
Abstract
Background
A thermostable alcohol dehydrogenase from Thermoanaerobacter brockii (TbSADH) has been repurposed to perform asymmetric reduction of a series of prochiral ketones with the formation of enantio-pure secondary alcohols, which are crucial chiral synthons needed in the preparation of various pharmaceuticals. However, it is incapable of asymmetric reduction when applied to bulky ketones. Recently, mutations at two key residues A85 and I86 were shown to be crucial for reshaping the substrate binding pocket. Increased flexibility of the active site loop appears to be beneficial in the directed evolution of TbSADH towards difficult-to-reduce ketones.
Methods
Using the reported mutant A85G/I86A as template, double-code saturation mutagenesis (DCSM) was applied at selected residues lining the substrate binding pocket with a 2-membered reduced amino acid alphabet.
Results and conclusions
The mutant A85G/I86A was first tested for activity in the reaction of the model substrate (4-chlorophenyl)-(pyridin-2-yl)methanone, which showed a total turnover number (TTN) of 3071. In order to further improve the turnovers, a small and smart mutant library covering a set of mutations at Q101, W110, L294, and C295 was created. Eventually, a triple-mutant A85G/I86A/Q101A was identified to be a superior catalyst that gave S-selective product with 99% ee and 6555 TTN. Docking computations explain the source of enhanced activity. Some of the best variants are also excellent catalysts in the reduction of other difficult-to-reduce ketones.

Similar content being viewed by others
Background
Optically active alcohols are essential synthons for the preparation of numerous pharmaceutical intermediates and fine chemicals. Both transition metals and biocatalysts are competent for the enantioselective synthesis of chiral alcohols. However, in the scenario of transforming difficult-to-reduce ketones, such as bulky diaryl ketones and ketones having sterically similar alpha- and alpha′-substituents, it is difficult to achieve high enantioselectivity by transition metal catalysis (Corey and Helal 1998; Morris 2009; Noyori 2002; Sandoval et al. 2008). As a complementary strategy to the traditional approach, it has been demonstrated that alcohol dehydrogenases (ADHs) can reduce such prochiral ketones with very high chemo-, regio-, and stereoselectivity, thereby enabling a sustainable synthetic route to chiral alcohols, which includes biodegradability of the biocatalyst (Garcia-Urdiales et al. 2005; Huisman et al. 2010; Nie et al. 2018; Sun et al. 2016a; Xu et al. 2018; Liang et al. 2018).
As an attractive ADH in biotechnology, the thermostable ADH from Thermoanaerobacter brockii (TbSADH) has already been repurposed to perform asymmetric reduction of prochiral ketones with the formation of enantio-pure secondary alcohols by our group and others (Heiss et al. 2001; Li et al. 2017; Musa et al. 2009; Qu et al. 2018; Sun et al. 2016a, b). TbSADH is an NAD(P)H- and Zn-dependent enzyme, and its reduction mechanism has been elucidated. In general, the carbonyl oxygen of the unsymmetrical ketone substrate coordinates to the Zn ion, and a hydride from the cofactor NADPH attacks the carbonyl carbon atom from either the Re or Si face, producing the corresponding optically active secondary alcohol (Li et al. 2017; Moa and Himo 2017; Nealon et al. 2015; Qu et al. 2018).
The crystal structure of TbSADH shows that the substrate binding site consists of two hydrophobic pockets, one smaller than the other (Fig. 1) (Korkhin et al. 1998). The substrate binds in an orientation determined by the best fit of the two alkyl groups attached to the carbonyl into these two pockets, dictating the stereochemical outcome (Keinan et al. 1986; Moa and Himo 2017; Nealon et al. 2015). The NAD(P)H cofactor then transfers its pro-R hydride to either the Re or Si face of the ketone, producing secondary alcohols with (S)- or (R)-configuration, respectively. To date, nearly all of the residues situated at the two pockets (shown in Fig. 1) have been analyzed by many different research groups, leading to the documentation of mutational effects on activity, enantioselectivity, and substrate specificity (Additional file 1: Table S1). In particular, recent studies have demonstrated that mutations at residues A85 and I86 can influence the motion of the active site loop, thereby increasing the plasticity of the binding pocket and thus raising the ability to accept non-natural substrates (Li et al. 2017; Liu et al. 2019).

The overall structure of the TbSADH catalytic pocket (PDB code 1YKF), consisting of a smaller (green) and a bigger (cyan) pockets. NADPH is shown by white sticks, whereas Zinc atom is depicted by a gray sphere. The zinc-coordinating residues are colored in magenta and are presented in lines
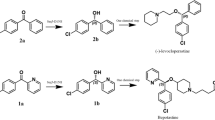
We sought to perform the biocatalytic reaction of difficult-to-reduce ketones such as 1 with formation of (S)-2, which is a crucial chiral synthon needed in the preparation of antiallergic drugs Bepotastine (Kida et al. 2010; Takahashi et al. 2004) and (S)-carbinoxamine (Barouh et al. 1971; Maclaren et al. 1955) (Scheme 1). Despite the fact that wild-type (WT) TbSADH showed no activity towards 1, we set out to expand its substrate scope by means of directed evolution.

ADH-catalyzed asymmetric reduction of ketone 1
Directed evolution of enzymes has proved to be a powerful technique to endow biocatalysts with novel catalytic repertoires (Bommarius 2015; Reetz 2011, 2013a; Sheldon and Pereira 2017; Arnold 2018; Denard et al. 2015; Sun et al. 2019). In recent studies, protein engineering of TbSADH was applied for various purposes. When aiming at the optimization of selectivity and activity, the combinatorial active-site saturation test (CAST) along with iterative saturation mutagenesis (ISM) at sites lining the binding pocket constitutes a viable protein engineering technique (Reetz 2017; Reetz and Carballeira 2007; Reetz et al. 2006, 2010). In this regard, CAST-based double-code saturation mutagenesis (DCSM), in which the reduced amino acid alphabet comprises two members as building blocks, has been shown to be efficient in manipulating enzyme stereoselectivity and activity (Sun et al. 2016c).
In continuation of the preceding study, we now report that DCSM is well-suited for significantly elevating the activity of TbSADH in the reduction of the challenging ketone 1 with no trade-off in enantioselectivity. Specifically, we focus saturation mutagenesis on the reported sites A85 and I86. Docking analysis was carried out on the best mutant to gain insight into the origin of altered catalytic profiles.
Results and discussion
Investigation of key residues A85 and I86
As delineated in the background information, two key residues A85 and I86 emerged as possible sites for mutagenesis. We therefore directly tested the reported mutants A85G, I86A, and A85G/I86A as catalysts in the reduction of the non-natural ketone 1. It is of interest to note that both of the two single mutants displayed weakly catalytic activity, while the double-mutant A85G/I86A showed a notably better conversion rate (Table 1). To further explore the essential role of A85 and I86 in the substrate specificity for ketone 1, the two residues were deleted individually and combinatorially. As a result, variants ∆A85 (“∆” indicates deletion), ∆I86, and ∆A85/∆I86 showed no measurable activity towards ketone 1, even though the mutants were expressed well in protein level (Additional file 1: Figure S1), suggesting the importance of proper amino acids at the positions 85 and 86 in accommodating the non-natural ketone 1.
Double-code saturation mutagenesis of TbSADH
The double-mutant A85G/I86A showed moderate performance for ketone 1 with excellent (S)-enantioselectivity (99% ee), but we wanted to further improve its activity while maintaining high (S)-selectivity. Accordingly, the other residues Q101, W110, L294, and C295 at the binding pocket were considered as candidates (Fig. 1 and Additional file 1: Table S1). Notably, aside from A85 and I86, mutations at sites W110, L294 as well as C295 have also been reported to be important in tuning enantioselectivity and/or activity (Additional file 1: Table S1). The above selected residues were then mutated by applying the DCSM strategy to the double-mutant A85G/I86A as template, using the two small amino acids alanine and serine as combinatorial building blocks due to their steric properties. After checking the activity, several mutants with improved activity towards ketone 1 were indeed obtained, all retaining the high enantioselectivity for (S)-2 shown by the double-mutant A85G/I86A (Table 2). Among them, mutants A85G/I86A/Q101A and A85G/I86A/C295A showed particularly excellent enantioselectivity (> 99% ee) and high conversion (> 90%) in the reduction of ketone 1.
Characterization of best mutants with total turnover numbers (TTNs) determination and kinetic parameters
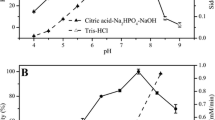
The above two selected TbSADH mutants together with A85G, I86A, and A85G/I86A were then characterized by TTN determination, which is an important parameter in catalysis (Hagen 2006). As shown in Table 3 and Fig. 2, A85G and I86A increased the TTN marginally for ketone 1, whereas the respective double-mutant A85G/I86A increased the TTN ca. sixfold. In sharp contrast, the triple-mutant A85G/I86A/Q101A led to an unusually high TTN of 6555, a 20-fold improvement over the single mutant A85G, and twofold over A85G/I86A. In parallel, the conversion rate for ketone 1 was also improved in a stepwise fashion (Fig. 2).

Total turnover numbers (TTNs) and the conversion rate of TbSADH mutants in the reaction of ketone 1. Reactions were done in triplicate
After protein purification, the best TbSADH mutant A85G/I86A/Q101A as well as A85G/I86A were subsequently characterized by enzyme kinetics. As a result, the variant A85G/I86A/Q101A displays a higher substrate affinity (Km 0.079 mM), and the catalytic efficiency (kcat/Km = 366.08 s−1 mM−1) is increased fourfold compared to A85G/I86A (kcat/Km = 89.47 s−1 mM−1) (Table 4). The apparent consistency between TTN determination and catalytic kinetics suggests that the one round ISM step yielded a remarkable improvement. Overall, the elevating conversion rates, TTN values, and kinetic parameters indicate additive mutational effects (Reetz 2013b) occurred in this iteratively enzyme engineering study.
Docking analysis for explaining the source of improved TTN
In order to gain insight into the possible origin of the evolved TTN, ketone 1 was docked into a homology model of mutant A85G/I86A/Q101A (Additional file 1: Table S2). The highest ranked docking pose for ketone 1 preferentially orientates in such a way that the π-face of the C=O bond undergoes nucleophilic attack to yield the (S)-product (Fig. 3). The protein–ligand interaction analysis suggested that this preferred binding mode results in π–π interactions between the phenyl substituent of the substrate and the side chains of nearby residues W110 and Y267 (Additional file 1: Figure S2), enforcing the phenyl group of the substrate to stay in the bigger pocket and therefore leaving the pyridine group of the substrate in the smaller pocket. Moreover, hydrogen bonding interaction was observed between the substrate carbonyl oxygen atom and the side chain of S39 (Additional file 1: Figure S2). We also performed the docking analysis using a homology model of mutant A85G/I86A; the interface energy of ketone 1 as evaluated by Rosetta energy units (REU) is less stable (− 19.74 REU) than that of A85G/I86A/Q101A (− 22.05 REU, Additional file 1: Table S2), indicating that the latter mutant confers better binding affinity towards ketone 1. The binding affinity diversity is supported by the kinetic parameters, the mutant A85G/I86A/Q101A has a lower Km value relative to A85G/I86A (Table 4).

Highest ranked docking pose for ketone 1 in mutant A85G/I86A/Q101A. a Overlay of the substrate binding pocket. b (S)-selective docking pose. The mutated residues are shown in magenta. The docking poses meet the reactivity requirements and are consistent with the formation of (S)-2
Testing the best mutants as catalysts in the reduction of other bulky ketones
In order to evaluate the substrate scope of three of the best mutants and WT, a series of diaryl ketones were subjected to reduction (Table 5). Compounds 3a–3f are characterized by a pyridine ring and a phenyl substituent, while 3g and 3h contain two phenyl groups (Scheme 2). WT shows extremely low activity towards 3b, 3c, and 3d, which all can be transformed by A85G/I86A/C295A, suggesting that this variant have a wider substrate scope. Notably, the variants all greatly increased the enzyme activity for 3h; however, it was reduced to the (R)-alcohol by WT, A85G/I86A, and A85G/I86A/Q101A; interestingly, A85G/I86A induces opposite stereoselectivity. This suggested that the substitution of diaryl ketones greatly affects enzyme activity and enantioselectivity. In order to gain insight whether such a drastic change of stereoselectivity and activity is general, we also performed the reduction of purely aliphatic (3i–3k) and also aryl–alkyl ketones (3l–3n) (Scheme 2). In the case of the aliphatic ketones, 3j was all reduced with excellent reversed enantioselectivity compared to WT. As for the aromatic ketone 3n, all the variants showed improved conversion and enantioselectivity (> 99% ee).

Asymmetric reduction of ketones 3a–3n
Conclusions and perspectives
Conversion of difficult-to-reduce ketones to enantio-pure secondary alcohols is of particular interest in some applications, and is also one of the challenges in organic synthesis. This work has shown that such challenge can be partly met by applying directed evolution to the robust enzyme TbSADH. Using the previously reported key mutants A85G and I86A as the starting templates and ketone 1 as the substrate, the double-mutant A85G/I86A enabled a 99% (S)-enantioselectivity and 3071 TTN, amounting to six- and ninefold improvements compared to the single mutants A85G and I86A, respectively. In order to further improve the catalytic efficiency (TTN) while conserving the high level of (S)-selectivity, structure-guided saturation mutagenesis using reduced amino acid alphabets based on DCSM strategies was carried out at sites lining the binding pocket (CAST residues). Eventually, the best mutant A85G/I86A/Q101A displaying 99% ee in favor of the (S)-enantiomer and up to 6555 TTN was obtained. The substrate docking analysis shed light on the possible source of stereoselectivity and activity. This work highlights the ease of elevating enzyme activity without trade-off in enantioselectivity by the DCSM strategy (Sun et al. 2016c).
Materials and methods
Materials
PrimeSTAR DNA polymerase and restriction enzyme Dpn I were obtained from TAKARA and NEB, respectively. The oligonucleotide synthesis and DNA sequencing were conducted by GENEWIZ technology. All reagents and chemicals were purchased from commercial sources and used without further purification. The gene coding TbSADH optimized for expression in E. coli was cloned into pRSFDuet-1 and constructed by previous co-worker (Agudo et al. 2013).
Model generation and substrate docking
All protein and ligand structures were prepared in the Schrödinger Maestro software (Schrödinger Suite 2015). The X-ray structure of ADH from Thermoanaerobacter brockii was used as the basis for model creation (Korkhin et al. 1998). The initial structure of the mutants A85G/I86A and A85G/I86A/Q101A was generated employing the PyMol program (http://www.pymol.org) using the WT model as a template, and was then relaxed/repacking using Rosetta program suite version 3.10. Docking ketone 1 into the two mutants was performed using Rosetta docking program (Combs et al. 2013). A total of 50 models were run, from which the top 5 poses lowest in ligand–protein interface_score energy with Rosetta energy units were selected for comparison (Additional file 1: Table S2). The Rosetta docking command lines are also supplied in Additional file 1.
Site-directed mutagenesis
Site-directed mutagenesis of TbSADH was performed using the megaprimer approach35 with PrimeSTAR DNA polymerase. The PCR conditions for short fragment: 95 °C for 5 min, (95 °C for 30 s, 56 °C for 30 s, 72 °C for 40 s), × 32 cycles, 72 °C for 10 min. For mega-PCR: 95 °C for 5 min, (95 °C for 30 s, 60 °C for 30 s, 72 °C for 6 min), × 26 cycles, 72 °C for 10 min. The PCR products were analyzed on agarose gel by electrophoresis. Then, 0.5 μL Dpn I was added to 10 μL PCR reaction mixture for the digestion at 37 °C for 3 h. After Dpn I digestion, the PCR products were directly transformed into electro-competent E. coli BL21(DE3) to create the mutants. The primers are shown in Additional file 1: Table S3.
Activity assay and determination of total turnover numbers (TTN)
The protein expression of TbSADH mutants was induced by 0.1 mM IPTG at 20 °C for 16 h. The cells were harvested, washed once with phosphate buffer (50 mM, pH 7.4), and then lysed by sonication. The crude extract was obtained by centrifugation and lyophilized under vacuum to form the crude enzyme powder. The reduction reactions were conducted by mixing phosphate buffer solution (50 mM, pH 7.4), NADP+ (1 mM), isopropanol (10% v/v), crude enzyme powder of the TbSADH mutants (5 mg), and ketone 1 (5 mM) in 0.5 mL Eppendorf tube. TTN was defined as the molar number of the product yield divided by the catalyst concentration. The procedure for TTN determination was the same as the activity assay except for using 10 mM ketone 1 and 0.05 mg/mL purified enzyme. The TbSADH mutants with His-Tag were purified by Nickel-affinity chromatography and analyzed by SDS-PAGE as previously reported (Sun et al. 2016a). Protein concentration was measured using the Bradford method. The reaction mixture was stirred at 30 °C, 1000 rpm for 24 h. The product and remaining substrate were extracted using equal volume of ethyl acetate for HPLC analysis to measure the conversion and ee values (Additional file 1: Table S4). All reactions were carried out in triplicate.
Determination of kinetic parameters
The reductive activity was measured by monitoring the consumption of NADPH from the decrease in absorbance at 340 nm using a molar absorption coefficient of 6220 M−1 cm−1. Purified enzyme was added to phosphate buffer (50 mM, pH 7.4) with a total volume of 200 μL containing substrate 1, NADPH, and 2.5% acetonitrile. One unit of enzyme activity was defined as the amount of enzyme catalyzing the conversion of 1 μmol NADPH per minute. The kinetic parameters were obtained by measuring the initial velocities of the enzyme reaction and curve-fitting according to the Michaelis–Menten equation. For ketone 1, 1 mM NADPH and ketone 1 in the range of 0.01–1 mM were used for the enzyme kinetic assays. For NADPH, the activity assay was performed in a mixture containing a varying concentration of NADPH (0.01–0.1 mM) and 1 mM ketone 1.
Substrate scope investigation
General procedure was carried out as follows: NADP+ (1 mM), isopropanol (10% v/v), crude enzyme powder of the TbSADH variants (10 mg/mL), and ketone substrate (10 mM) were mixed in a phosphate buffer solution (0.5 mL, 50 mM, pH 7.4). The reaction mixture was stirred at 30 °C, 1000 rpm for 24 h. Then the product and remaining substrate were extracted using an equal volume of ethyl acetate for HPLC analysis and n-butanol for GC analysis to obtain the conversion and ee values. The absolute configuration was determined by comparison of the elution order with literature data for HPLC analysis and performed with Chiralcel OD-H column (4.6 mm × 250 mm × 5 μm) and Chiralpak ADH column (4.6 mm × 250 mm × 5 μm). Some of them were determined based on authentic racemic mixture and enantiomer product for GC analysis, and performed with Hydrodex-β-TBDAc (25 × 0.25 mm ID). Analytic conditions of HPLC and GC are listed in Additional file 1: Tables S4 and S5. All reactions were carried out in triplicate.
Availability of data and materials
The data and materials in this work are available from the corresponding author on reasonable request.
Abbreviations
- ADH:
-
alcohol dehydrogenase
- TbSADH:
-
an alcohol dehydrogenase from Thermoanaerobacter brockii
- DCSM:
-
double-code saturation mutagenesis
- TTN:
-
total turnover number
- WT:
-
wild-type
- CAST:
-
combinatorial active-site saturation test
- REU:
-
Rosetta energy units
- HPLC:
-
high-performance liquid chromatography
References
Agudo R, Roiban GD, Reetz MT (2013) Induced axial chirality in biocatalytic asymmetric ketone reduction. J Am Chem Soc 135(5):1665–1668
Arnold FH (2018) Directed evolution: bringing new chemistry to life. Angew Chem Int Ed Engl 57(16):4143–4148
Barouh V, Dall H, Patel D, Hite G (1971) Stereochemical aspects of antihistamine action. 4. absolute configuration of carbinoxamine antipodes. J Med Chem 14(9):834–836
Bommarius AS (2015) Biocatalysis: a status report. Annu Rev Chem Biomol Eng 6:319–345
Combs SA, Deluca SL, Deluca SH, Lemmon GH, Nannemann DP, Nguyen ED, Willis JR, Sheehan JH, Meiler J (2013) Small-molecule ligand docking into comparative models with Rosetta. Nat Protoc 8(7):1277–1298
Corey EJ, Helal CJ (1998) Reduction of carbonyl compounds with chiral oxazaborolidine catalysts: a new paradigm for enantioselective catalysis and a powerful new synthetic method. Angew Chem Int Ed Engl 37(15):1986–2012
Denard CA, Ren H, Zhao H (2015) Improving and repurposing biocatalysts via directed evolution. Curr Opin Chem Biol 25:55–64
Garcia-Urdiales E, Alfonso I, Gotor V (2005) Enantioselective enzymatic desymmetrizations in organic synthesis. Chem Rev 105(1):313–354
Hagen J (2006) Industrial catalysis: a practical approach. Wiley, New York
Heiss C, Laivenieks M, Zeikus JG, Phillips RS (2001) Mutation of cysteine-295 to alanine in secondary alcohol dehydrogenase from Thermoanaerobacter ethanolicus affects the enantioselectivity and substrate specificity of ketone reductions. Bioorg Med Chem 9(7):1659–1666
Huisman GW, Liang J, Krebber A (2010) Practical chiral alcohol manufacture using ketoreductases. Curr Opin Chem Biol 14(2):122–129
Keinan E, Hafeli EK, Seth KK, Lamed R (1986) Thermostable enzymes in organic-synthesis. 2. asymmetric reduction of ketones with alcohol-dehydrogenase from Thermoanaerobium-brockii. J Am Chem Soc 108(1):162–169
Kida T, Fujii A, Sakai O, Iemura M, Atsumi I, Wada T, Sakaki H (2010) Bepotastine besilate, a highly selective histamine H(1) receptor antagonist, suppresses vascular hyperpermeability and eosinophil recruitment in in vitro and in vivo experimental allergic conjunctivitis models. Exp Eye Res 91(1):85–91
Korkhin Y, Kalb AJ, Peretz M, Bogin O, Burstein Y, Frolow F (1998) NADP-dependent bacterial alcohol dehydrogenases: crystal structure, cofactor-binding and cofactor specificity of the ADHs of Clostridium beijerinckii and Thermoanaerobacter brockii. J Mol Biol 278(5):967–981
Li G, Maria-Solano MA, Romero-Rivera A, Osuna S, Reetz MT (2017) Inducing high activity of a thermophilic enzyme at ambient temperatures by directed evolution. Chem Commun 53(68):9454–9457
Liang C, Nie Y, Mu X, Xu Y (2018) Gene mining-based identification of aldo-keto reductases for highly stereoselective reduction of bulky ketones. Bioresour Bioprocess 5:33
Liu B, Qu G, Li J-K, Fan W, Ma J-A, Xu Y, Nie Y, Sun Z (2019) Conformational dynamics-guided loop engineering of an alcohol dehydrogenase: capture, turnover and enantioselective transformation of difficult-to-reduce ketones. Adv Synth Catal. https://doi.org/10.1002/adsc.201900249
Maclaren WR, Bruff WC, Eisenberg BC, Weiner H, Martin WH (1955) A clinical comparison of carbinoxamine maleate, tripelennamine hydrochloride, and bromodiphenhydramine hydrochloride in treating allergic symptoms. Ann Allergy 13(3):307–312
Moa S, Himo F (2017) Quantum chemical study of mechanism and stereoselectivity of secondary alcohol dehydrogenase. J Inorg Biochem 175:259–266
Morris RH (2009) Asymmetric hydrogenation, transfer hydrogenation and hydrosilylation of ketones catalyzed by iron complexes. Chem Soc Rev 38(8):2282–2291
Musa MM, Lott N, Laivenieks M, Watanabe L, Vieille C, Phillips RS (2009) A single point mutation reverses the enantiopreference of Thermoanaerobacter ethanolicus secondary alcohol dehydrogenase. ChemCatChem 1(1):89–93
Nealon CM, Musa MM, Patel JM, Phillips RS (2015) Controlling substrate specificity and stereospecificity of alcohol dehydrogenases. ACS Catal 5(4):2100–2114
Nie Y, Wang S, Xu Y, Luo S, Zhao Y, Xiao R, Montelione GT, Hunt JF, Szyperski T (2018) Enzyme engineering based on X-ray structures and kinetic profiling of substrate libraries: alcohol dehydrogenases for stereospecific synthesis of a broad range of chiral alcohols. ACS Catal 8(6):5145–5152
Noyori R (2002) Asymmetric catalysis: science and opportunities (Nobel lecture). Angew Chem Int Ed Engl 41(12):2008–2022
Qu G, Lonsdale R, Yao P, Li G, Liu B, Reetz MT, Sun Z (2018) Methodology development in directed evolution: exploring options when applying triple-code saturation mutagenesis. ChemBioChem 19(3):239–246
Reetz MT (2011) Laboratory evolution of stereoselective enzymes: a prolific source of catalysts for asymmetric reactions. Angew Chem Int Ed Engl 50(1):138–174
Reetz MT (2013a) Biocatalysis in organic chemistry and biotechnology: past, present, and future. J Am Chem Soc 135(34):12480–12496
Reetz MT (2013b) The importance of additive and non-additive mutational effects in protein engineering. Angew Chem Int Ed Engl 52(10):2658–2666
Reetz MT (2017) Recent advances in directed evolution of stereoselective enzymes. In: Alcalde M (ed) In directed enzyme evolution: advances and applications. Springer, Stuttgart, pp 69–99
Reetz MT, Carballeira JD (2007) Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat Protoc 2(4):891–903
Reetz MT, Wang LW, Bocola M (2006) Directed evolution of enantioselective enzymes: iterative cycles of CASTing for probing protein-sequence space. Angew Chem Int Ed Engl 45(8):1236–1241
Reetz MT, Prasad S, Carballeira JD, Gumulya Y, Bocola M (2010) Iterative saturation mutagenesis accelerates laboratory evolution of enzyme stereoselectivity: rigorous comparison with traditional methods. J Am Chem Soc 132(26):9144–9152
Sandoval CA, Li Y, Ding K, Noyori R (2008) The hydrogenation/transfer hydrogenation network in asymmetric reduction of ketones catalyzed by [RuCl2(binap)(pica)] complexes. Chem Asian J 3(10):1801–1810
Schrödinger Suite 2015-1 (2015) LLC, New York
Sheldon RA, Pereira PC (2017) Biocatalysis engineering: the big picture. Chem Soc Rev 46(10):2678–2691
Sun Z, Lonsdale R, Ilie A, Li G, Zhou J, Reetz MT (2016a) Catalytic asymmetric reduction of difficult-to-reduce ketones: triple-code saturation mutagenesis of an alcohol dehydrogenase. ACS Catal 6(3):1598–1605
Sun Z, Li G, Ilie A, Reetz MT (2016b) Exploring the substrate scope of mutants derived from the robust alcohol dehydrogenase TbSADH. Tetrahedron Lett 57(32):3648–3651
Sun Z, Lonsdale R, Li G, Reetz MT (2016c) Comparing different strategies in directed evolution of enzyme stereoselectivity: single-versus double-code saturation mutagenesis. ChemBioChem 17(19):1865–1872
Sun Z, Liu Q, Qu G, Feng Y, Reetz MT (2019) The utility of B-factors in protein science: interpreting rigidity, flexibility and internal motion and engineering thermostability. Chem Rev 119:1626–1665
Takahashi H, Ishida-Yamamoto A, Iizuka H (2004) Effects of bepotastine, cetirizine, fexofenadine, and olopatadine on histamine-induced wheal- and flare-response, sedation, and psychomotor performance. Clin Exp Dermatol 29(5):526–532
Xu G, Wang Y, Tang M, Zhou J, Zhao J, Han R, Ni Y (2018) Hydroclassified combinatorial saturation mutagenesis: reshaping substrate binding pockets of KpADH for enantioselective reduction of bulky–bulky ketones. ACS Catal 8(9):8336–8345
Acknowledgements
Not applicable.
Funding
This work was financially supported by CAS Pioneer Hundred Talents Program (No. 2016-053), National Natural Science Foundation of China (No. 31700698), the Key Research Program of the Chinese Academy of Sciences (No. KFZD-SW-212 and KFZD-SW-215), the Natural Science Foundation of Tianjin (No. 18JCYBJC24600), Open Project Funding of the State Key Laboratory of Bioreactor Engineering (No. 2017OPEN02), the Key Projects in the Tianjin Science & Technology Pillar Program (No. 15PTCYSY00020).
Author information
Authors and Affiliations
Contributions
ZS designed the concept of this study. BL carried out the experimental procedures. GQ and YJ performed theoritical analysis and drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors approved the consent for publishing the manuscript to bioresources and bioprocessing.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1.
Additional tables and figures including: Table S1. Summary of the TbSADH residues involved in this work. Table S2. Rosetta docking results. Table S3. List of primers for site-directed saturation mutagenesis. Table S4. Analytical conditions of HPLC. Table S5. Analytical conditions of GC. Figure S1. SDS-PAGE analysis of the protein expression of the variants △A85 (“△” indicates deletion), △I86 and △A85/△I86. As shown, all the three variants are well expressed. M: molecular markers. Lines 2~4, 5~7, 8~10 and 11~13 represent: whole cell lysate, soluble protein, inclusion body and empty vector, respectively. BL21(DE3) harboring the empty vector pRSFDuet-1 was used as a negative control. The expected size of TbSADH (37.58 KDa) is indicated by black arrow. Figure S2. The non-bonded interactions between ketone 1 and mutant A85G/I86A/Q101A, investigated by PLIP tool (Salentin et al. 2015). And computational command lines for running Rosetta Docking.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Qu, G., Liu, B., Jiang, Y. et al. Laboratory evolution of an alcohol dehydrogenase towards enantioselective reduction of difficult-to-reduce ketones. Bioresour. Bioprocess. 6, 18 (2019). https://doi.org/10.1186/s40643-019-0253-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40643-019-0253-9