SiO2 in H2O–NaCl fluids, which also shows decreasing solubility with increasing NaCl (Fig. 17). Second, the solubility of the MgO component in saline fluids increases with increasing NaCl concentration probably (Macris et al. 2020) through formation of Mg–Cl type complexes in the fluid. In fact, Macris et al. (2020) proposed that mixed OH, Cl species (MgClOH) existed in such saline fluids. They suggested two possible, but in their words, nonunique solution mechanisms for forsterite (Mg2SiO4) in H2O + NaCl fluids to rationalize the reported solubility data at 1 GPa and 800° and 900 °C, respectively:
and

Modified from Macris et al. (2020)
A. Mg2SiO4 solubility in H2O–NaCl fluid as a function of mol fraction of NaCl at 1 GPa and at temperatures indicated. B. Mg/Si ratio of H2O–NaCl fluid in equilibrium with forsterite + enstatite at 1 GPa and 800° and 900 °C as a function of NaCl concentration of the fluid.
The speciation proposed in Eqn. (15a, b) has not been determined directly, and other reactions can also be written. However, they may serve to illustrate how Mg–Cl bonding in fluid complexes may account for the enhanced solubility of forsterite in NaCl-bearing fluids.
The concept illustrated for forsterite solubility in H2O–NaCl fluid should perhaps also apply to solution of MgSiO3 in such fluids. However, if so, as a portion of the Mg2+ in MgSiO3 would be tied up in the Mg-bearing fluid complexes, the Mg/Si ratio of the crystalline would, if anything, be expected to decrease from that of the MgSiO3 stoichiometry and perhaps lead to formation of SiO2 polymorphs. Such an evolution contrasts with the reported incongruent solution of MgSiO3 (enstatite) in H2O–NaCl fluids to produce Mg2SiO4 (forsterite) + fluid. The latter behavior would be analogous the solubility behavior of MgSiO3 (enstatite) in pure H2O at similar temperature are pressure conditions (Zhang and Frantz 2000). Clearly, these relationships require further confirmation by direct determination of the complexes formed in the H2O–NaCl fluids in these systems.
2.2.5 H2O–Al2O3(–NaCl–KOH–SiO2) in aqueous fluid
Given that Al2O3 typically is the second- or third-most abundant rock-forming oxide in most igneous and metamorphic rocks, characterization of its solubility behavior in fluids is important. Moreover, although it is commonly assumed that Al2O3 is the least soluble in pure H2O among the major rock-forming major oxides (e.g., Carmichael 1969), evidence from rocks indicates that Al2O3 can be quite mobile under some circumstances (e.g., Kerrick 1990; McLelland et al. 2002).
2.2.5.1 Al2O3 solubility in aqueous fluid
Examination of Al2O3 solubility in aqueous fluids is constrained by the pressure–temperature stability field of corundum, which in the Al2O3–H2O system is limited at low temperature by transformation to diaspore and H2O, which takes place between ~ 500 and 600 °C in the 1–4 GPa pressure range, for example (Kennedy 1959). At higher temperature, the Al2O3 solubility in aqueous fluid in the Al2O3–H2O system, which is in the ppm range, is a positive and linear function of pressure (Becker et al. 1983; see Fig. 18);
where Al2O3 (fluid) is in ppm.

Solubility of corundum (Al2O3) in aqueous fluid in the Al2O3–H2O system as a function of pressure at 670–700 °C. Modified after Becker et al. (1983)
A simple solution model for Al2O3 in aqueous solutions such as
has been proposed (Pokrovski and Helgeson 1995). However, the equilibrium constant for this reaction in the 50–220 MPa pressure range reached a minimum between 250 and 300 °C before increasing as the temperature is increased further (Walther 1997).Footnote 4 This changing temperature-dependent solubility behavior may lead to the suggestion that more than one solution mechanism of Al2O3 in aqueous solution is possible such as, for example;
in addition to equilibrium (17).
2.2.5.2 Al2O3 solubility in aqueous fluid in more complex systems with and without halogens
In order to mimic better natural conditions, SiO2 and alkali metals need to be added to the Al2O3–H2O system (Currie 1968; Anderson and Burnham 1983; Manning 2007; Wohlers et al. 2011; Schmidt et al. 2014). The influence of SiO2 alone on Al2O3 solubility in aqueous fluid is between 3.3 and 4.8 times greater than the Al2O3 solubility in the Al2O3–H2O system without SiO2 (Becker et al. 1983; Manning 2007; Tropper and Manning 2007). It should be noted, however, that whereas the Si content of such fluid was 0.3 ± 0.1 molal, that of Al was 0.008 ± 0.007 molal. In other words, for all practical purposes, the solute in SiO2-bearing fluids in those experiments was essentially all silicate and did not indicate enhanced Al2O3 solubility in aqueous SiO2-bearing aqueous solution.
By adding KOH or NaCl to H2O fluid, the Al2O3 solubility increases by several orders of magnitude compared with the Al2O3 solubility in pure H2O (Pascal and Anderson 1989; Walther 1997, 2001; Wohlers and Manning 2009; Newton and Manning 2006; see also Fig. 19). This solubility is a positive function of the KOH and NaCl concentrations at given temperature and pressure (Pascal and Anderson 1989; Azaroual et al. 1996; Newton and Manning 2006).

A. Aluminum solubility in alkaline fluid, mAl, as a function of KOH concentration in the Al2O3–H2O–KOH system at 50–200 MPa pressure and 400 °C. B. Aluminum solubility in H2O–NaCl fluid as a function of NaCl concentration in the system Al2O3–H2O–NaCl at 800 °C and 1 GPa.
Addition of NaCl to corundum + quartz increases the Al2O3 solubility further compared with Al2O3 solubility in the quartz-free system (Newton and Manning 2008; see also Fig. 20). Here, the molality, mAl2O3, is a complex and positive function of both the SiO2 and NaCl concentrations, which has been described with the empirical expression (Newton and Manning 2008):
where \({m}_{\mathrm{Al}2\mathrm{O}3}^{0}\) is the molality in NaCl-free fluid. A somewhat different expression was given for more NaCl-rich solutions.

The solubility of Al2O3 in fluids in the NaAlSi3O8–H2O system is another example of effects on solubility of added components at high temperature and pressure (Currie 1968; Anderson and Burnham 1983; Woodland and Walther 1987; Schmidt et al. 2014). The total aluminosilicate solubility in the NaAlSi3O8–H2O system is on the order of 1 wt%. However, the dissolution of NaAlSi3O8 in H2O fluid is slightly incongruent as first observed by Currie (1968), who reported that Na/Al in the aqueous solution is greater than 1 (Fig. 21). Incongruent dissolution of NaAlSi3O8 in a fluid with excess Na and Si over that of the NaAlSi3O8 stoichiometry, as also reported more recently by Mysen and Shang (2003) from experiments in closely related systems, implies that an Al-rich crystalline phase should be formed. In the system NaAlSi3O8–H2O, this phase could be corundum (Al2O3) or an Al-rich silicate phase such as sillimanite or kyanite (AlSi2O5), for example. However, neither Currie (1968) nor Anderson and Burnham (1965, 1983) reported any crystalline phase in their run product. This matter remains, therefore, unresolved.

Modified from Currie (1968)
Solubility in the system NaAlSi3O8–H2O A. Concentration of Al in fluid as a function of pressure at temperatures indicated. B. Concentration of Na in fluid as a function of pressure at temperatures indicated.
Addition of NaCl to the NaAlSi3O8–H2O system results in decreased solubility in the fluid (Fig. 22). Moreover, the solubility in aqueous solution approaches congruent as the pressure is increased (Shmulovich et al. 2001). In this regard the NaAlSi3O8 solubility behavior in saline solutions resembles the solubility in pure H2O. We note, however, that the results of Shmulovich et al. (2001) differ some from those reported by Tagirov et al. (2002) who reported decreased NaAlSi3O8 solubility with increased NaCl at low NaCl concentration in aqueous fluids and increased solubility at high concentration (Fig. 23). This behavior led Tagirov et al. (2002) to propose different Al-bearing species depending on the NaCl concentration (Fig. 23). In this model, at low NaCl concentration, the Al-species is \(\mathrm{Al}(\mathrm{OH}{)}_{4}^{-}\). With increasing NaCl concentration, the activity of NaCl is sufficient to stabilize and NaAl(OH)3Cl0 species in the fluid, which was proposed to explain the increased NaAlSi3O8 solubility at high NaCl concentrations.

Solubility of NaAlSi3O8 in H2O–NaCl fluid in the system NaAlSi3O8–H2O–NaCl as a function of NaCl concentrations at 0.9 GPa and temperatures shown on individual curves. Modified after Shmulovich et al. (2001)

Modified from Tagirov et al. (2002)
Al speciation in H2O–NaCl fluid n as a function of Al and Na concentration in the system albite–aragonite–quartz–H2O–NaCl at 400 °C and 50 MPa in slightly acidic solution with pH = 7.1–4.8.
2.3 Solubility of minor and trace elements in fluids
Transport of trace elements in fluids often is dominated by fluids rich in H2O and chloride. Such transport can be particularly important in subduction zone settings where magma can carry unique trace element signatures caused by their transport in aqueous fluids from a dehydrating subducting slab to the overlying mantle wedge where partial melting takes place (Mysen and Boettcher 1975; Wyllie 1982; Ayers and Watson 1993a; Elliott et al. 1997; Iizuka and Mysen 1998; Brenan et al. 1998; Baier et al. 2008; Till et al. 2012; D'Souza and Canil 2018). A number of relevant solubility data exist. Here, we will provide a few important examples.
2.3.1 Titanium solubility
Rutile is often employed to deduce petrogenetic history of igneous rocks (e. g., Foley et al. 2000). The Ti concentration in fluids at high temperature and pressure is critical for stabilization of rutile in source regions of magma. Such data are important because rutile governs the abundance of a number of geochemically important trace elements has been used to account for the low abundance of HFSE for example (Ayers and Watson 1993b; Brenan et al. 1994; Stalder et al. 1998; Keppler 2017). The Ti concentration in zircon also has been used as a geothermometer (Watson et al. 2006).
The solubility of TiO2 in pure H2O is quite low, perhaps around 10 ppm or so under conditions of the lower crust and upper mantle. The Ti solubility in the TiO2–H2O fluids increases slightly with increasing temperature and pressure, but remains in the tens of ppm range (Antignano and Manning 2008; Mysen 2012; see also Fig. 24). Raman spectra of the TiO2–H2O solutions at temperatures and pressures similar to those of the solubility experiments by Antignano and Manning (2008) indicate that TiO2 in pure H2O solutions exists in or near sixfold coordination with oxygen (Mysen 2012).

Titanium concentration in in aqueous fluid in equilibrium with rutile in the system TiO2–H2O A. as a function of temperature at 1 GPa pressure, and B. as a function of pressure at 800 °C Modified after Antignano and Manning (2008)
The TiO2 solubility in aqueous solution in the TiO2–SiO2–H2O system is not appreciably different from the solubility in Si-free TiO2–H2O system (Antignano and Manning 2008). However, by adding an Na-containing compound to such systems, the TiO2 solubility in aqueous fluids is greatly enhanced (Hayden and Manning 2011; Mysen 2012). For example, the Ti solubility in such fluids increased from a few tens of ppm in the TiO2–H2O system to 0.3–0.4 wt% when NaAlSi3O8 is added (Hayden and Manning 2011) and to about 0.6 wt% by adding NaCl to the TiO2–H2O system (Tanis et al. 2016). The Ti solubility in NaF–H2O fluids increases by an additional 50–100% compared with the Ti solubility in H2O–NaCl fluids (Tanis et al. 2016).
From the in situ Raman spectra of the fluids containing Na-silicate compounds, Ti4+ is in fourfold coordination with oxygen, which, of course, contrasts with the approximately sixfold coordination of Ti4+ in TiO2–H2O solution in similar temperature and pressure ranges (Mysen 2012). From the vibrational spectra of TiO2-saturated aqueous solutions with Na and Si added to the system, a solubility reaction such as (Mysen 2012);
was found to describe the solubility behavior of Ti4+. The Ti4+ forms, therefore, an oxycomplex in the form of a Q0-like species in which Ti4+ is in fourfold coordination (equivalent to TiO44−). In Eq. (20), the QSio(HNa) formulation is meant to indicate that both H+ and Na+ form bonding with nonbridging oxygen in isolated SiO4 tetrahedra, whereas in the Q1Si(Na) complex, Na+ alone forms bonding with nonbridging oxygen in the slightly more polymerized dimers (Q1).
It is possible, but has not been documented as yet, that any alkali-bearing compound would cause TiO2 solution behavior analogous to that in Eq. (20). The solution mechanism in Eq. (19) is, therefore, greatly different from Ti4+ in solution in pure H2O where Ti4+ is in sixfold coordination with oxygen. From the temperature dependence of equilibrium (20), it is evident that the ∆H is lower by up to about 50% in the (Na + Al)-bearing systems compared with the ∆H from the simpler Na-silicate + TiO–H2O system (Mysen 2012).
The trace element signatures of magma formed by partial melting of the mantle wedge above subducting plates to a considerable extent reflect contributions to the peridotite geochemistry from fluids derived from the slab itself (e.g., Zheng 2019). The extensive depletion of high field strength elements (HFSE) in island arc magmas is particularly notable (Keppler 2017). Those geochemical features have been ascribed to the presence of rutile (TiO2) during partial melting of the peridotite wedge (Brenan et al. 1994; Foley et al. 2000). Given the generally low TiO2 concentration in typical mantle peridotite (e.g., Putirka et al. 2011) and the absence, therefore, of rutile in common peridotite, whether or not rutile is present during partial melting of a mantle wedge may depend on the extent to which its TiO2 content mantle wedge source region of partial melts may have been altered by ingress of fluid from a dehydrating subducting slab. This possibility, in turn, would depend on the availability of alkali metals in the fluid derived from the slab because alkali metals appear to be critical factors controlling the TiO2 solubility of the fluid as evidenced in the experimental data regarding greatly enhanced Ti solubility when forming oxytitanate complexes in aqueous fluid discussed above. The TiO2 concentration in such aqueous fluids can vary by nearly 3 orders of magnitude depending on such compositional factors (Mysen 2012)! Therefore, if the subducting slab were of felsic composition, the fluid derived from it would be alkali-rich and can contain significant proportions of TiO2, whereas were the fluid derived from dehydrating mafic and ultramafic rocks, the fluids would contain less alkalies and, therefore, will have less TiO2 in solution. One might propose, therefore, that the extent to which rutile exists in the mantle wedge undergoing partial melting to yield island arc magma with attendant HFSE depletion of the partial melt, depends on the geochemistry of the source of the fluids that contributed to the mantle wedge composition.
2.3.2 Other trace elements in fluids
The principles that govern the Ti solubility in simple aqueous solutions as well as compositionally more complex solution environment may also aid in our understanding of how other trace elements dissolve in aqueous solutions. These trace elements may include other HFSE such as Zr, Hf, Nb, and Ta, transition metals including Cr and Mo, and actinides such as U and Th. In other words, their solubility in aqueous solutions could be greatly enhanced by formation of oxycomplexes that are charge compensated by alkali metals or possibly alkaline earths (Keppler and Wyllie 1991; Peiffert et al. 1996; Ulrich and Mavrogenes 2008; Bali et al. 2011, 2012; Wilke et al. 2012; Watenphul et al. 2014; Mysen 2012; Keppler 2017). In addition, for some of these trace elements (e.g., uranium, thorium, molybdenum, niobium, and tantalum), redox conditions also can affect the solubility in important ways (e. g., Bailey and Ragnarsdottir 1994; Peiffert et al. 1996). Salinity also can be important (Rustioni et al. 2021).
2.3.2.1 Zirconium Solubility
The solubility of ZrO2 in fluids in the ZrO2–H2O system at pressures and temperatures corresponding to the deep crust and upper mantle is at the ppm level (Wilke et al. 2012; Mysen 2015c). This solubility (Fig. 25) resembles that of TiO2 in the TiO2–H2O system under similar temperature and pressure conditions (Fig. 24) with a simple solution mechanism such as
with the equilibrium constant;
where m is molality. From linear relationship between ln K and 1/T (kelvin), the ∆H = 43 ± 16 kJ/mol for the solution reaction illustrated in Eq. (21). This enthalpy resembles the 50–60 kJ/mol value for Ti solution in the TiO2–H2O system (Mysen 2012).

Modified from Mysen (2015c)
Zirconium solubility in aqueous solutions as a function of temperature and pressure for the various systems indicated on individual dashed lines.
The Zr solubility, much as the Ti solubility, is quite sensitive to added components in the fluid. For example, addition of Na+ to aqueous solutions results in Zr solubility increases by approximately an order of magnitude (Fig. 25). The simplest way to describe the solution mechanism of ZrO2 under these conditions may be expressed as (Mysen 2015c):
In this environment, Zr4+ is in fourfold coordination with oxygen as evidenced by the Raman spectra of such fluids recorded, while the fluid and coexisting Zr-bearing crystalline materials were at the high temperature and pressure of interest (Mysen 2015c). However, from existing X-ray and Raman spectroscopic data of such fluids (Wilke et al. 2012; Mysen 2015c), several more complex reactions involving zirconosilicate or separate silicate and zirconate complexes could be considered. Given the structural interpretation Raman spectra of the fluids in ZrO2–SiO2–NaOH–H2O, some Si–OH bonding in addition to Zr4+ in fourfold oxygen coordination is likely with one reaction that is consistent with all structural data is (Mysen 2015c):
This structural behavior of Zr4+ differs its solution mechanism in the simple ZrO2–H2O fluid system, where the vibrational spectra have been interpreted to indicate oxygen coordination numbers in excess of 6 (Mysen 2015c).
In summary, the key to enhanced solubility of HFSE in aqueous solutions is the stabilization of oxycomplexes associated with alkali metals or, perhaps alkaline earths. The exact form in which the metal cation is added to the solution may not be so important. It is likely, for example, that the more electropositive the metal cation is, the greater is its effect, and the greater is the solubility of the oxycomplex in aqueous fluids. One might speculate, therefore, that much as was discussed for Ti solubility above, fluid in equilibrium with felsic magma will be alkali metal rich and, therefore, form Zr-bearing oxycomplexes with greater solubility in aqueous solutions than fluids in equilibrium with mafic igneous rocks where the more electronegative alkaline earths are less likely to stability the oxycomplex.
2.3.2.2 Molybdenum solubility
The solubility of molybdenum in aqueous, saline solutions is in the 100–10,000 ppm range (Ulrich and Mavrogenes 2008; Bali et al. 2012; Hurtig and Williams-Jones 2014). It is a strong function of both oxygen fugacity and solution salinity (Bali et al. 2012).
The Mo solubility increases by about 2 orders of magnitude when the fO2 increases by about 4 orders of magnitude. The solubility of oxidized Mo increases by about an order of magnitude when the NaCl concentration increases from 0 to about 15 wt% (Fig. 26).

Molybdenum solubility in H2O–NaCl solutions. A. As a function of oxygen fugacity in 5 wt% NaCl saline solution at 2.61 GPa and 700 °C. B. As a function of NaCl concentration at 2.61 GPa and 700 °C at the oxygen fugacity of the NNO buffer. Modified after Bali et al. (2012)
2.3.2.3 Trace element solubility and sulfur in aqueous solution
Sulfur in aqueous solution can exist in multiple oxidation states, which can affect its influence on the solubility of trace elements in S-bearing fluids. The sulfur species are H2S, SO2, SO3, and HSO3 (Binder and Keppler 2011; Eldridge et al. 2018). An additional sulfur species, S3−, originally proposed by Pokrovski and Dubrovinski (2011) has been suggested to be an important intermediate species stabilizing transition metals (Tossell 2012; Pokrovski et al. 2015). In the numerical simulations by Tossell (2012), the simple reaction:
has a negative free energy change at high temperature (− 110 kJ/mol at 450 °C, for example), while at ambient temperature the ∆G of the reaction is positive (25 kJ/mol). From this information, it follows that the S3− would be stabilized with high temperature.
Reduced sulfur in aqueous solution can have particular influence on solubility of metals such as Au, Ag, Cu, Mo, and Zn (Gibert et al. 1998; Trigub et al. 2017; Pokrovski et al. 2008; Frank et al. 2011; Tagirov and Seward 2010; Zhang et al. 2012). The solution mechanisms of these elements in some ways resemble one another, and only the solution behavior of Au will be summarized here.
The solubility of Au with reduced sulfur in aqueous solution, is positively correlated with concentration of H2S (Fig. 27A; see also Trigub et al. 2017). The Au solubility also increases rapidly with increasing pH (Fig. 27B).

A. Solubility of Au in H–O–S solutions as a function of H2S content, mH2S, at 450 °C and 100 MPa and pH = 3 ± 0.2. B. Solubility of Au in H–O–S solutions as a function of pH at 450 °C and 100 MPa with molality of H2S, mH2S = 1, and hydrogen fugacity, fH2 = 0.1 MPa. C. Speciation of Au-sulfide complexes in H–O–S solutions as a function of pH and Au concentration. Modified after Pokrovski et al. (2008) and Trigub et al. (2017)
The Au solution mechanism has been described with an expression of the type (Pokrovski et al. 2008);
Pokrovski et al. (2008) concluded that AuHS° complexes dominated with pH < 5, whereas at higher pH conditions, the dominant Au species was Au(HS)2− (Fig. 27C). The existence of such sulfur species also has been inferred from Au L3-edge X-ray absorption (Trigub et al. 2017).
2.4 Structure and properties of fluids
Physical and chemical properties of fluids, including their solvent capacity, vary with fluid composition as well as type and proportion of oxide solutes. The properties, in turn, reflect the fluid structure and the solution mechanism(s) of the solute(s). It is necessary, therefore, to ascertain how fluid structure varies with composition of solvent and solute, temperature, and pressure. With this information, modeling transport properties and processes of fluids and fluid–rock interaction in the Earth's interior becomes a tractable problem.
2.4.1 Structure of H2O fluid
Under most conditions, H2O affects the physics and chemistry of rock-forming materials more than other fluid components and species in the C–O–H–N–S system (Kohlstedt et al. 2006; Kushiro 1972; Whittington et al. 2000; Bouhifd et al. 2006; Grove et al. 2012). These effects include interaction between H2O dissolved in magmatic liquids as well as in crystalline materials, and the extent and efficiency with which fluids migrate through rock matrices. These and other effects reflect the structure of H2O and the interaction between its structural elements and the materials with which H2O interacts.
The structure of H2O is comprised of monomers, dimers, and sometimes even more polymerized species under the temperature and pressure conditions of the Earth's interior (Gorbaty and Kalinichev 1995; Hoffmann and Conradi 1997; Katayama et al. 2010). In these structures, many of the individual H2O molecules are linked together with hydrogen bonding, the proportions of which vary with temperature and pressure (Schneider et al. 1958; Hoffmann and Conradi 1997; Sahle et al. 2013). The density of H2O fluid is also linked to the proportions of those structural entities, and, therefore, to temperature and pressure.
The latter structural features have been interpreted from the proton NMR spectra of H2O (Hoffmann and Conradi 1997), recorded spectra from ambient conditions to 40 MPa and 600 °C. In these spectra, the chemical shift of 1H is sensitive to both temperature and pressure (Fig. 28). The discontinuity on the curves in Fig. 28 reflects the crossing of the liquid–vapor curve of H2O.

Modified from Hoffman and Conradi (1997)
1H NMR chemical shift of O A. as a function of temperature, and. B. As a function of pressure. Translation to extent of hydrogen bonding is based on relationship between chemical shift and H2O fluid density.
The 1H chemical shift decreased with increasing temperature and increased with increasing pressure (Fig. 28). This spectral evolution reflects decreasing abundance of hydrogen bonded structure the higher the temperature and an increased abundance of hydrogen bonding with increasing pressure (Hoffmann and Conradi 1997; see also Fig. 29). For example, from 1H NMR spectra of pure H2O, Hoffmann and Conradi (1997) estimated the proportion of hydrogen bonding decreasing from about 80% of the H2O structure at ambient temperature and pressure to less than 10% of the H2O structure at 600 °C in in the 30–40 MPa pressure range as illustrated in Fig. 29. The influence of pressure under isothermal condition is a 10–30% hydrogen bond-fraction increase between ambient pressure and 30 GPa.

Schematic representation of the structure of H2O at low temperature with about 80% of the H2O molecules connected with hydrogen bonding (long arrow between H and O atoms) and high temperature with about 10% hydrogen bonding and the result of H2O molecules isolated from one another
This structural model developed from the NMR data is consistent with that of results from X-ray and neutron diffraction, which also have been interpreted to indicate that the extent of hydrogen bonding in H2O fluid increased with increasing pressure (Sahle et al. 2013; Soper and Ricci 2000). Similar conclusions were reached from high-temperature/high-pressure Raman spectra of fluid and supercritical H2O fluid (Walrafen et al. 1988; Frantz et al. 1993; Foustoukos and Mysen 2012).
Pressure and temperature not only affect hydrogen bonding in the H2O structure, Katayama et al. (2010) found increased coordination numbers for the H2O molecule so that at pressures near 4 GPa the number reached 9 (Fig. 30) based on X-ray diffraction data recorded along the pressure–temperature trajectory of the melting curve of H2O to 17 GPa and 850 K. This coordination number (9) means that each H2O molecule is surrounded by 9 other H2O molecules. Katayama et al. (2010) also commented that this coordination number is typical for simple liquids such as noble gases (8–9). At pressure above about 4 GPa, no further coordination changes were reported. Those higher-pressure X-ray data were interpreted to show a decreased nearest-neighbor distance at pressures above about 4 GPa.

Oxygen coordination number of H2O as a function of pressure. Modified rom Katayama et al. (2010)
2.4.2 Structure of H2O–NaCl fluid
Radial distribution functions derived from neutron diffraction using (H2O, D2O) + NaCl fluids show the nearest H2O molecules about 2 Å from the Cl− anion (Botti et al. 2004). The oxygen in the H2O molecules was located about 3 Å from the Cl− anion. The average solvation number for H2O from both the Cl–H and Cl–O distances is 5.8 (Heuft and Meijer 2003). The larger fraction of the H2O is in hydration shells surrounding Cl− compared with the number of H2O molecules surrounding Na+.
2.4.3 Structure and thermodynamics of H2O–C–OH fluids
The two C-bearing species considered here are CO2 and CH4 as these are the two main C-bearing species relevant to rock-forming processes in the Earth. Carbon dioxide dominates under redox conditions above that defined by the magnetite–wüstite (MW) buffer, whereas under more reducing conditions, CH4 is the main species.
In the modern Earth, CO2 likely is the principal species in the upper mantle, whereas under deeper mantle conditions, the fO2 may be sufficiently low (and fH2 high) for CH4 to be the main species. During the first few tens of millions of years of the Earth's history, redox conditions were at and below the IW oxygen buffer (Righter and Drake 1997; Gessmann and Rubie 2000) such that CH4 was the principal C-bearing fluid species in the Earth.
2.4.3.1 H2O–CO2
Fluids in the H2O–CO2 system comprise molecular CO2, CO32−, together with HCO3− groups at least to pressures below about 1.6 GPa (Frantz 1998; Schmidt 2014; Mysen 2015a). At higher pressure, Martinez et al. (2004) concluded that the bicarbonate, HCO3−, was not stable in the fluid. At 200 MPa, molecular CO2 becomes increasingly important with increasing temperature as do the CO32− groups (Frantz 1998). From experiments in the H2O–CO2 fluid system at higher pressure (Schmidt 2014), the CO32− and CO2 abundance decreases with increasing pressure, whereas that of the HCO3− shows an increase. These pressure effects on C–O–H speciation in H2O–CO2 fluids diminish with decreasing temperature.
Property measurements of H2O–CO2 fluid have focused on thermodynamic properties such as activity-composition and volume relations. From volume data, activity and activity coefficients of the fluid species have been obtained (Frost and Wood 1997; Deering et al. 2016) because activity coefficient of component i, γi, is linked to its partial molar volume and the volume of pure i, \(\overline{V }\) i and Vi, respectively so that:
In this equation, R is the gas constant, T is temperature, and P is pressure.
Activity-composition relations of H2O-CO2 fluids also have been obtained by combining decarbonation and dehydration reactions such as, for example (Aranovich and Newton 1999);
and
The results of Aranovich and Newton (1999), using this method (Fig. 31), were quite similar to those reported by Duan and Zhang (2006) from numerical simulation of the mixing behavior in H2O–CO2 fluids (solid lines in Fig. 31).

2.4.3.2 H2O–CH4
From the experimental data available for silicate-saturated H2O-CH4 fluids in equilibrium with-saturated silicate melts, molecular CH4 coexist with CH3 groups. These latter groups substitute for oxygen in the silicate tetrahedra of silicate dissolved in the fluid (Mysen et al. 2011). An equilibrium reaction of the type;
where the superscript, n, denotes the number of bridging oxygen in the silicate species described with the Qn-notation.
Equilibrium (32) shifts to the right with increasing temperature, which results in ∆H = 16 ± 5 kJ/mol for the reaction. The ∆H-value of equilibrium (32) for the fluid is about 1/3 of that in coexisting melt (Mysen 2015b). This enthalpy difference likely reflects the greater deviations from ideal mixing in silicate melts compared with silicate-saturated H2O–CH4 fluid at high temperature and pressure.
Volume of mixing is among the few property measurements available for H2O–CH4 fluids (Fig. 32). There is a distinctly nonlinear volume evolution as a function of H2O–CH4 fluid composition (Shmonov et al. 1993). The results of the numerical simulation of H2O–CH4 fluid volumes by Zhang et al. (2007) (solid lines in Fig. 32) are in very good agreement with the experimental data of Shmonov et al. (1993).

Modified from Zhang et al. (2007)
Molar volumes of H2O–CH4 fluids as a function of H2O/CH4 ratio at 400 °C and pressures indicated on curves from experiments by Shmonov et al. (1993) and compared with calculated molar volumes by Zhang et al. (2007). Lines are results from Zhang et al. (2007), whereas data points from experiments by Shmonov et al. (1993).
2.4.4 Structure and thermodynamics of H2O–S–O–H fluid
Sulfur, the third-most important fluid species in many igneous processes (Symonds et al. 1994), can occur both in reduced, S2−, and oxidized, SO2 and SO3, forms depending on redox conditions during magmatic processes. Reduced sulfur species dominate with fO2 conditions more reducing than near that of the NNO buffer (O'Neill and Mavrogenes 2002). Oxidized sulfur is the main species under more oxidizing conditions (Scaillet et al. 1998).
This fO2-dependent redox ratio of sulfur means, for example, that igneous rocks more mafic than andesite will have essentially all sulfur in melts and exsolving gases in sulfide form (H2S) because the oxygen fugacity during their formation and evolution of such more mafic magma typically is less than that defined by the NNO oxygen buffer (Carmichael and Ghiorso 1990). On the other hand, more silica-rich igneous rocks such as andesite, dacite, and rhyolite, which typically are formed at greater oxygen fugacity conditions than that of the NNO oxygen buffer during their formation (Carmichael and Ghiorso 1990), have essentially all their sulfur in oxidized form, SO2 and SO3, or their hydrated form, sulfuric acid (Scaillet et al. 1998; Jugo 2009). These latter sulfur species can become important components of fluids formed by degassing of such felsic magma.
From the temperature dependence of the equilibrium constant for the reaction
the ∆H is 1442 ± 63 kJ/mol (Binder and Keppler 2011) with no discernible pressure dependence. In contrast, the reaction describing oxidation from SO2 to SO3
is both temperature and pressure dependent. From the temperature dependence, the ∆H decreases (becomes more negative) from − 160 ± 50 kJ/mol to − 308 ± 9 kJ/mol between 150 and 250 MPa, for example (Binder and Keppler 2011).
2.5 Fluid migration and mass transport; permeability and porosity
Transport of mass in the Earth for the most part takes place via movement of fluids and magma. In this section, we will discuss how some of the properties of fluids affect their migration through crystalline rocks and how fluid properties can affect rock-forming properties and processes. The extent and ease of fluid migration, in turn, depend on the rock porosity, which has been linked to permeability via Archie's Law (Archie 1942);
In this equation, k is permeability, \(\phi\) is the fluid fraction (porosity), d is grain size, and C is a constant. The value of the superscript, n, commonly is reported to be between 1 and 3 (Dullien 1992), although for natural fluids and magma, values less than 1 are often reported for best fit to experimental data (Wark and Watson 1998; Price et al. 2006; Shimojuku et al. 2012).
Archie's Law [Eq. (35)] assumes that there is only one grain size, but in rocks, more often than not, this is not the case. For example, with two different grain sizes, 1 and 2, in the following relationships describes the relations between porosity and grain size (Wark and Watson 2000):
Additional variables include different surface energies of different crystallographic surfaces.
Fluids in the Earth's interior often are dominated by H2O + CO2 and also can include chloride and sulfur compounds. The latter components can have substantial impact on the fluid transport capacity both in terms of their efficiency as solvents as well as the permeability of such fluids in a crystalline rock matrix (Watson et al. 1990; Holness 1992; Huang et al. 2020).
Migration of fluids through a rock matrix has been the subject of experimental study (Mysen et al. 1978; Cohen and Watson 1996; Wark and Watson 1998; Nakamura and Watson 2001). For example, in early experiments to determine the velocity of H2O passing through a crystalline peridotite under conditions relevant to fluid migration from a dehydrating subducting plate into the overlying peridotite mantle wedge, migration velocity of this aqueous fluid were reported to be on the order a few mm/hr (Mysen et al. 1978). This rate (mm/hr), from laboratory experiments conducted under hydrostatic or near hydrostatic conditions, differs significantly from that inferred from earthquake swarms in the Marianas and Izu-Bonin arcs, where White et al. (2019) interpreted seismic data to be consistent with fluid movement from a dehydrating slab into overlying mantle to be on the order of km/hr. These different migration rates may be because in the experiments by Mysen et al. (1978), aqueous fluid migrated along grain boundaries in a hydrostatic medium, whereas it is possible that the rate interpreted from the earthquake swarms exists in an environment under shear where fluid could migrate along in shear zones above subducting plate with much less resistance to fluid movements. Migration rate in such a setting would be much faster than grain boundary travel in a hydrostatic environment. This difference may account for the different fluid transport rate in experiments (Mysen et al. 1978) compared with rates under natural conditions (White et al. 2019).
To conduct experiments to determine fluid migration velocity under controlled conditions directly relevant to fluid flow in the Earth, grain size and grain size distribution, whether or not different minerals exist in the mineral assemblage, proportion and composition of fluid, porosity of the crystalline assemblage, and interfacial energies between solids and between solids and fluid must be controlled (Jurewicz and Watson 1985; Korenagi and Kelemen 1998; Wark and Watson 2000; Mu et al. 2016; Iwamori et al. 2007; Huang et al. 2020). Fluid migration in the Earth's interior often can take place in a stress field such as existing near the interface of subducting plates and the overlying mantle wedge (e. g., Hacker et al. 2003). The orientation of fluid and melt pockets in a stress field varies with the magnitude of the stress field so that increasing differential stress results in increasing deviation from the direction of the stress field (Daines and Kohlstedt 1997). Deformation of the fluid/rock system will be the result (Wanamaker and Kohlstedt 1991; Walte et al. 2011).
2.5.1 Fluid wetting angle
A major variable affecting fluid migration through a rock matrix is the wetting angle or dihedral angle. This angle, often represented by the symbol, θ, is the angle at the junction between two adjoining solid and liquid (fluid or melt) (Fig. 33).

Schematic illustration of dihedral angle, Θ, in an isotropic medium of solid and fluid
The key factor determining the wetting angle of liquids in a solid matrix and, therefore, permeability, porosity, and ultimately migration rate is the energy of the solid–solid and solid–liquid interfaces of the crystalline assemblage with which the liquid is in contact. With a fixed solid–solid interface energy, γss, the main factor governing the wetting angle becomes the energy of the solid–liquid interface, γsl, because:
Under the simple conditions described with Eq. (37), for θ < 60°, the fluid will form an interconnected network, whereas with θ > 60° it will not.
The ratio of the two interfacial energies, \(\frac{{\gamma }_{\mathrm{ss}}}{{\gamma }_{\mathrm{sl}}}\), and, therefore, the dihedral or wetting angle, is linked to the solubility in the fluid phase of one or more of the components in the solid (Takei and Shimizu 2003). In the environment such as expressed with Eq. (37), the wetting angle is proportional to the solubility of the components of the solid materials in the fluid. For example, using experimental data on wetting angles and dissolved components in aqueous fluid in contact with olivine at mantle pressures and temperature, a tripling of the solute concentration in the aqueous solvent results in a 25% lowering of the wetting angle of this fluid as pressure, and, therefore, solute concentration, is increased from 1 to 8 GPa (Fig. 34).

Modified from Takei and Shimizu (2003)
A. Solubility in aqueous fluid in forsterite–H2O system and B. dihedral angle (expressed as the ratio, ρ = 1/(2cos(θ/2)) as a function of pressure.
The relationship between solubility and wetting angle such as in Fig. 34 exists because the concentration and speciation of components dissolved in aqueous fluids near the interface of fluid with a mineral such as olivine, for example, increasingly resemble each other as solute concentration increases with increasing pressure. In the case of forsterite + H2O, this evolution exists because the solubility of mantle components such as MgO and SiO2 in aqueous fluids increases with increasing pressure (Zhang and Frantz 2000; Newton and Manning 2002; Kawamoto et al. 2004). The local structure of the dissolved silicate components also becomes increasingly similar to that of the adjoining olivine crystals at the fluid/olivine interface (Mysen et al. 2013). This evolving structural similarity of aqueous fluid and forsterite results in lowering of γsl and, therefore, a decreased θ.
2.5.1.1 Wetting angle and composition of fluid and crystalline matrix
Dihedral angle of aqueous fluid has been determined for crustal rock-forming minerals such as quartz, plagioclase, calcite, and dolomite (Watson and Brenan 1987; Hay and Evans 1988; Laporte and Watson 1991; Holness 1992, 1993, 1995; Nakamura and Watson 2001; Yoshino et al. 2002). Wetting behavior by H2O fluids in contact with mantle mineral assemblages has been determined for olivine, pyroxenes, and garnet and their high-pressure polymorphs (Watson et al. 1987, 1990; Mibe et al. 2003; Ono et al. 2002; Mibe et al. 1998; Yoshino et al. 2007; Matsukage et al. 2017; Liu et al. 2018).
In the quartz–H2O system, the dihedral angle, θ, is slightly above 60° at upper crustal pressures, but decreases rapidly with increasing pressure to those of the deep continental crust and uppermost mantle (Fig. 35; see also Watson et al. 1990; Holness 1992). The θ also decreases as a near linear function of temperature and at 1 GPa pressure where it crosses the 60° threshold at temperatures near 1100 °C (Fig. 35B). This temperature-dependent dihedral angle could be linked to the rapidly increasing quartz solubility in aqueous fluid as the temperature approaches supercriticality, which is just below 1100 °C at pressures near 1 GPa (see also Kennedy et al. 1962, for discussion of phase relations in the SiO2–H2O system).

Modified from Watson et al. (1990)
A. Dihedral angle in dunite–H2O at 1200 °C as a function of pressure. B. Dihedral angle in dunite–H2O as a function of temperature at 1 GPa.
By adding CO2 to H2O, the dihedral angle, θ, in the quartz-H2O–CO2 system, for example, this angle increases rapidly with increasing CO2/(CO2 + H2O) of the fluid at fixed temperature and pressure (Watson and Brenan 1987; Holness 1992; Holness and Graham 1995). The dihedral angle is near 100° for the SiO2–CO2 system (Fig. 36). This much larger dihedral angle for fluid in the SiO2–CO2 system compared with the SiO2–H2O system is consistent with the much lower solubility of SiO2 in CO2 fluid than in H2O fluid (Newton and Manning 2000).

Modified from Watson and Brenan (1987)
Dihedral angle in quartz-H2O–CO2 at 1 GPa and 950°-1150 °C as a function of fluid composition.
Fluid salinity also can affect the dihedral angle such as observed, for example, in the quartz-H2O–NaCl system (Watson and Brenan 1987; Laporte and Watson 1991; Holness 1992). The SiO2 solubility in H2O–NaCl fluids is, however, a complex function of pressure and NaCl concentration (Newton and Manning 2000). It is no surprise, therefore, that the dihedral angle in this system also is a complex function of salinity of the fluid (Fig. 37).

Evolution of dihedral angle in quartz + H2O + NaCl at 800 °C as a function of pressure. Modified after Holness (1992)
Plagioclase is a major part of mineral assemblages in most crustal rocks. That importance notwithstanding, experimental data on fluid wetting angles in plagioclase + fluid systems are not common. In one study with H2O fluid in contact with anorthite-rich plagioclase (Yoshino et al. 2002), the θ decreased with increasing anorthite component in the plagioclase. Within the pressure–temperature stability field of anorthite in the CaAl2Si2O8–H2O system (Boettcher 1970; see also Fig. 38), the pressure above which the θ < 60° decreases from about 1.2 GPa and 700 °C to about 0.8 GPa and 1200 °C. Above these temperature and pressure conditions, the θ = 60° isopleth intersects the incongruent melting curve of anorthite + H2O to yield corundum + melt (Boettcher 1970). It must be emphasized, however, that the experimental data in Fig. 38 extend from pure anorthite to only about 95% of the anorthite component in plagioclase. Extrapolation to lower An component concentration in plagioclase, therefore, is uncertain. It is, in fact, likely that the dihedral angle might decrease as the plagioclase becomes more albite-rich, because the solubility of NaAlSi3O8 in H2O fluid is likely greater than that of CaAl2Si2O8 in H2O (Anderson and Burnham 1983; Newton and Manning 2007). Such different solubilities, which depend on plagioclase composition, would mean that the increasing dihedral angle of in the plagioclase–H2O system in the initial high-An component composition range is likely to change to lowering of the angle between plagioclase and aqueous fluid as the plagioclase becomes more albitic.

Considerable experimental data exist for wetting behavior of aqueous fluid in contact with olivine. In the olivine–H2O system under crustal temperature and pressure conditions, the dihedral angle exceeds 60° (Mibe et al. 1998, 1999). However, this angle decreases relatively rapidly with increasing temperature. The angle decrease is particularly rapid at pressure and temperature conditions where the olivine–H2O system approach and perhaps exceed the supercritical temperature and pressure conditions (Mibe et al. 1998, 1999; Yoshino et al. 2007; Huang et al. 2020; see also Fig. 39).

Modified from Yoshino et al. (2007)
Dihedral angle of aqueous fluid in the olivine–H2O system as a function of pressure at temperatures indicated.
Addition of CO2 to aqueous fluid in equilibrium with olivine results in wetting angle changes that are qualitatively similar to adding CO2 to fluids in contact with other silicate minerals such as quartz, for example (Watson and Brenan 1987; Huang et al. 2020). In all cases, the dihedral angle increases as a systematic function of increasing CO2 concentration in the fluid (Huang et al. 2020). Most likely, decreasing Mg2SiO4 solubility in Mg2SiO4–H2O–CO2 fluids with increasing CO2/(CO2 + H2O) governs this dihedral angle evolution.
In the olivine–H2O–NaCl system, in contrast to the olivine–H2O–CO2 system, the dihedral angle decreases rapidly from above 70° in pure H2O at 1 GPa and 800 °C to less than 60° with 10 mol% NaCl and less in solution. However, little or no angle change was reported with 10–50% NaCl in the aqueous fluid (Liu et al. 2018). This dihedral angle trend with increasing salinity of aqueous fluid likely reflects complex solubility behavior of olivine in NaCl–H2O fluids perhaps involving a combination of chloride complexing together with formation of silicate complexes. A complex such as MgClOH suggested by Macris et al. (2020) for MgO dissolution in fluids in the MgO–H2O–NaCl system is one possibility because forsterite solubility in saline fluids increases with increasing chloride concentration (Macris et al. 2020). However, more complex species, perhaps including Si4+, could also exist in the H2O–NaCl fluids, but absent direct structural information, this (and perhaps other) possibilities cannot be evaluated with confidence.
The fluid wetting angles in pyroxene–H2O and garnet–H2O systems are significantly greater than those of aqueous fluid in contact with the main upper mantle mineral phase, olivine (Ono et al. 2002; Mibe et al. 2003; Liu et al. 2018). The angle in both systems remains at or above 60° at least to pressures near 5 GPa, but decreases rapidly as conditions approach those of the critical endpoint in the eclogite–H2O system.
The relationship between wetting angle, pressure, temperature, and solubility in H2O-rich fluid in a mantle environment would imply that the migration rate of aqueous fluid in a mantle wedge overlying a dehydrating subducting slab, will increase with increasing depth because the solubility of fluids in peridotite–H2O systems increases with increasing pressure such as discussed above (e g., Kawamoto et al. 2004; Melakhova et al. 2007). Furthermore, given that aqueous fluids in subduction zones commonly are saline and increasing salinity in model peridotite–H2O–chloride systems enhances the solubility in the fluid (Macris et al. 2020), this situation would further enhance the mobility of aqueous fluids in the mantle wedge above subducting plates.
2.5.1.2 Fluid wetting angles and rock properties
The temperature and pressure effects on wetting angles and connectivity of aqueous fluids in contact with minerals in the Earth's interior can have profound effects on geochemical properties of fluid-bearing upper mantle materials and mineral assemblages (Watson 1991; Brenan 1993; Iizuka and Mysen 1998; Bebout et al. 1999; Kawamoto et al. 2014). Wetting angle and fluid connectivity also can affect geophysical properties of fluid-bearing rock systems (Wiens et al. 2006; Reynard et al. 2011; Yoshino and Katsura 2013; Ogawa et al. 2014). Moreover, as wetting angle governs connectivity and wetting angle varies with temperature, pressure, and fluid composition, geochemical and geophysical properties that may be linked to fluid in rocks would also depend on those variables.
Geochemical properties of materials that have experienced fluid infiltration include trace and major element diffusion and abundance as well as possible isotopic changes (Watson 1991; Brenan 1993; Iizuka and Mysen 1998; Brenan et al. 1998; Lupulescu and Watson 1999; Manning 2004; Foustoukos and Mysen 2012; Dalou et al. 2015; Labidi et al. 2016). For example, diffusivity depends on volume fraction and composition of permeating fluids. The diffusivity of halogens through a rock sample containing H2O, for example, can vary by orders of magnitude depending on the volume of H2O even at small fluid concentrations (Fig. 40; see also Brenan 1993). Similarly, Watson (1991) documented how the diffusion constant for Fe in H2O and (H2O + CO2)-bearing mineral systems depends on both the proportion of fluid and its H2O/CO2 ratio.

Modified from Brenan (1993)
Chlorine diffusion in quartz-H2O as a function of total fluid concentration.
The evolution of fluid-sensitive trace elements such as Be, B, and Li in subduction zone settings is another example of fluid infiltration causing geochemical changes. Here, Brenan et al. (1998) determined their abundance and abundance ratio in aqueous fluids that were derived from dehydration of hydrous minerals (lawsonite and amphibole) in the subducting plate. They commented that decreased B/Be abundance ratio in the fluid with depth in subduction zones reflects decreasing H2O concentration in the subducting slab with depth (Poli and Schmidt 2002). What most likely happens is decreasing H2O/CO2 ratio in the fluid with increasing depth. That decrease could result in decreasing B/Be ratio in the fluid because of different solubilities of B and Be as a function of the H2O/CO2 ratio of this fluid, which, in turn would change mineral/fluid partition coefficients.
Electrical conductivity and seismic velocity are two important geophysical properties often employed to estimate fluid (and melt) distribution in the Earth. Electrical conductivity as a function of fluid fraction, salinity, and fluid connectivity have been calibrated experimentally (Shimojuku et al. 2012; Guo et al. 2016; Sun et al. 2020; Huang et al. 2021). Seismic velocities in subduction zones also have been used to estimate total H2O content (Carlson and Miller 2003; Hacker and Abers 2004). Fluid connectivity may also help explain relationships between electrical conductivity and seismic properties such as observed in subduction zones, for example (Fig. 41; see also Pommier 2014).

There exist high-conductivity layers in the Earth's deep crust (Guo et al. 2018). Layers of high electrical conductivity also have been reported from subduction zones (Wanamaker et al. 2009; Guo and Keppler 2019). This high electrical conductivity could result from on the order of 1% aqueous fluid with significant salinity. From experimental data on electrical conductivity in forsterite + H2O mixtures without chloride, Huang et al. (2021) concluded that the high electrical conductivity often reported from the mantle wedge above subducting plates could be accommodated by 5–10 volume % aqueous fluid in the wedge (Fig. 42). Of course, were the fluid saline, as is often suggested (Kawamoto et al. 2014; Kumagai et al. 2014), the volume fraction of fluid could be smaller. A significantly smaller fraction of aqueous fluid (perhaps less than 1%) would be consistent with modeling results from Iwamori (2007), for example. Water concentrations in the 0.5–1 wt% range in the source regions of andesitic magma in this mantle wedge would also be consistent with results of melting experiments on hydrous peridotite mantle (Till et al. 2012).

Modified from Huang et al. (2021)
Electrical conductivity of forsterite + H2O as a function of fluid volume. Also shown is a range of conductivity anomalies in mantle wedges above subducting plates.
3 Concluding remarks
Fluids and magmatic liquids are the dominant transport media in the Earth. Complete miscibility between fluid and melt in silicate–H2O systems can be found at pressures and temperatures in excess of about 1 GPa and 800 °C for granite–H2O. These pressure and temperature coordinates increase as a system becomes more mafic and reaches conditions of the lowermost upper mantle for peridotite–H2O. Under such conditions, fluids and melts are indistinguishable.
Fluids in the Earth dominantly are compositions in the system C–O–H–N–S. Other volatiles that sometimes occur in significant proportions include halogens, and in particular F and Cl, and noble gases. Halogens can affect physical and chemical properties of both magma and crystalline materials, whereas noble gases likely do not affect most properties significantly.
Several of the C–O–H–N–S components can exist in different oxidation states within the redox range of the silicate Earth. Oxidized species in fluids are H2O, CO2, N2, SO2, and SO3. Anionic complexes such as OH−, CO32−, HCO3−, and SO42− groups often coexist with the molecular species in terrestrial fluids and dissolved in magmatic liquids. Reduced terrestrial fluid are CH3−,NH2−, NH3, S2−, and HS− with those reduced anionic species often coexisting with the reduced molecular species, H2, CH4, NH3, and H2S. H2O dominates most environments whether under oxidizing or reducing conditions.
Among the typical fluid species, H2O tends to be the most efficient solvent of major, minor, and trace elements at high temperature and pressure. The solution capacity of aqueous fluids sometimes is enhanced further by dissolved halogens and sulfur. In contrast, addition of CO2 or nitrogen species to aqueous fluids has the opposite effect.
The solubility in aqueous solutions of minor and trace elements such as, for example, Ti, Zr, and Hf as well as other HFSE can be significantly affected by alkali metals by forming metal oxyanion complexes. Formation of aluminate complexes will enhance the solubility of Al2O3 in aqueous fluids in a similar manner. Such complexes can be 5–6 orders of magnitude more soluble in aqueous solutions compared with the solubility of the elements in their cation or simple oxide form. It is also likely that the solubility of such complexes increases the more electropositive the metal cation associated with the oxyanion complex(es).
Fluid-mediated transport is accomplished with fluid passing through cracks and through percolation channels along grain boundaries. Percolation velocity is linked to permeability, which, in turn, is governed by rock porosity. Finally, porosity is controlled by wetting angles, θ, at the interface between fluid and the mineral surfaces of surrounding rocks. This angle is negatively correlated with the solubility of silicate components in the fluids. When θ < 60°, the fluid will wet all grain boundaries of an isotropic crystalline material thus leading to enhanced mass transport ability, whereas when greater than 60°, grain boundary wetting does not occur and fluid-mediated transport is diminished. With anisotropic crystal structures, the wetting angles for individual crystal surfaces will vary depending on the properties of the specific surface.
For fluids, the compositions of which are dominated by H2O, CO2, and salts such as chlorides, the θ is the greatest for CO2 fluids and the smallest for brines (H2O + salt). Essentially all CO2 fluids in contact with silicate minerals exhibit θ > 60° and would not, therefore, result in wetting of grain boundaries. CO2-rich fluids are not, therefore, efficient mass transport media in the Earth. This could be the situation during granulite metamorphism, for example, because the principal fluid component in granulite facies rocks tends to be CO2-rich (e.g., Touret et al. 2011). In the continental upper mantle, CO2 also is major fluid component, so wetting by fluid in such tectonic settings is not likely. With H2O and H2O + chlorides, however, the θ < 60° so that complete wetting of grain boundaries is common. This situation exists under lower grade metamorphism and during fluid transport in of subduction zones (typically < 100 km depth).
Geophysical and geochemical anomalies in the Earth's interior can be linked to the presence of fluids and, in particular, the extent to which fluids wet grain boundaries. For example, the geochemistry of the mantle wedge above subduction zones can be affected in this manner. Similarly, fluid infiltration will lead to enhanced electrical conductivity and enhanced seismic wave attenuation such as often reported near convergent plate boundaries.
Availability of data and materials
Not applicable.
Notes
In the Qn-notation, the superscript, n, denotes the number of bridging oxygen in the silicate species. This means that the equivalent species for Q0, Q1, and Q2 are SiO4, SiO3.5, and SiO3, respectively. It also means that the greater the value of n, the more polymerized is the silicate network of the Qn-species.
Equation (9) does not consider the speciation of dissolved SiO2 in aqueous fluid, but whether this is done or not, any decrease in the fugacity of H2O, fH2O, would shift such a reaction to the left.
This structural feature has been documented in silicate melts with the aid of MAS NMR spectroscopy of silicate melts, for example (Lee and Stebbins, 2003).
The 250˚-300˚C for the proposed minimum solubility is at temperatures below the lower temperature limit of corundum in the Al2O3–H2O system (Kennedy, 1959). Walther (1997) reported, however, corundum with fluid to temperatures as low as 272˚C. The apparent conflict with the phase equilibrium data of Kennedy (1959) does not seem to be resolved.
Abbreviations
- GPa:
-
Gigapascal
- MPa:
-
Megapascal
- θ :
-
Wetting angle
- ϕ :
-
Porosity
- γ :
-
Surface energy
- H :
-
Enthalpy
- D i fluid/melt :
-
Partition coefficient of i between fluid and melt
- T :
-
Temperature
- P :
-
Pressure
References
Allegre C, Manhes G, Lewin E (2001) Chemical composition of the Earth and the volatility control on planetary genetics. Earth Planet Sci Lett 185:49–69
Anderson GM, Burnham CW (1965) The solubility of quartz in supercritical water. Amer J Sci 263:494–511
Anderson GM, Burnham CW (1967) Reactions of quartz and corundum with aqueous chloride and hydroxide solutions and high temperatures and pressures. Amer J Sci 265:12–27
Anderson GM, Burnham CW (1983) Feldspar solubility and the transport of aluminum under metamorphic conditions. Amer J Sci 283A:283–297
Antignano AM, Manning CE (2008) Rutile solubility in H2O, H2O–SiO2, and H2O–NaAlSi3O8 fluids at 0.7–2.0 GPa and 700–1000 °C: implications for mobility of nominally insoluble elements. Chem Geol 255:283–293
Aranovich LY, Newton RC (1999) Experimental determination of CO2–H2O activity-composition relations at 600–1000 °C and 6–14 kbar by reversed decarbonation and dehydration reactions. Amer Mineral 84:1319–1332
Archie GE (1942) The electrical resistivity log as an aid in determining some reservoir characteristics. Trans AIME 146:54–62
Aubaud CF, Pineau F, Hekinian R, Javoy M (2005) Degassing of CO2 and H2O in submarine lavas from the society hotspot. Earth Planet Sci Lett 235:511–527
Audetat A, Edmonds M (2020) Magmatic-hydrothermal fluids. Elements 16:401–406
Ayers JC, Wason EB (1993a) Rutile solubility and mobility in supercritical aqueous fluids. Contrib Mineral Petrol 114:321–330
Ayers JC, Watson EB, Harrison TM, Miller CF, Ryerson FJ (1993b) Apatite/fluid partitioning of rare-earth elements and strontium: experimental results at 1.0 GPa and 1000 °C and application to models of fluid-rock interaction. Chem Geol 110:299–314
Azaroual M, Pascal ML, Roux J (1996) Corundum solubility and aluminum speciation in KOH aqueous solutions at 400 °C from 0.5 to 2.0 kbar. Geochim Cosmochim Acta 60:4601–4614
Baier J, Audétat A, Keppler H (2008) The origin of the negative niobium tantalum anomaly in subduction zone magmas. Earth Planet Sci Lett 267:290–300
Bailey EH, Ragnarsdottir KV (1994) Uranium and thorium solubilities in subduction zone fluids. Earth Planet Sci Lett 124:119–129
Bali E, Audetat A, Keppler H (2011) The mobility of U and Th in subduction zone fluids: an indicator of oxygen fugacity and fluid salinity. Contrib Mineral Petrol 161:597–613. https://doi.org/10.1007/s00410-010-0552-9
Bali E, Keppler H, Audetat A (2012) The mobility of W and Mo in subduction zone fluids and the Mo–W–Th–U systematics of island arc magmas. Earth Planet Sci Lett 351:195–207
Bassett WA, Shen AH, Bucknum M, Chou IM (1994) A new diamond cell for hydrothermal studies to 2.5 GPa and from −190 °C to 1200 °C. Rev Sci Instrum 64:2340–2345
Bebout GE, Ryan JG, Leeman WP, Bebout AE (1999) Fractionation of trace elements by subduction zone metamorphism-effect of convergent-margin thermal evolution. Earth Planet Sci Lett 171:63–81
Bebout GE, Fogel ML, Cartigny P (2013) Nitrogen: highly volatile yet surprisingly compatible. Elements 9:333–338
Becker KH, Cemič L, Langer KEOE (1983) Solubility of corundum in supercritical water. Geochim Cosmochim Acta 47:1573–1578
Beermann O, Botcharnikov RE, Nowak M (2015) Partitioning of sulfur and chlorine between aqueous fluid and basaltic melt at 1050 degrees C, 100 and 200 MPa. Chem Geol 418:132–157
Binder B, Keppler H (2011) The oxidation state of sulfur in magmatic fluids. Earth Planet Sci Lett 301:190–198
Boettcher AL (1970) The system CaO–Al2O3–SiO2–H2O at high pressures and temperatures. J Petrol 11:337–379
Botcharnikov RE, Linnen LE, Wilke M, Holtz F, Jugo PJ, Berndt J (2011) High gold concentrations in sulphide-bearing magma under oxidizing conditions. Nature Geosci 4:112–115
Botcharnikov RE, Holtz F, Behrens H, Sanchez-Valle C, Gaillard F, Ghosh S, Mezger M (2015) Solubility and fluid-melt partitioning of H2O and Cl in andesitic magmas as a function of pressure between 50 and 500 MPa. Chem Geol 418:117–131
Botti AF, Bruni F, Inmberti S, Ricci MA, Soper AKS (2004) Ions in water: the microscopic structure of a concentrated HCl solution. J Chem Phys 121:7840–7848
Bouhifd MA, Whittington A, Roux J, Richet P (2006) Effect of water on the heat capacity of polymerized aluminosilicate glasses and melts. Geochim Cosmochim Acta 70:711–722
Brenan JM (1993) Diffusion of chlorine in fluid-bearing quartzite: effects of fluid composition and total porosity. Contrib Mineral Petrol 115:215–224
Brenan JM, Shaw HF, Phinney DL, Ryerson FJ (1994) Rutile-aqueous fluid partitioning of Nb, Ta, Hf, Zr, U and Th: implications for high field strength element depletions in island-arc basalts. Earth Planet Sci Lett 128:327–329
Brenan JM, Ryerson FJ, Shaw HF (1998) The role of aqueous fluids in the slab-to-mantle transfer of boron, beryllium, and lithium during subduction: experiments and models. Geochim Cosmochim Acta 62:3337–3347
Buckermann WA, Muller-Warmuth W, Frischat GH (1992) A further 29Si MAS NMR study on binary alkali silicate glasses. Glastechn Ber 65:18–21
Bureau H, Keppler H (1999) Complete miscibility between silicate melts and hydrous fluids in the upper mantle: experimental evidence and geochemical implications. Earth Planet Sci Lett 165:187–196
Bureau H, Keppler H, Metrich N (2000) Volcanic degassing of bromine and iodine: experimental fluid/melt partitioning data and applications to stratospheric chemistry. Earth Planet Sci Lett 183:51–60
Burnham CW, Holloway JR, Davis NF (1969) Thermodynamic properties of water to 1000 °C and 10,000 bars. Geol Soc America Spec Paper 132:96p
Busigny V, Cartigny P, Philippot P (2011) Nitrogen in ophiolitic meta-gabbros: a reevaluation of modern nitrogen fluxes in subduction zones and implication for the early Earth atmosphere. Geochim Cosmochim Acta 75:7502–7221
Carlson RL, Miller DJ (2003) Mantle wedge water contents estimated from seismic velocities in partially serpentinized peridotites. Geophys Res Lett. https://doi.org/10.1029/2002GL016600
Carmichael DM (1969) On the mechanism of prograde metamorphic reactions in quartz-bearing pelitic rocks. Contrib Mineral Petrol 20:244–267
Carmichael ISE, Ghiorso MS (1990) Controls on oxidation-reduction relations in magmas. In: Nicholls J, Russell JK (eds) Modern methods of igneous petrology: understanding magmatic processes. The Mineralogical Society of America, Washington, DC, pp 191–212
Chevychelov VY, Botcharnikov RE, Holtz F (2008) Experimental study of chlorine and fluorine partitioning between fluid and subalkaline basaltic melt. Dokl Earth Sci 422:1089–1092
Chu X, Ague JJ (2013) Phase equilibria for graphitic metapelite including solution of CO2 in melt and cordierite: implications for dehydration, partial melting and graphite precipitation. J Metamorph Geol 31:843–862
Cody GD, Mysen BO, Lee SK (2005) Structure vs. composition: a solid state 1H and 29Si NMR study of quenched glasses along the Na2O-SiO2-H2O join. Geochim Cosmochim Acta 69:2373–2384
Cohen TH, Watson EB (1996) Permeability evolution of partially molten mantle with textural maturation (abstr). AGU 1996 fall meeting, American Geophysical Union, Washington, DC, United States, p 840
Connolly JAD (2005) Computation of phase equilibria by linear programming: a tool for geodynamic modeling and its application to subduction zone decarbonation. Earth Planet Sci Lett 236:524–541
Cruz MF, Manning CE (2015) Experimental determination of quartz solubility and melting in the system SiO2–H2O–NaCl at 15–20 kbar and 900–1100 degrees C: implications for silica polymerization and the formation of supercritical fluids. Contrib Mineral Petrol 170(4):1–17. https://doi.org/10.1007/s00410-015-1187-7
Currie KL (1968) On the solubility of albite in supercritical water in the range 400 to 600 °C and 750 to 3500 bars. Amer J Sci 266:321–341
D’Souza RJ, Canil D (2018) The partitioning of chalcophile elements between sediment melts and fluids at 3 GPa, 950–1050 °C with implications for slab fluids in subduction zones. Earth Planet Sci Lett 49:215–225
Daines MJ, Kohlstedt DL (1997) Influence of deformation on melt topology in peridotites. J Geophys Res 102:10257–10271
Dalou C, Mysen BO (2015) The effect of H2O on F and Cl solubility and solution mechanisms of in aluminosilicate melts at high pressure and high temperature. Amer Mineral 100:633–643
Dalou C, Le Losq C, Mysen BO, Cody GD (2015) Solubility and solution mechanisms of chlorine and fluorine in aluminosilicate melts at high pressure and high temperature. Amer Mineral 100:2272–2283
Deering CE, Cairns EC, McIsaac JD, Read AS, Marriott RA (2016) The partial molar volumes for water dissolved in high-pressure carbon dioxide from T = (318.28 to 369.40) K and pressures to P=35 MPa. J Chem Thermodyn 93:337–346
Dolejs D, Zajacz Z (2018) Halogens in silicic magmas and their hydrothermal systems. In: Harlov DE, Aranovich L (eds) The role of halogens in terrestrial and extraterrestrial geochemical processes. Springer, Cham, pp 431–543
Duan Z, Zhang Z (2006) Equation of state of the H2O, CO2, and H2O–CO2 systems up to 10 GPa and 2573.15K: molecular dynamics simulations with ab initio potential surface. Geochim Cosmochim Acta 70:2311–2324
Dullien FAL (1992) Porous media: fluid transportation and flow structure. Academic Press, San Diego
Eggler DH, Baker DR (1982) Reduced volatiles in the system C–H–O: implications to mantle melting, fluid formation and diamond genesis. In: Reidel D (ed) High-pressure research in geophysics. Kluwer Academic Publishers, Boston/Dordrecht, pp 237–250
Eggler DH, Kadik AA (1979) The system NaAlSi3O8–H2O–CO2: I. Compositional and thermodynamic relations of liquids and vapors coexisting with albite. Amer Mineral 64:1036–1049
Eldridge DL, Mysen BO, Cody GD (2018) Experimental estimation of the bisulfite isomer quotient as a function of temperature using Raman spectroscopy: implications for sulfur isotope fractionations in aqueous sulfite solutions. Geochim Cosmochim Acta 220:309–328
Elliott T, Plank T, Zindler A, While W, Bourdon B (1997) Element transport from slab to volcanic front at the Mariana arc. J Geophys Res 102:14991–15019
Evans KA, Tomkins AG (2020) Metamorphic fluids in orogenic settings. Elements 16:381–387
Foley SE, Barth EMG, Jenner CA (2000) Rutile/melt partition coefficients for trace elements and an assessment of the influence of rutile on the trace element characteristics of subduction zone magmas. Geochim Cosmochim Acta 64:993–938
Fournier RO, Potter RW (1982) An equation correlating the solubility of quartz in water from 25° to 900 °C at pressures up to 10,000 bars. Geochim Cosmochim Acta 46:1969–1973
Foustoukos DI, Mysen BO (2012) D/H isotope fractionation in the H2–H2O system at supercritical water conditions: compositional and hydrogen bonding effects. Geochim Cosmochim Acta 86:88–102
Franck EU (1973) Concentrated electrolyte solutions at high temperatures and pressures. J Solut Chem 2:339–353
Frank MR, Simon AC, Pettke T, Candela PA, Piccoli PM (2011) Gold and copper partitioning in magmatic-hydrothermal systems at 800 °C and 100 MPa. Geochim Cosmochim Acta 75:2470–2482
Frantz JD (1998) Raman spectra of potassium carbonate and bicarbonate aqueous fluids at elevated temperatures and pressures: comparison with theoretical simulations. Chem Geol 152:211–225
Frantz JD, Dubessy J, Mysen BO (1993) An optical cell for Raman spectroscopic studies of supercritical fluids and its application to the study of water to 500 C and 2000 bar. Chem Geol 106:9–26
Frost DJ, McCammon CA (2008) The redox state of the Earth’s mantle. Ann Rev Earth Planet Sci 36:389–420
Frost DJ, Wood BJ (1997) Experimental measurements of the properties of H2O–CO2 mixtures at high pressures and temperatures. Geochim Cosmochim Acta 61:3301–3310
Gennaro EA, Paonita G, Iacono-Marziano Y, Moussallam Y, Pichavant M, Peters N, Martel C (2020) Sulphur behavior and redox conditions in Etnean magmas during magma differentiation and degassing. J Petrol 61(10):egaa095. https://doi.org/10.1093/petrology/egaa095
Gessmann CK, Rubie DC (2000) The origin of the depletions of V, Cr and Mn in the mantles of the Earth and Moon. Earth Planet Sci Lett 184:95–187
Gibert F, Pascal ML, Pichavant M (1998) Gold solubility and speciation in hydrothermal solutions: experimental study of the stability of hydrosulphide complex of gold (AuHS°) at 350 to 450 °C and 500 bars. Geochim Cosmochim Acta 62:2931–2947
Goldsmith JR, Newton RC (1977) Scapolite-plagioclase stability relations at high pressures and temperatures in the system NaAlSi3O8-CaAl2Si2O8-CaCO3-CaSO4. Amer Mineral 62:1063–1081
Gorbaty YE, Kalinichev AG (1995) Hydrogen bonding in supercritical water. 1. Experimental results. J Phys Chem 99:5336–5340
Grove TL, Till CB, Krawczynski MJ (2012) The role of H2O in subduction zone magmatism. Ann Rev Earth Planet 40:413–439
Guo H, Keppler H (2019) Electrical conductivity of NaCl-bearing aqueous fluids to 900 °C and 5GPa. J Geophys Res Solid Earth 124(2):1397–1411. https://doi.org/10.1029/2018jb016658
Guo X, Chen Q, Ni H (2016) Electrical conductivity of hydrous silicate melts and aqueous fluids: measurement and applications. Sci China Earth Sci 59:889–900
Guo H, Audetat A, Dolejs D (2018) Solubility of gold in oxidized, sulfur-bearing fluids at 500–850 °C and 200–230 MPa: a synthetic fluid inclusion study. Geochim Cosmochim Acta 222:655–670
Hacker BR, Abers GA (2004) Subduction factory 3: an excel worksheet and macro for calculating the densities, seismic wave speeds, and H2O contents of minerals and rocks at pressure and temperature. Geochem Geophys Geosyst,. https://doi.org/10.1029/2003GC000614
Hacker BR, Abers GA, Peacock SM (2003) Subduction factory: 1. Theoretical mineralogy, densities, seismic wave speeds, and H2O contents. J Geophys Res B 108:2029. https://doi.org/10.1029/2001JB001127
Hallam M, Eugster HP (1976) Ammonium silicate stability relations. Contrib Mineral Petrol 57:227–244
Harlov DE (2015) Apatite: a fingerprint for metasomatic processes. Elements 11:171–176
Hay RS, Evans B (1988) Intergranular distribution of pure fluid and the nature of high-angle grain boundaries in limestone and marble. J Geophys Res 93:8959–8974
Hayden LA, Manning CE (2011) Rutile solubility in supercritical NaAlSi3O8–H2O fluids. Chem Geol 284:74–81
Henry DJ, Daigle NM (2018) Chlorine incorporation into amphibole and biotite in high-grade iron-formations: interplay between crystallography and metamorphic fluids. Amer Mineral 103:55–68
Heuft JM, Meijer EJ (2003) Density functional theory based nilecylar-dynamics study of aqueous chloride solvation. J Chem Phys 119:11788–11791
Hoffmann MM, Conradi MS (1997) Are there hydrogen bonds in supercritical water? J Amer Chem Soc 119:3811–3817
Holness MB (1992) Equilibrium dihedral angles in the system quartz-CO2-H2O-NaCl at 800 °C and 1–15 kbar: the effects of pressure and fluid composition on the permeability of quartzites. Earth Planet Sci Lett 114:171–184
Holness MB (1993) Temperature and pressure dependence of quartz-aqueous fluid dihedral angles: the control of adsorbed H2O on the permeability of quartzites. Earth Planet Sci Lett 117:363–377
Holness MB (1995) The effect of feldspar on quartz-H2O–CO2 dihedral angles at 4 kbar, with consequences for the behaviour of aqueous fluids in migmatites. Contrib Mineral Petrol 118:356–364
Holness MB (1997) Surface chemical controls on pore-fluid connectivity in texturally equilibrated materials. In: Jamtveit B, Yardley BWD (eds) Fluid flow and transport in rocks: mechanisms and effects. Springer, Dordrecht, pp 149–169
Holness MB, Graham CM (1995) P-T-X effects on equilibrium carbonate-H2O-CO2-NaCl dihedral angles: constraints on carbonate permeability and the role of deformation during fluid infiltration. Contrib Mineral Petrol 119:301–313
Hsu Y-J, Zajacz Z, Ulmer P, Heinrich CA (2019) Chlorine partitioning between granitic melt and H2O-CO2-NaCl fluids in the Earth’s upper crust and implications for magmatic-hydrothermal ore genesis. Geochim Cosmochim Acta 261:171–190
Huang Y, Nakatani T, Nakamura M, McCammon CA (2020) Experimental constraint on grain-scale fluid connectivity in subduction zones. Earth Planet Sci Lett 552:116610. https://doi.org/10.1016/j.epsl.2020.116610
Huang Y, Guo H, Nakatani T, Uesugi K, Nakamura M, Keppler H (2021) Electrical conductivity in texturally equilibrated fluid-bearing forsterite aggregates at 800 °C and 1 GPa: implications for the high electrical conductivity anomalies in mantle wedges. J Geophys Res B 126(4):e2020JB021343. https://doi.org/10.1029/2020JB021343
Hunt JD, Manning CE (2012) A thermodynamic model for the system SiO2–H2O near the upper critical end point based on quartz solubility experiments at 500–1100 °C and 5–20 kbar. Geochim Cosmochim Acta 86:196–213
Hurtig NC, Williams-Jones AE (2014) An experimental study of the solubility of MoO3 in aqueous vapour and low to intermediate density supercritical fluids. Geochim Cosmochim Acta 136:169–193
Hustoft JW, Kohlstedt DL (2006) Metal-silicate segregation in deforming dunitic rocks. Geochem Geophys Geosyst. https://doi.org/10.1029/2005GC001048
Iacono-Marziano G, Morizet Y, Le Trong E, Gaillard F (2012) New experimental data and semi-empirical parameterization of H2O-CO2 solubility in mafic melts. Geochim Cosmochim Acta 97:1–23
Iizuka Y, Mysen BO (1998) Experimental study on dehydration and silica metasomatism in the subduction zones. Terra Nova 10(1):28
Iveson AA, Wesbster JD, Rowe MC, Neill OK (2017) Major element and halogen (F, Cl) mineral-melt-fluid partitioning in hydrous rhyodacitic melts at shallow crustal conditions. J Petrol 58:2465–2492
Iwamori H (2007) Transportation of H2O beneath the Japan arcs and its implications for global water circulation. Chem Geol 239:182–198
Iwamori H, Richardson HC, Maruyama S, Santosh M (2007) Numerical modeling of thermal structure, circulation of H2O, and magmatism-metamorphism in subduction zones; implications for evolution of arcs. Gondwana Res 11:109–119
Jambon A (1994) Earth degassing and large-scale geochemical cycling of volatile elements. Rev Mineral Geochem 30:479–517
Jugo PJ (2009) Sulfur content at sulfur saturation in oxidized magmas. Geology 37:415–418
Jugo PJ, Wilke M, Botcharnikov RE (2010) Sulfur K-edge XANES analysis of natural and synthetic basaltic glasses: implications for S speciation and S content as function of oxygen fugacity. Geochim Cosmochim Acta 74:5926–5938
Jurewicz SR, Watson EB (1985) The distribution of partial melt in a granitic system: the application of liquid phase sintering theory. Geochim Cosmochim Acta 49:1109–1121
Kaminsky FV, Wirth R (2017) Nitrides and carbonitrides from the lowermost mantle and their importance in the search for Earth’s “lost” nitrogen. Amer Mineral 102:1667–1676
Katayama Y, Saitoh HT, Ikeda T, Aoki K (2010) Structure of liquid water under high pressure up to 1.7 GPa. Phys Rev B 81(1):014109. https://doi.org/10.1103/PhysRevB.81.014109
Kawamoto T, Matsukage KN, Mibe K, Isshiki M, Nishimura K, Ishimatsu OS (2004) Mg/Si ratios of aqueous fluids coexisting with forsterite and enstatite based on phase relations in the Mg2SiO4-SiO2-H2O system. Amer Mineral 89:1433–1437
Kawamoto TN, Yoshikawa Y, Kumagai MHT, Mirabueno M, Okuno, Kobayashi, M (2013) Mantle wedge infiltrated with saline fluids from dehydration and decarbonation of subducting slab. Proc Natl Acad Sci USA 110:9663–9668
Kawamoto T, Mibe K, Bureau H, Reguer S, Mocuta C (2014) Large-ion lithophile elements delivered by saline fluids to the sub-arc mantle. Earth Planet Space 66:1–11. https://doi.org/10.1186/1880-5981-66-61
Kennedy GC (1950) Pressure-volume-temperature relations in water at elevated temperatures and pressures. Amer J Sci 248:540–564
Kennedy GC (1959) Phase relations in the system Al2O3-H2O at high temperature and pressure. Amer J Sci 257:563–573
Kennedy GC, Wasserburg GJ, Heard HC, Newton RC (1962) The upper three-phase region in the system SiO2-H2O. Amer J Sci 260:501–521
Keppler H (1996) Constraints from partitioning experiments on the composition of subduction-zone fluids. Nature 380:237–240
Keppler H (2017) Fluids and trace element transport in subduction zones. Amer Mineral 102:5–20
Keppler H, Wyllie PJ (1991) Partitioning of Cu, Sn, Mo, W, U, and Th between melt and aqueous fluid in the systems haplogranite-H2O-HCl and haplogranite-H2O-HF. Contrib Mineral Petrol 109:139–150
Kerrick DM (1990) The Al2SiO5 polymorphs. Rev Mineral Geochem 22:406
Kessel R, Ulmer P, Pettke T, Schmidt MW, Thompson AB (2005) The water–basalt system at 4 to 6 GPa: phase relations and second critical end-point in a K-free eclogite at 700 to 1400 °C. Earth Planet Sci Lett 237:873–892
Kilinc IA, Burnham CW (1972) Partitioning of chloride between a silicate melt and coexisting aqueous phase from 2 to 8 kilobars. Econ Geol 67:231–235
Klein-BenDavid O, Pettke T, Kessel R (2011) Chromium mobility in hydrous fluids at upper mantle conditions. Lithos 125:122–130 (Oslo)
Kohlstedt DL, Keppler H, Smyth JR (2006) The role of water in high-temperature rock deformation. Rev Mineral Geochem 62:377–396
Konzett J, Fei Y (2000) Transport and storage of potassium in the Earth’s upper mantle and transition zone: an experimental study to 23 GPa in simplified and natural bulk compositions. J Petrol 41:583–603
Konzett J, Ulmer P (1999) The stability of hydrous potassic phases in lherzolitic mantle—an experimental study to 9.5 GPa in simplified and natural bulk compositions. J Petrol 40:629–652
Korenaga J, Kelemen PB (1998) Melt migration through the oceanic lower crust: a constraint from melt percolation modeling with finite solid diffusion. Earth Planet Sci Lett 156:1–11
Kravchuk IF, Kotelnikov AR, Senin VG (2004) Partitioning of volatile (Cl, F, and S) and rare alkali (Rb and Cs) elements in the system aluminosilicate melt-fluid. Geochem Int 42:1071–1077
Kumagai Y, Kawamoto T, Yamamoto J (2014) Evolution of carbon dioxide-bearing saline fluids in the mantle wedge beneath the northeast Japan Arc. Contrib Mineral Petrol 168:1–13
Kushiro I (1972) Effect of water on the composition of magmas formed at high pressures. J Petrol 13:311–334
Labidi J, Shahar A, Losq Le, Hillgren V, Mysen BO, Farquhar J (2016) Experimentally determined sulfur isotope fractionation between metal and silicate and implications for planetary differentiation. Geochim Cosmochim Acta 175:181–194
Laporte D, Watson EB (1991) Direct observation of near-equilibrium pore geometry in synthetic quartzites at 600 °C–800 °C and 2–10.5 kbar. J Petrol 99:873–878
Lee SK, Stebbins JF (2003) Nature of cation mixing and ordering in Na-Ca silicate glasses and melts. J Phys Chem B 107:3141–3148
Liu X, Matsukage KN, Li Y, Takahashi E, Suzuki T, Xiong X (2018) Aqueous fluid connectivity in subducting oceanic crust at the mantle transition zone conditions. J Geophys Res B 123:6562–6573
Lupulescu A, Watson EB (1999) Low melt fraction connectivity of granitic and tonalitic melts in a mafic crustal rock at 800 °C and 1 GPa. Contrib Mineral Petrol 134:202–216
Macris CA, Newton RC, Wykes J, Pan R, Manning CE (2020) Diopside, enstatite and forsterite solubilities in H2O and H2O-NaCl solutions at lower crustal and upper mantle conditions. Geochim Cosmochim Acta 279:119–142
Maekawa H, Maekawa T, Kawamura K, Yokokawa T (1991) The structural groups of alkali silicate glasses determined from 29Si MAS-NMR. J Non-Cryst Solids 127:53–64
Manning CE (1994) The solubility of quartz in H2O in the lower crust and upper mantle. Geochim Cosmochim Acta 58:4831–4840
Manning CE (2004) The chemistry of subduction-zone fluids. Earth Planet Sci Lett 223:1–16
Manning CEE (2007) Solubility of corundum plus kyanite in H2O at 700 °C and 10 kbar: evidence for Al-Si complexing at high pressure and temperature. Geofluids 7:258–269
Manning CE, Aranovich LY (2014) Brines at high pressure and temperature: Thermodynamic, petrologic and geochemical effects. Precambrian Res 253:6–16
Manning CE, Frezzotti ML (2020) Subduction-zone fluids. Elements 16:395–400
Manning CE, Shock EL, Sverjensky DA (2013) The chemistry of carbon in aqueous fluids at crustal and upper-mantle conditions: experimental and theoretical constraints. Rev Mineral Geochem 75:109–148
Martinez I, Sanchez-Valle C, Daniel RB (2004) High-pressure and high-temperature Raman spectroscopy of carbonate ions in aqueous solution. Chem Geol 207:47–58
Matsukage KN, Hashimoto M, Nishihara Y (2017) Morphological stability of hydrous liquid droplets at grain boundaries of eclogite minerals in the deep upper mantle. J Mineral Petrol Sci 112:346–358
McDonough WF, Sun SS, Arndt NT, Shirey S (1995) The composition of the Earth. Chem Geol 120:223–253
McLelland J, Morrison J, Selleck B, Cunningham B, Olson C, Schmidt K (2002) Hydrothermal alteration of late- to post-tectonic Lyon Mountain granitic gneiss, Adirondack mountains, New York: origin of quartz-sillimanite segregations, quartz-albite lithologies, and associated Kiruna-type low-Ti Fe oxide deposts. J Metamorph Geol 20:175–190
McMillan P (1984) Structural studies of silicate glasses and melts—applications and limitations of Raman spectroscopy. Amer Mineral 69:622–644
Melekhova E, Schmidt MW, Ulmer P, Pettke T (2007) The composition of liquids coexisting with dense hydrous magnesium silicates at 11–13.5GPa and the endpoints of the solidi in the MgO–SiO2–H2O system. Geochim Cosmochim Acta 71:3348–3360
Metrich N, Rutherford MJ (1992) Experimental study of chlorine behavior in hydrous silicic melts. Geochim Cosmochim Acta 56:607–616
Mibe K, Fujii YA (1999) Control of the location of the volcanic front in island arcs by aqueous fluid connectivity in the mantle wedge. Nature 401:259–262
Mibe K, Fujii T, Yasuda A (1998) Connectivity of aqueous fluid in the Earth’s upper mantle. Geophys Res Lett 25:1233–1236
Mibe K, Fujii T, Masuda A (2002) Composition of aqueous fluid coexisting with mantle minerals at high pressure and its bearing on the differentiation of the Earth’s mantle. Geochim Cosmochim Acta 66:2273–2285
Mibe K, Yoshino T, Ono S, Yasuda A, Fujii T (2003) Connectivity of aqueous fluid in eclogite and its implications for fluid migration in the Earth's interior. J Geophys Res 108. https://doi.org/10.1029/2002JB001960
Mibe K, Kanzaki M, Kawamoto T, Matsukage KN, Fei Y, Ono S (2007) Second critical endpoint in the peridotite-H2O system. J Geophys Res B. https://doi.org/10.1029/2005jb004125
Mibe K, Kawamoto T, Matsukage KN, Fei Y, Ono S (2011) Slab melting versus slab dehydration in subduction-zone magmatism. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.1010968108
Moecher DP, Essene EJ (1990) Phase equilibria for calcic scapolite, and implications of variable Al-Si disorder for P-T-XCO2, and a-X relations. J Petrol 31:997–1024
Moretti R, Metrich N, Arienzo I, Di Renzo V, Aiuppa A, Allard P (2018) Degassing vs. eruptive styles at Mt. Etna Volcano (Sicily, Italy); Part I, volatile stocking, gas fluxing, and the shift from low-energy to highly explosive basaltic eruptions. Chem Geol 482:1–17
Morey GW, Hesselgesser JM (1951) The solubility of quartz and some other substances in superheated steam at high pressures. Amer Soc Mech Engin Trans 73(7):865–875
Morrissey LJ, Tomins AJ (2020) Evaporite-bearing orogenic belts produce ligand-rich and diverse metamorphic fluids. Geochim Cosmochim Acta 275:163–187
Mu S, Faul UH (2016) Grain boundary wetness of partially molten dunite. Contrib Mineral Petrol 171(5):1–15. https://doi.org/10.1007/s00410-016-1250-z
Mysen BO (2010) Speciation and mixing behavior of silica-saturated fluid at high temperature and pressure. Amer Mineral 95:1807–1816
Mysen BO (2012) High-pressure-temperature titanium solution mechanisms in silicate-saturated aqueous fluids and hydrous silicate melts. Amer Mineral 97:1241–1251
Mysen BO (2015a) Carbon speciation in silicate-C-O-H as a function of redox conditions: an experimental study, in-situ to 1.7 GPa and 900 °C. Amer Mineral 100:872–882
Mysen BO (2015b) Redox-controlled solution mechanisms and hydrogen isotope fractionation in silicate-COH melt+fluid to upper mantle temperature and pressure. J Geophys Res B 120:7440–7459. https://doi.org/10.1002/2015JB011954
Mysen BO (2015c) An in-situ experimental study of Zr4+ transport capacity of water-rich fluids in the temperature and pressure range of the deep crust and upper mantle. Prog Earth Planet Sci. https://doi.org/10.1186/s40645-015-0070-5
Mysen BO (2018) Mass transfer in the Earth’s interior: fluid-melt interaction in aluminosilicate-C–O–H–N systems at high pressure and temperature under oxidizing conditions. Proc Earth Planet Sci 5(6):1–18. https://doi.org/10.1186/s40645-017-0161-6
Mysen BO, Boettcher AL (1975) Melting of a hydrous mantle: II. Geochemistry of crystals and liquids formed by anatexis of mantle peridotite at high pressures and high temperatures as a function of water, carbon dioxide and hydrogen activities. J Petrol 16:549–593
Mysen BO, Richet P (2019) Silicate glasses and melts, 2nd edn. Elsevier, New York, p 720
Mysen BO, Shang J (2003) Fractionation of major elements between coexisting H2O-saturated silicate melt and silicate-saturated aqueous fluids in aluminosilicate systems at 1–2 GPa. Geochim Cosmochim Acta 67:3925–3936
Mysen BO, Kushiro I, Fujii T (1978) Preliminary experimental data bearing on the mobility of H2O in crystalline upper mantle. Carnegie Inst Wash Year Book 78:793–797
Mysen BO, Virgo D, Seifert FA (1982) The structure of silicate melts: implications for chemical and physical properties of natural magma. Rev Geophys 20:353–383
Mysen BO, Cody GD, Morrill PL (2009) Solution behavior of reduced C-O-H volatiles in silicate melts at high pressure and temperature. Geochim Cosmochim Acta 73:1696–1710
Mysen BO, Kumamoto K, Cody GD, Fogel ML (2011) Solubility and solution mechanisms of C–O–H volatiles in silicate melt with variable redox conditions and melt composition at upper mantle temperatures and pressures. Geochim Cosmochim Acta 75:6183–6199
Mysen BO, Mibe K, Chou I-M, Bassett WA (2013) Structure and equilibria among silicate species in aqueous fluids in the upper mantle: Experimental SiO2-H2O and MgO–SiO2–H2O data recorded in-situ to 900 °C and 5.4 GPa. J Geophys Res 118:6076–6085
Nagashima S, Katsura T (1973) The solubility of sulfur in Na2O-SiO2 melts under various oxygen partial pressures at 1100 °C, 1250 °C, and 1300 °C. Bull Chem Soc Japan 46:3099–3103
Nakamura Y, Kushiro I (1974) Composition of the gas phase in Mg2SiO4-SiO2-H2O at 15 kbar. Year Book Carnegie Inst Wash 73:255–258
Nakamura M, Watson EB (2001) Experimental study of aqueous fluid infiltration into quartzite: implications for the kinetics of fluid redistribution and grain growth driven by interfacial energy reduction. Geofluids 1:73–89
Newton RC, Manning CE (2000) Quartz solubility in H2O-NaCl and H2O-CO2 solutions at deep crust-upper mantle pressures and temperatures: 2–15 kbar and 500–900 °C. Geochim Cosmochim Acta 64:2993–3006
Newton RC, Manning CE (2002) Solubility of enstatite+forsterite in H2O in deep crust/upper mantle conditions: 4 to 15 kbar and 700 to 900 °C. Gechim Cosmochim Acta 66:4165–4176
Newton RC, Manning CE (2003) Activity coefficient and polymerization of aqueous silica at 800 °C,12 kbar, from solubility measurements and SiO2-buffering mineral assemblages. Contrib Mineral Petrol 146:135–146
Newton RC, Manning CE (2006) Solubilities of corundum, wollastonite and quartz in H2O–NaCl solutions at 800 °C and 10 kbar: interaction of simple minerals with brines at high pressure and temperature. Geochim Cosmochim Acta 70:5571–5582
Newton RC, Manning CE (2007) Solubility of grossular, Ca3Al2Si3O12, in H2O–NaCl solutions at 800 °C and 10 kbar, and the stability of garnet in the system CaSiO3–Al2O3–H2O–NaCl. Geochim Cosmochim Acta 71:5191–5202
Newton RC, Manning CE (2008) Solubility of corundum in the system Al2O3–SiO2–H2O–NaCl at 800 °C and 10 kbar. Chem Geol 249:250–261
Newton RC, Manning CE (2016) Evidence for SiO2-NaCl complexing in H2O–NaCl solutions at high pressure and temperature. Geofluids 16:342–348
Nowak M, Behrens H (1995) The speciation of water in haplogranitic glasses and melts by in-situ, near-infrared spectroscopy. Geochim Cosmochim Acta 59:3445–3450
Ogawa Y, Ichiki M, Kanda W, Mishina M, Asamori K (2014) Three-dimensional magnetotelluric imaging of crustal fluids and seismicity around Naruko volcano NE Japan. Earth Planet Space 66:1–13
Ohtani E (2019) The role of water in Earth’s mantle. Nat Sci Rev 7:224–232
O’Neill HSC (1991) The origin of the Moon and the early history of the earth—A chemical model. Part 1: the Moon. Geochim Cosmochim Acta 55:1135–1158
O’Neill HSC, Mavrogenes JA (2002) The sulfide capacity and sulfur content at sulfide saturation of silicate melts at 1400 °C and 1 bar. J Petrol 43:1049–1087
O’Neill DD, Rubie DC (1998) Oxide-metal equilibria to 2500 °C and 25 GPa: implications for core formation and the light component in the Earth’s core. J Geophys Res 103:12239–12260
Ono S, Mibe M, Yoshino T (2002) Aqueous fluid connectivity and pyrope aggregates: water transport in the deep mantle by a subducted oceanic crust without any hydrous minerals. Earth Planet Sci Lett 203:895–903
Oppenheimer C (2003) Volcanic degassing. In: Rudnick RL (ed) The Crust. Elsevier, Amsterdam, pp 123–166
Orville PM (1975) Stability of scapolite in the system Ab-An-NaCl-CaCO+ at 4 kb and 750 °C. Geochim Cosmochim Acta 39:1091–1105
Papale P, Moretti R, Barbato D (2006) The compositional dependence of the saturation surface of H2O+CO2 fluids in silicate melts. Chem Geol 229:78–95
Pascal ML, Anderson GM (1989) Speciation of Al, Si, and K in supercritical solutions: experimental study and interpretation. Geochim Cosmochim Acta 53:1843–1856
Peiffert C, Cuney M, Nguyen-Trung C (1996) Uranium in granitic magmas part 2: experimental determination of uranium solubility and fluid-melt partitioning coefficients in the uranium oxide-haplogranite-H2OL-NaX (X=Cl, F) system at 770 °C 2 kbar. Geochim Cosmochim Acta 60:1515–1529
Philippot P, Agrinier P, Scambelluri M (1998) Chlorine cycling during subduction of altered oceanic crust. Earth Planet Sci Lett 161:33–44
Plessen B, Harlov DE, Henry D, Guidotti CV (2010) Ammonium loss and nitrogen isotopic fractionation in biotite as a function of metamorphic grade in metapelites from western Maine, USA. Geochim Cosmochim Acta 74:4759–4771
Pokrovski GS, Dubessy J (2015) Stability and abundance of the trisulfur radical ion S3− in hydrothermal fluids. Earth Planet Sci Lett 411:298–309
Pokrovski GS, Dubrovinsky LS (2011) The S3− ion is stable in geological fluids at elevated temperatures and pressures. Science 331:1052–1054
Pokrovski GS, Helgeson HC (1995) Thermodynamic properties of aqueous species and the solubilities of minerals at high pressures and temperatures; the system Al2O3-H2O-NaCl. Amer J Sci 295:1255–1342
Pokrovski GS, Borisova AY, Harrichoury J-C (2008) The effect of sulfur on vapor-liquid fractionation of metals in hydrothermal systems. Earth Planet Sci Lett 266:345–362
Poli S, Schmidt MW (2002) Petrology of subducted slabs. Ann Rev Earth Planet Sci 30:207–235
Pommier A (2014) Geophysical assessment of migration and storge conditions of fluids in subduction zones. Earth Planet Sp 66:1–11
Price JD, Wark DA, Watson EB, Smith AM (2006) Grain-scale permeabilities of faceted polycrystalline aggregates. Geofluids 6:302–318
Putirka K, Ryerson F, Perfit M, Ridley WI (2011) Mineralogy and composition of the oceanic mantle. J Petrol 52:279–313
Reynard B, Mibe K, van de Moortele B (2011) Electrical conductivity of the serpentinised mantle and fluid flow in subduction zones. Earth Planet Sci Lett 307:387–394
Richardson FD, Fincham CJB (1954) Sulphur in silicate and aluminate slags. J Iron Steel Inst 178(1):4–15
Righter K, Drake MJ (1997) Metal-silicate equilibrium in a homogeneously accreting earth: new results for Re. Earth Planet Sci Lett 146:541–553
Riley GN, Kohlstedt DL (1991) Kinetics of melt segregation in upper mantle-type rocks. Earth Planet Sci Lett 105:500–521
Rustioni G, Audetat A, Keppler H (2021) The composition of subduction zone fluids and the origin of the trace element enrichment in arc magmas. Contrib Mineral Petrol 176(7):1–19. https://doi.org/10.1007/s00410-021-01810-8
Sahle C, Niskanen J, Schmidt C, Setfanski J, Gilmore K, Forov Y, Jahn S, Wilke M, Sternemann C (2013) Microscopic structure of water at elevated pressures and temperatures. Proc Natl Acad Sci USA 110(16):6301–6306. https://doi.org/10.1073/pnas.1220301110
Scaillet B, Clemente B, Evans BW, Pichavant M (1998) Redox control of sulfur degassing in silicic magmas. J Geophys Res B 103:23937–23949
Scambelluri M, Philippot P (2001) Deep fluids in subduction zones. Lithos 55:213–227
Scheuermann PP, Tan C, Seyfried WE (2018) Quartz solubility in the two-phase region of the NaCl-H2O system: an experimental study with application to the Piccard hydrothermal field mid-Cayman rise. Geochem Geophys Geosyst 19:3570–3582
Schmidt C (2014) Raman spectroscopic determination of carbon speciation and quartz solubility in H2O + Na2CO3 and H2O + NaHCO3 fluids to 600 °C and 1.53 GPa. Geochim Cosmochim Acta 145:281–296
Schmidt C, Wohlers A, Marquardt K, Watenphul A (2014) Experimental study on the pseudobinary H2O + NaAlSi3O8 at 600–800 °C and 0.3-2.4 GPa. Chem Geol 388:40–47
Schneider WG, Bernstein HJ, Pople JA (1958) Proton magnetic resonance chemical shift of free (gaseous) and associated (liquid) hydride molecules. J Chem Phys 28:601–607
Shen A, Keppler H (1995) Infrared spectroscopy of hydrous silicate melts to 1000 °C and 10 kbar: direct observation of H2O speciation in a diamond cell. Amer Mineral 80:1335–1338
Shen A, Keppler H (1997) Direct observation of complete miscibility in the albite-H2O system. Nature 385:710–712
Shi X, Mao S, Hu J, Zhang J, Zheng J (2019) An accurate model for the solubilities of quartz in aqueous NaCl and/or CO2 solutions at temperatures up to 1273 K and pressures up to 20,000 bar. Chem Geol 513:73–87
Shimojuku A, Yoshino T, Yamazaki D, Okudaira T (2012) Electrical conductivity of fluid-bearing quartzite under lower crustal conditions. Phys Earth Planet Int 198:1–8
Shinohara H, Iiyama JT, Matsuo M (1989) Partition of chlorine compounds between silicate melt and hydrothermal solutions: I. Partition of NaCl-KCl. Geochim Cosmochim Acta 53:2617–2630
Shmonov VM, Sadus RJ, Franck EU (1993) High-pressure phase equilibria and supercritical PVT data of the binary water plus methane mixture to 7 23 K and 200 MPa. J Phys Chem 97:9054–9059
Shmulovich K, Graham C, Yardley B (2001) Quartz, albite and diopside solubilities in H2O-NaCl and H2O-CO2 fluids at 0.5-0.9 GPa. Contrib Mineral Petrol 141:95–108
Signorelli S, Carroll MR (2000) Solubility and fluid-melt partitioning of Cl in hydrous phonolitic melts. Geochim Cosmochim Acta 64:2851–2862
Signorelli S, Carroll MR (2002) Experimental study of Cl solubility in hydrous alkaline melts: constraints on the theoretical maximum amount of Cl in trachytic and phonolitic melts. Contrib Mineral Petrol 143:209–218
Soper AK, Ricci MA (2000) Structures of high-density and low-density water. Phys Rev Lett 84:2881. https://doi.org/10.1103/PhysRevLett.84.2881
Sowerby J Keppler (1998) Water speciation in rhyolite glass and melt. In: Annual report 1997, bayerisches forschungsinstitut fur experimentelle geochemie und geophysik, Bayreuth. pp 70–73
Stalder R, Foley SF, Drey GP, Horn L (1998) Mineral-aqueous fluid partitioning of trace elements at 900–1200 °C and 3.0–5.7 GPa: new experimental data for garnet, clinopyroxene, and rutile, and implications for mantle metasomatism. Geochim Cosmochim Acta 62:1781–1801
Stalder R, Ulmer P, Thompson AB, Gunther D (2001) High pressure fluids in the system MgO–SiO2–H2O under upper mantle conditions. Contrib Mineral Petrol 140:607–618
Sun WQ, Dai LD, Li HP, Hu HY, Jiang JJ, Wang MQ (2020) Electrical conductivity of clinopyroxene-NaCl-H2O system at high temperatures and pressures: implications for high-conductivity anomalies in the deep crust and subduction zone. J Geophys Res B 125(4):e2019JB019093. https://doi.org/10.1029/2019jb019093
Sverjensky DA, Harrison AB, Azzolini D (2014) Water in the deep Earth: the dielectric constant and the solubilities of quartz and corundum to 60kb and 1200 °C. Geochim Cosmochim Acta 129:125–145
Symonds RB, Rose WI, Bluth GJS, Gerlach TM (1994) Volcanic gas studies: methods, results and applications. In: Carroll MR, Holloway JR (eds) Volatiles in Magmas. De Gruyter, Berlin, pp 1–66
Tagirov B, Seward TM (2010) Hydrosulfide/sulfide complexes of zinc to 250 °C and the thermodynamic properties of sphalerite. Chem Geol 269:301–311
Tagirov B, Schott J, Harrichourry JC, Salvi S (2002) Experimental study of aluminum speciation in fluoride-rich supercritical fluids. Geochim Cosmochim Acta 66:2013–2024
Takei Y, Shimizu I (2003) The effects of liquid composition, temperature, and pressure on the equilibrium dihedral angles of binary solid-liquid systems inferred from a lattice-like model. Phys Earth Planet Int 139:225–242
Tanis EA, Simon A, Zhang Y, Chow P, Xiao Y, Hanchar JM, Tschauner O, Shen G (2016) Rutile solubility in NaF-NaCl-KCl-bearing aqueous fluids at 0.5–2.79 GPa and 250–650 °C. Geochim Cosmochim Acta 177:170–181
Tao R, Zhang L, Tian M, Zhu K, Liu X, Liu J, Hoefer HE, Stagno V, Fei Y (2018) Formation of abiotic hydrocarbon from reduction of carbonate in subduction zones: constraints from petrological observation and experimental simulation. Geochim Cosmochim Acta 239:390–408
Taylor WR, Green DH (1989) The role of reduced C-O-H fluids in mantle partial melting. In: Ross J (ed) Kimberlites and related rocks—their composition, occurrence, origin, and emplacement. Blackwell Scientific Publishers, Carlton Australia, pp 592–602
Till CB, Grove TL, Wither AC (2012) The beginnings of hydrous mantle wedge melting. Contrib Mineral Petrol 163:669–688
Tossell JA (2012) Calculation of the properties of the S3- radical anion and its complexes with Cu+ in aqueous solution. Geochim Cosmochim Acta 95:79–92
Touret JLR, Huizenga J-M (2011) Fluids in granulites. Geol Soc Amer Mem 207:25–37
Trigub AL, Tagirov BR, Kvashnina KO, Lafuerza S, Filimonova ON, Nickolsky MS (2017) Experimental determination of gold speciation in sulfide-rich hydrothermal fluids under a wide range of redox conditions. Chem Geol 471:52–64
Trønnes RG (2002) Stability range and decomposition of potassic richterite and phlogopite end members at 5–15 GPa. Mineral Petrol 74:129–148
Tropper P, Manning CE (2007) The solubility of corundum in H2O at high pressure and temperature and its implications for Al mobility in the deep crust and upper mantle. Chem Geol 240:54–60
Ulmer P (2001) Partial melting in the mantle wedge—the role of H2O in the genesis of mantle-derived ‘arc-related’ magmas. Phys Earth Plane Int 127:215–232
Ulrich T, Mavrogenes J (2008) An experimental study of the solubility of molybdenum in H2O and KCl–H2O solutions from 500 °C to 800 °C and 150 to 300 MPa. Geochim Cosmochim Acta 72:2316–2330
van Keken PU, Hacker BR, Syracuse EM, Abers GA (2011) Subduction factory: 4. Depth-dependent flux of H2O from subducting slabs worldwide. J Geophys Res B Earth. https://doi.org/10.1029/2010JB007922
Wallace PJ, Edmonds M (2011) The sulfur budget in magmas: evidence from melt inclusions, submarine glasses, and volcanic gas emissions. Rev Mineral Geochem 73:215–246
Walrafen GE, Hokmabadi MS, Yang WH, Piermarini GJ (1988) High-temperature high-pressure Raman-spectra from liquid water. J Phys Chem 92:4540–4542
Walte NP, Rubie DC, Bons PD, Frost DJ (2011) Deformation of a crystalline aggregate with a small percentage of high-dihedral-angle liquid: implications for core-mantle differentiation during planetary formation. Earth Planet Sci Lett 305:124–134
Walther JV (1997) Experimental determination and interpretation of the solubility of corundum in H2O between 350 and 600 °C from 0.5 to 2.2 kbar. Geochim Cosmochim Acta 61:4955–4964
Walther JV (2001) Experimental determination and analysis of the solubility of corundum in 0.1 and 0.5 m NaCl solutions between 400 and 600 °C from 0.5 to 2.0 kbar. Geochim Cosmochim Acta 65:2843–2851
Walther JV, Schott J (1988) The dielectric constant approach to speciation and ion pairing at high temperature and pressure. Nature 332:635–638
Wanamaker BJ, Kohlstedt DL (1991) The effect of melt composition on the wetting angle between silicate melts and olivine. Phys Chem Mineral 18:26–36
Wannamaker PE, Caldwell TG, Jiracek GR, Maris V, Hill GJ, Ogawa BHM, Bennie SL, Heise W (2009) Fluid and deformation regime of an advancing subduction system at Marlborough, New Zealand. Nature 460:733–736
Wark DA, Watson EB (1998) Grain-scale permeabilities of texturally equilibrated, monomineralic rocks. Earth Planet Sci Lett 164:591–605
Wark DA, Watson EB (2000) Effect of grain size on the distribution and transport of deep-seated fluids and melts. Geophys Res Lett 27:2029–2032
Watenphul AC, Schmidt C, Jahn S (2014) Cr(III) solubility in aqueous fluids at high pressures and temperatures. Geochim Cosmochim Acta 126:212–227
Watson EB (1991) Diffusion in fluid-bearing and slightly-melted rocks: experimental and numerical approaches illustrated by iron transport in dunite. Contrib Mineral Petrol 107:417–434
Watson EB, Brenan JM (1987) Fluids in the lithosphere, 1. Experimentally-determined wetting characteristics of CO2-H2O fluids and their implications for fluid transport, host-rock physical properties, and fluid inclusion formation. Earth Planet Sci Lett 85:497–515
Watson EB, Lupulescu A (1993) Aqueous fluid connectivity and chemical transport in clinopyroxene-rich rocks. Earth Planet Sci Lett 117:279–294
Watson EB, Brenan JM, Baker DR (1990) Distribution of fluids in the continental mantle. In: Menzies MA (ed) Continental mantle. Clarendon Press, Oxford, pp 111–125
Watson EB, Wark DA, Thomas JB (2006) Crystallization thermometers for Zircon and Rutile. Contrib Mineral Petrol 151:413–433. https://doi.org/10.1007/s00410-006-0068-5
Watson EB (1990) Distribution of fluid in the continental mantle. In: Continental mantle, pp 111–125
Webster JD (1992) Water solubility and chlorine partitioning in Cl-rich granitic systems: effects of melt composition at 2 kbar and 800 °C. Geochim Cosmochim Acta 56:679–687
Webster JD, Botcharnikov RE (2011) Distribution of sulfur between melt and fluid in S-O-H-C-Cl-bearing magmatic systems at shallow crustal pressures and temperatures. Rev Mineral Geochem 73:247–283
Webster JD, Kinzler RJ, Mathez EA (1999) Chloride and water solubility in basalt and andesite melts and implications for magma degassing. Geochim Cosmochim Acta 63:729–738
Webster JD, Tappen CM, Mandeville CW (2009) Partitioning behavior of chlorine and fluorine in the system apatite-melt-fluid: II. Felsic silicate systems at 200 MPa. Geochim Cosmochim Acta 73:559–581
Weill DF, Fyfe WS (1964) The solubility of quartz in H2O in the range 1000–4000 bars and 400–550 °C. Geochim Cosmochim Acta 28:1243–1255
Wendlandt HG, Glemser O (1964) The reaction of oxides with water at high pressures and temperatures. Ang Chim 3:47–54
White LT, Rawlinson N, Lister GS, Waldhauser F, Hejrani B, Thompson DA, Tanner D, Colin G, Tkalčić H, Morgan JP (2019) Earth’s deepest earthquake swarms track fluid ascent beneath nascent arc volcanoes. Earth Planet Sci Lett 521:25–36
Whittington AP, Richet P, Holtz F (2000) Water and the viscosity of depolymerized aluminosilicate melts. Geochim Cosmochim Acta 64:3725–3736
Wiens DA, Seama N, Conder JA (2006) Mantle structure and flow patterns beneath active back-arc basins inferred from passive seismic and electromagnetic methods. In: Christie DM, Fisher CR, Lee SM, Givens S (eds) Back-arc spreading systems: geological, biological, chemical, and physical interactions. American Geophysical Union Geophysical Monograph Series, Washington
Wilk M, Schmidt C, Dubrail J, Appel K, Borchert M, Kvashnina K, Manning CE (2012) Zircon solubility and zircon complexation in H2O+Na2O+SiO2±Al2O3 fluids at high pressure and temperature. Earth Planet Sci Lett 349–350:15–25
Winkler HGF (1965) Petrogenesis of metamorphic rocks. Springer Verlag, New York, p 220
Wohlers A, Manning CE (2009) Solubility of corundum in aqueous KOH solutions at 700 °C and 1 GPa. Chem Geol 262:310–317
Wohlers A, Manning CE, Thompson AB (2011) Experimental investigation of the solubility of albite and jadeite in H2O, with paragonite plus quartz at 500 and 600 °C, and 1–2.25 GPa. Geochim Cosmochim Acta 75:2924–2939
Woodland AB, Walther JV (1987) Experimental determination of the solubility of the assemblage paragaonite, albite, and quartz in supercritical H2O. Geochim Cosmochim Acta 51:365–372
Wyllie PJ (1982) Subduction products according to experimental prediction. Geol Soc Amer Bull 93:468–476
Xie Z, Walther JV (1993) Quartz solubilities in NaCl solutions with and without wollastonite at elevated temperatures and pressure. Geochim Cosmochim Acta 57:1947–1955
Xiong X, Zhao Z, Zhu J, Rao B, Lai M (1998) Partitioning of F between aqueous fluids and albite granite melt and its petrogenetic and metallogenetic significance. Chin J Geochem 17:303–310
Yardley BWD, Bodnar RJ (2014) Fluids in the continental crust. Geochem Perspect 3:1–127
Yardley BWD, Graham JT (2002) The origins of salinity in metamorphic fluids. Geofluids 2:249–256
Yoshino T, Katsura T (2013) Electrical conductivity of mantle minerals: role of water in conductivity anomalies. Ann Rev Earth Planet Sci 41:605–628
Yoshino T, Mibe K, Yasuda A, Fujii T (2002) Wetting properties of anorthite aggregates: implications for fluid connectivity in continental lower crust. J Geophys Res B. https://doi.org/10.1029/2000JB000440
Yoshino T, Nishihara Y, Kaeato S (2007) Complete wetting of olivine grain boundaries by a hydrous melt near the mantle transition zone. Earth Planet Sci Lett 256:466–474
Zajacz Z (2015) The effect of melt composition on the partitioning of oxidized sulfur between silicate melts and magmatic volatiles. Geochim Cosmochim Acta 158:223–244
Zajacz Z, Candela P, Piccoli P, Sanchez-Valle C (2013) Controls on the composition of magmatic volatiles in the crust: implications for ore genesis and volcanic degassing. Mineral Mag 77(5):2577. https://doi.org/10.1180/minmag.2013.077.5.26
Zhang Y-G, Frantz JD (2000) Enstatite-forsterite-water equilibria at elevated temperatures and pressures. Amer Mineral 85:918–925
Zhang C, Duan Z, Zhang Z (2007) Molecular dynamics simulation of the CH4 and CH4-H2O systems up to 10 GPa and 2573 K. Geochim Cosmochim Acta Acta 71:2036–2055
Zhang L, Audétat A, Dolejš D (2012) Solubility of molybdenite (MoS2) in aqueous fluids at 600–800 °C, 200MPa: a synthetic fluid inclusion study. Geochim Cosmochim Acta 77:175–185. https://doi.org/10.1016/j.gca.2011.11.015
Zheng Y-F (2019) Subduction zone geochemistry. Geosci Front 10:1223–1254
Zotov N, Keppler H (2002) Silica speciation in aqueous fluids at high pressures and high temperatures. Chem Geol 184:71–82
Acknowledgements
Critical and detailed reviews by Eiji Ohtani and an unidentified reviewer are greatly appreciated. The support by our library in the literature search is greatly appreciated.
Funding
This review was written with the complete support from Carnegie Institution of Washington.
Author information
Authors and Affiliations
Contributions
As I am the sole author, I contributed 100% to the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The author declares that he has no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mysen, B. Fluids and physicochemical properties and processes in the Earth. Prog Earth Planet Sci 9, 54 (2022). https://doi.org/10.1186/s40645-022-00516-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40645-022-00516-0