
Abstract
Background
In addition to the risk of developing ventilator-induced lung injury, patients with ARDS are at risk of developing hyperoxic injury due the supra-physiological oxygen supplementation clinically required to reverse hypoxemia. Alterations of endogenous surfactant system participate in the pulmonary dysfunction observed in ARDS. Administration of exogenous surfactant could have protective effects during hyperoxia.
Methods
Male BALB/c mice (8–10 weeks), a strain highly sensitive to hyperoxia, received the exogenous surfactant-containing protein SP-B and SP-C by intranasal instillation 12 h before starting 24 h of exposure to hyperoxia in an inhalation chamber and were compared to mice receiving hyperoxia alone and to controls subjected to normoxia.
Results
Compared to the hyperoxia group, the administration of exogenous surfactant was able to reduce lung inflammation through a reduction in the influx of neutrophils and inflammatory biomarkers such as TNF, IL-17, and HMGB1 expression. The antioxidant activity prevented oxidative damage by reducing lipid peroxidation and protein carbonylation and increasing superoxide dismutase activity when compared to the hyperoxia group.
Conclusion
Our results offer new perspectives on the effects and the mechanism of exogenous surfactant in protecting the airway and lungs, in oxygen-rich lung microenvironment, against oxidative damage and aggravation of acute inflammation induced by hyperoxia.
Similar content being viewed by others


Background
Studies have shown that alterations of the endogenous surfactant system contribute to pulmonary dysfunction and atelectasis in ARDS [1]. These alterations include decrease of pool size and functional modifications of endogenous surfactant. Mechanical ventilation by itself can lead to the oxidation of surfactant components [1]. A meta-analysis of studies in adult patients with ARDS showed that exogenous surfactant (ES) administration may improve oxygenation in the first 24 h but does not mitigate mortality and long-term oxygenation (> 120 h) [2]. Important concerns have been raised, however, regarding the practical administration of surfactant and its ability to reach the alveoli [3]. Recently, our research team showed that ES administration decreased oxidative stress in the lungs of mice exposed to cigarette smoke [4].
Hyperoxia can promote changes in the function of surfactant subtypes [5] and is able to induce inflammation and oxidative stress [6]. In humans, an administration of high concentrations of oxygen has shown that the lung is susceptible to oxygen toxicity after breathing pure oxygen for only 17 h [7]. Several studies have demonstrated a higher risk of mortality in patients treated with high concentrations of oxygen, but the mechanisms are unclear [8,9,10,11]. Several studies using knock-out mice showed that deficiencies in several surfactant proteins (SP-A, SP-B, SP-C, and SP-D) result in higher inflammation induced by hyperoxia [12,13,14] and upregulation of cytokines, such as the high mobility group box protein 1 (HMGB1) [15]—a pro-inflammatory cytokine expressed by lung epithelial and endothelial cells and alveolar macrophages [16], and which attracts neutrophils, releases cytokines as TNFα, and inhibits macrophages migration [17]. SP-D can also inhibit the secretion of TNF, IFNγ, and IL-6 [18]; decreases peroxidation of surfactant lipid mixtures [19]; and reduces recruitment of inflammatory cells to lungs after infection [20], while SP-A inhibits IL-10 production and decreases recruitment of macrophages [21]. It has been shown that the inhibition of extracellular HMGB1 attenuated hyperoxia-induced inflammatory acute lung injury followed by reduction of leukocyte and polymorphonuclear cells [16], while other experimental models confirm that the depletion of HMGB1 reduces acute lung injury [22, 23]. None of these experimental models, however, was originally designed to investigate in vivo the combined effects of surfactant on hyperoxia-induced inflammation.
This study evaluated the effect of exogenous surfactant administration to prevent hyperoxic acute lung injury in BALB/c adult mice. This mice strain was used due to its sensitivity to hyperoxia [6].
Methods
Animals
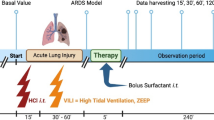
Male BALB/c mice (8–10 weeks, 20–25 g) were purchased from the Animal Science Center (ASC) of the Federal University of Ouro Preto (Ouro Preto, MG, Brazil) in a controlled-environment with cycled lighting (12 h light/12 h dark, lights on at 6:00 PM), with controlled temperature (21–22± 2 °C) and relative humidity (50 ± 10%). The animals received food and water ad libitum. The experimental design was approved by the Ethics Committee for Animal Research of UFOP (No. 2015/14).
The animals were divided into four groups: Control group (CG)—control mouse was exposed to normoxia in air room. Surfactant group (SG)—mice were challenged with ES by intranasal instillation 12 h before hyperoxia. Hyperoxia group (HG)—the animals were exposed to 100% oxygen in a chamber for 24 h. Hyperoxia-surfactant group (HSG)—mice were challenged with ES by intranasal instillation 12 h before hyperoxia followed by exposure to 100% oxygen in a chamber for 24 h.
Hyperoxia protocol
A cylinder containing 8000 L (180 Kgf/cm2, White Martins, Praxair Inc., São Paulo, Brazil) medical O2 was coupled to the Bourdon tube and the Thorpe tube (0–15 L / min.). A silicone conduit was connected to the Thorpe tube and the oxygen inhalation chamber (20x15x30cm), as described previously [6]. At the end of the oxygen exposure, the animals were euthanized by an overdose of ketamine (130 mg/kg) and xylazine (0.3 mg/kg).
Surfactant administration
The ES (The CUROSURF brand surfactant (Chiesi Farmaceutici S.p.A., Parma, Italy) was administered by intranasal instillation 12 h before exposure to hyperoxia in only one dose of 2.0 mL/kg/day (50 μL) [4].
Bronchoalveolar lavage fluid
The thorax of each animal was opened for collection of the bronchoalveolar lavage fluid (BALF). The left lung was clamped, the trachea was cannulated and the right lung was washed with 3 × 500 μL of saline solution. A 250 μL/sample was centrifuged at 1000 rpm for 1 min (cytospin technique—g Force). The total count of cells in the BALF was performed using a Neubauer chamber and the differential count using Panotic stained with Fast (Laborclin, Paraná, BR). The identification of inflammatory cells and differential counts was performed on slides by light microscopy [24].
Tissue processing and homogenization
After BALF collection, the right ventricle of each animal was perfused with saline to remove blood from the lungs. The right lung was clamped so that only the left lung was perfused with buffered 4% formalin (pH 7.2) via the trachea. The material was then processed and stained with hematoxylin and eosin for histological analysis. The right lung was removed and stored in labeled tubes. Afterwards, the right lung was homogenized in phosphate buffer (pH 7.5) and centrifuged at 10.000 rpm for 10 min. Supernatant was collected, and the samples were stored (− 80 °C) for biochemical analyses.
Immunoassays for inflammatory markers in BALF
BALF were used for the analyses of TNF, IL-17, and CCL5. Immunoassays (Peprotech kits, Ribeirão Preto, Brazil) were performed in 96-well plates on which 100 μl of monoclonal antibody to the protein (or peptide) of interest was added and samples were diluted in PBS containing 0.1% bovine serum albumin-BSA (Sigma-Aldrich, St Louis, MO). After incubation for 12 h at room temperature, the plates were blocked with 300 μl/well of a PBS solution containing 1% BSA for 1 h at 37 °C. Samples were applied in a volume of 25 μl to each well. The reaction was read on ELISA reader at 490 nm.
Antioxidant enzymes and biomarkers of oxidative damage
All chemicals were purchased from Sigma-Aldrich Chemical Co., (Sigma-Aldrich Inc., St. Louis, MO, USA). All measurements described below were performed on lung homogenates using a spectrophotometer (Beckman model DU 640; Fullerton, CA) or a microplate reader (Bio-Rad model 550, Hercules, CA). Catalase (CAT) activity was calculated from the rate of decrease in the concentration of hydrogen peroxide (U/mg protein), which was determined from the absorbance at 240 nm. The SOD activity was measured according to the Marklund method [25] which is based on the ability of the enzyme to inhibit the auto-oxidation of pyrogallol. Absorbances were read on ELISA reader at a wavelength of 570 nm. The oxidative damage was determined by levels of malondialdehyde (MDA) measured during an acid-heating reaction with thiobarbituric acid and was determined from the absorbance at 535 nm (described by Buege et al.) [26]. The carbonylation of proteins was performed (according to Levine et al. [27]). The total protein analysis was performed by the Bradford method [28].
Morphometric stereological analysis
The volume density analyses of the alveolar septum (Vv [sa]) and the alveolar spaces (Vv [a]) were performed in a test system composed of 16 points and a known test area. The test system was coupled to a monitor attached to a microscope. The number of points (Pp) that reached the alveolar septa (Vv [sa]) and the alveolar spaces (Vv [a]) were evaluated according to the total number of points in a test system (Pt). The reference volume was estimated by the point-of-use counting of the test point (Pt) systems. A total area of 1.94 mm2 was analyzed to determine the volume densities of the alveolar septa (Vv [sa]) and the alveolar spaces (Vv [a]) in sections stained with H&E, respectively [24, 29].
Immunohistochemical assay
Histological sections were stained with HMGB1 (EPR3507) (Abcam, UK) by immunohistochemistry. Morphometric analysis of the sections stained by immunohistochemistry was performed in 20 random fields of the slides photographed at a magnification of × 20 using ImageJ 1.6.0 software. In each field, the total number of nuclei and the number of nuclei labeled for the antibody used were counted, and the ratio of labeled nuclei/total nuclei was calculated [30].
Blood collection
A blood aliquot of each animal was collected by means of cardiac puncture in polypropylene tubes with 15 μL of anticoagulants to evaluate hematological parameters. Bc2800vet Auto Hematology Analyzer (Mindray® Bio-Medical Electronics Co. Ltda, Shenzhen, China) was used for the analyses. Futhermore, to perform the blood smear, 2 μL of blood was pipetted (automatic pipette) at one end of a sterile histological slide. They were stained with the Quick Panoptic Kit (Laborclin, Pinhais, Paraná, Brazil). A total of 100 leukocytes were counted per slide, which were differentiated into monocytes, neutrophils, and lymphocytes [31].
Statistical analysis
The sample size was calculated using a statistical power of 95% and a level of significance of 5% (BioEstat 5.3). The variable used to calculate power was alveolar macrophages, we were looking for 30% reduction. Data were expressed as mean ± SD. Evaluation of data normality was performed using the Kolmogorov-Smirnov test. The univariate analysis of variance (ANOVA one-way) followed by Newman-Keuls post-test was used for parametric data. The Kruskal-Wallis test followed by Dunn’s post-test was used for nonparametric data. A significant difference was considered when p < 0.05. All analyses were performed using GraphPad Prism software version 5.00 for Windows 7, GraphPad Software (San Diego, CA, USA).
Results
Total and differential leukocyte count in BALF
Hyperoxia promoted a reduction of total leukocytes (ANOVA; P < 0.0001) and macrophages (ANOVA; P < 0.0001) compared to control and surfactant groups (p < 0.05), though the absolute levels of neutrophils (ANOVA; P = 0.0068) were significantly higher than in control group animals (p < 0.05). The exogeneous surfactant administration decreased the levels of neutrophils and increased the levels of macrophages in animals exposed to hyperoxia (p < 0.05). Lymphocytes were not different than from the control group. By comparison to the control, surfactant alone did not induce any change in the level of inflammatory cells (Table 1).
Inflammatory markers in BALF
The levels of TNF, CCL5 and IL-17 in BALF were analyzed to assess the effects of exogeneous surfactant administration. Hyperoxia resulted in an increase in the levels of TNF (ANOVA; p = 0.0003) and IL-17 (ANOVA; p = 0.0006) when compared to control and surfactant groups (p < 0.05). These levels decreased with the administration of surfactant compared to hyperoxia group (p < 0.05). In addition, the exposure to hyperoxia resulted in a decrease of CCL5 (ANOVA; p = 0.0028) compared to control and surfactant group (p < 0.05). Surfactant administered before hyperoxia was not able to restore the levels of CCL5 compared to hyperoxia group. By comparison to control, surfactant alone did not induce any change in inflammatory markers in BALF (Table 2).
Redox status analyses
The analysis of lung homogenate revealed important protective effects of the surfactant on hyperoxia-induced pulmonary damage. The exposure to 100% oxygen resulted in increased levels of the malondialdehyde (ANOVA; p = 0.0143) and protein carbonyl content (ANOVA; p = 0.0167) when compared to control and surfactant groups (p < 0.05). The levels of malondialdehyde and protein carbonyl were reduced compared to animals exposed to hyperoxia (p < 0.05). The SOD activity (ANOVA; p = 0.0125) decreased in the presence of 100% oxygen but increased with administration of exogenous surfactant (p < 0.05). Regarding CAT activity (ANOVA; p = 0.1750), there were no difference among the experimental groups. By comparison to control, surfactant alone did not induce any change in relation the redox status in lung homogenate in our experimental model of exposed to hyperoxia (Table 2).
Morphometric analyses of pulmonary parenchyma
The stereological analyses showed no significant difference among groups for Vv [a] (ANOVA; p = 0.1994) and Vv [sa] (ANOVA; P = 0.4955) as observed in Fig. 1. We investigated the expression of HMGB1 in lung parenchyma by immunohistochemistry. Alone, hyperoxia led to a significant increase of HMGB1 expression (ANOVA; P = 0.0013) in lung parenchyma compared to control and surfactant groups (p < 0.05). The administration of exogenous surfactant was able to reduce the levels of HMGB1 in lung parenchyma of animals exposed to hyperoxia (p < 0.05). By comparison to control, surfactant alone did not induce any change the levels of HMBG1 (Fig. 2).

Stereological analyses of lung sections. a Volume density of alveolar septa and b volume density of alveolar airspace. c Histological section of lung parenchyma stained with HE. Barr = 50 μm. CG control group, SG surfactant group, HG hyperoxia group, HSG hyperoxia and surfactant group

Immunohistochemistry for HMGB1. a Histological section of lung parenchyma stained by immunohistochemical technique. Barr = 100 μm. The arrows point to marked nuclei. b Ratio between the number of nuclei labeled for HMGB1 antibody and the total number of nuclei. Data are expressed as mean ± SD (n = 9). (a) represents a significant difference between the groups when compared to the CG; the letter (b) is significant difference between the groups when compared to the SG, and letter (c) represents a significant difference between the groups when compared to the HG. Data were analyzed by one-way ANOVA followed by Newman-Keuls’s post-test
Hematological parameters
The exposure to hyperoxia increased the number of erythrocytes (ANOVA; p = 0.0024), hematocrit (ANOVA; p = 0.0061), hemoglobin (ANOVA; p = 0.0127) when compared to control and surfactant groups (p < 0.05), while surfactant administered before hyperoxia prevented most of the effects when compared to hyperoxia group (p < 0.05). There was a decrease in leucocytes (ANOVA; p = 0.0044) when compared to control and surfactant groups (p < 0.05). Monocytes (ANOVA; p = 0.0374) and neutrophils (ANOVA; p < 0.0001) were lower in animals exposed to hyperoxia than in the control group (p < 0.05). Lymphocytes were not different than from control group (Table 3).
Discussion
In this study, we evaluated the preventive effects of exogenous surfactant administration on an experimental model of exposure to pure oxygen. Our results showed that administration of exogenous surfactant was able to reduce lung inflammation through the reduction of inflammatory biomarkers such as TNF and HMGB1, as well as antioxidant activity by reducing the redox imbalance caused by exposure to hyperoxia. In regards to hyperoxia, our results show similar levels of BAL inflammatory cells and TNF which have been previously associated with lung tissue injury (based on MMP2, MMP9, and inflammatory scores) in studies from our group [6].
Immunomodulatory and anti-inflammatory effect of exogenous surfactant exposure prior to hyperoxia
In the present study, we observed a lower number of monocyte and macrophages in hyperoxia conditions with concomitant low expression of CCL5, possibly because hyperoxia-induced apoptosis of alveolar macrophages (as in culture) mediated by mitogen-activated protein kinase pathway [32].
Macrophages are important producers of CCL5 [33], and the exposure to hyperoxia leads to programmed death of the alveolar macrophages. The absence of alveolar macrophages activated seems to polarize immune response toward T cells and dendritic cells [34].
In the present study, high levels of neutrophils were attracted into the lung, probably resulting from a chemoattraction guided by HMGB1-modulated IL-17. This could have important influence on oxidative damage, because neutrophils are a great source of reactive oxygen species [35]. In a previous study, a concomitant elevation of IL-17 levels and neutrophils in the airways induced by hyperoxia were observed [36]. A precursor study showed that hyperoxia-induced inflammatory acute lung injury can be attenuated by inhibition of extracellular HMGB1 [16]. In turn, IL-6 leads to higher STAT3 expression (including the stimulation of hSP-B transcription in respiratory epithelial cells) [37]. These cytokines promote naive T cells differentiation to Th-17, mediated by ROR gamma, STAT3 transcription factor [38] and HMGB1—an important promoter of Th17 cell differentiation through the elevation of ROR-γt mRNA expression [39].
Antioxidant effect of exogenous surfactant exposure prior hyperoxia insult
The antioxidant effect of SP-B was observed in the present study, in vivo, when the levels of SOD, TBARS, and protein carbonyl returned to normal after exposure to exogenous surfactant on hyperoxia-induced by inflammation. A recent study showed that SP-B exhibits good scavenging activities on HO−·and O2− [40]. We speculate that protection against hyperoxia-induced inflammatory acute lung injury is initiated by the ability of the surfactant to significantly reduce oxidant radicals. Without oxidative stress and lipid peroxidation, cell membranes remain intact and the primary immune barrier is maintained. In addition, normal levels of reactive oxygen species maintain cell regulation, without apoptosis and cell signaling directed toward lung inflammation [41,42,43]. In vitro studies showed, however, that both surfactants, endogenous and exogenous, may be subjected to oxidative inactivation [44] and the oxidative modification, and functional inactivation of SP-A is accentuated when SP-A underwent lipoperoxidation [45]. Further studies will be needed to assess the importance of this phenomenon.
Limitations of exogenous surfactant administration in adult patients with ARDS
Despite a strong biological rationale and positive results in pediatric patients, surfactant therapy has failed to demonstrate a mortality benefit in adult patients with ARDS [3]. One potential explanation for the lack of positive findings may stem from a lack of surfactant reaching the distal airways and alveoli as a result of suboptimal instilled dose volume. In recent computational and airway modeling experiments [46], it has been suggested that adult conducting airways have almost 100-fold more surface airway compared to neonatal airways and therefore require considerable amounts of instilled surfactant to become fully coated. Only after saturation of the proximal conducting airways can the distal airways be reached. Further mechanistic support for suboptimal delivery could be explained by highly concentrated formulation of surfactant used in adult studies. The same computational modeling [46] suggests that highly concentrated surfactant could further increase viscosity, thereby preventing distal delivery as may have been the case in two unsuccessful controlled trials in adult patients with ARDS by Spragg et al. [47, 48]. Furthermore, although we did not directly measure the lung distribution of surfactant, our experimental model of exogenous surfactant administration was modeled after the study by Ganguly et al. [49]. They demonstrated that intranasally administered particles deposited in non-target lung locations were translocated to peripheral sites in the lung therapeutically after surfactant application [49].
On the other hand, new studies with KO animals (HMGB1−/−) and/or pharmacological antagonists might have reinforced mechanistic relationships over association between surfactant treatment and changes in biological biomarkers in our experimental model or the association of hyperoxia and mechanical ventilation. This model does not include mechanical ventilation which would be present in the clinical setting. However, this allowed us to specifically identify the effects of surfactant by itself.
Conclusions
In summary, our study demonstrated that the administration of exogenous surfactant was able to reduce the inflammation and oxidative stress of the lungs induced by hyperoxia in our experimental model. Our results open new perspectives for research on the mechanism of exogenous surfactant in protecting the airway and lungs, in oxygen-rich lung microenvironment, against oxidative damage and aggravation of acute inflammation induced by hyperoxia.
Abbreviations
- ANOVA:
-
Analysis of variance
- ARDS:
-
Acute respiratory distress syndrome
- ASC:
-
Animal Science Center
- BALF:
-
Bronchoalveolar lavage fluid
- CAT:
-
Catalase
- CCL5:
-
Chemokine (C-C motif) ligand 5
- ES:
-
Exogenous surfactant
- HMGB1:
-
High mobility group box protein 1
- HO:
-
Hydroxyl radical
- IFN:
-
Interferon
- IL-10:
-
Interleukin-10
- IL-17:
-
Interleukin-17
- IL-6:
-
Interleukin-6
- MDA:
-
Malondialdehyde
- O2 − :
-
Superoxide radical
- ROR-γt:
-
Mutant transcription factor retinoic acid-related orphan receptor-gamma t
- SOD:
-
Superoxide dismutase
- STAT3:
-
Signal transducer and activator of transcription 3
- TBARS:
-
Thiobarbituric acid reactive substances
- TNF:
-
Tumor necrosis factor
- VILI:
-
Ventilator-induced lung injury
- Vv [a]:
-
Volume density of the alveolar space
- Vv [sa]:
-
Volume density of the alveolar septum
References
Zenri H, Rodriquez-Capote K, McCaig L, Yao LJ, Brackenbury A, Possmayer F et al (2004) Hyperoxia exposure impairs surfactant function and metabolism. Crit Care Med 32(5):1155–1160
Meng H, Sun Y, Lu J, Fu S, Meng Z, Scott M et al (2012) Exogenous surfactant may improve oxygenation but not mortality in adult patients with acute lung injury/acute respiratory distress syndrome: a meta-analysis of 9 clinical trials. J Cardiothorac Vasc Anesth 26(5):849–856. https://doi.org/10.1053/j.jvca.2011.11.006
Grotberg JB, Filoche M, Willson DF, Raghavendran K, Notter RH (2017) Did reduced alveolar delivery of surfactant contribute to negative results in adults with acute respiratory distress syndrome? Am J Respir Crit Care Med 195(4):538–540. https://doi.org/10.1164/rccm.201607-1401LE
Machado DF, Campos KKD, da Silva NP, de Oliveira Ramos C, Cangussu SD, de Paula Costa G et al (2018) The administration of surfactant decreased oxidative stress in lungs of mice exposed to cigarette smoke. Int Immunopharmacol 54:275–279. https://doi.org/10.1016/j.intimp.2017.11.023
Dombrowsky H, Tschernig T, Vieten G, Rau GA, Ohler F, Acevedo C et al (2006) Molecular and functional changes of pulmonary surfactant in response to hyperoxia. Pediatr Pulmonol 41(11):1025–1039. https://doi.org/10.1002/ppul.20443
Nagato AC, Bezerra FS, Lanzetti M, Lopes AA, Silva MA, Porto LC et al (2012) Time course of inflammation, oxidative stress and tissue damage induced by hyperoxia in mouse lungs. Int J Exp Pathol 93(4):269–278. https://doi.org/10.1111/j.1365-2613.2012.00823.x
Davis WB, Rennard SI, Bitterman PB, Crystal RG (1983) Pulmonary oxygen toxicity. Early reversible changes in human alveolar structures induced by hyperoxia. N Engl J Med 309(15):878–883. https://doi.org/10.1056/NEJM198310133091502
Page D, Ablordeppey E, Wessman BT, Mohr NM, Trzeciak S, Kollef MH et al (2018) Emergency department hyperoxia is associated with increased mortality in mechanically ventilated patients: a cohort study. Crit Care 22(1):9. https://doi.org/10.1186/s13054-017-1926-4
Helmerhorst HJ, Roos-Blom MJ, van Westerloo DJ, de Jonge E (2015) Association between arterial hyperoxia and outcome in subsets of critical illness: a systematic review, meta-analysis, and meta-regression of cohort studies. Crit Care Med 43(7):1508–1519. https://doi.org/10.1097/CCM.0000000000000998
Rincon F, Kang J, Maltenfort M, Vibbert M, Urtecho J, Athar MK et al (2014) Association between hyperoxia and mortality after stroke: a multicenter cohort study. Crit Care Med 42(2):387–396. https://doi.org/10.1097/CCM.0b013e3182a27732
Brenner M, Stein D, Hu P, Kufera J, Wooford M, Scalea T (2012) Association between early hyperoxia and worse outcomes after traumatic brain injury. Arch Surg 147(11):1042–1046. https://doi.org/10.1001/archsurg.2012.1560
Tokieda K, Iwamoto HS, Bachurski C, Wert SE, Hull WM, Ikeda K et al (1999) Surfactant protein-B-deficient mice are susceptible to hyperoxic lung injury. Am J Respir Cell Mol Biol 21(4):463–472. https://doi.org/10.1165/ajrcmb.21.4.3436
Nyp MF, Mabry SM, Navarro A, Menden H, Perez RE, Sampath V et al (2018) Lung epithelial-specific TRIP-1 overexpression maintains epithelial integrity during hyperoxia exposure. Physiol Rep 6(5). https://doi.org/10.14814/phy2.13585
Jain D, Atochina-Vasserman E, Kadire H, Tomer Y, Inch A, Scott P et al (2007) SP-D-deficient mice are resistant to hyperoxia. Am J Physiol Lung Cell Mol Physiol 292(4):L861–L871. https://doi.org/10.1152/ajplung.00145.2006
Entezari M, Javdan M, Antoine DJ, Morrow DM, Sitapara RA, Patel V et al (2014) Inhibition of extracellular HMGB1 attenuates hyperoxia-induced inflammatory acute lung injury. Redox Biol 2:314–322. https://doi.org/10.1016/j.redox.2014.01.013
Ding J, Cui X, Liu Q (2017) Emerging role of HMGB1 in lung diseases: friend or foe. J Cell Mol Med 21(6):1046–1057. https://doi.org/10.1111/jcmm.13048
Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ (2000) HMG-1 as a mediator of acute lung inflammation. J Immunol 165(6):2950–2954
Jain D, Atochina-Vasserman EN, Tomer Y, Kadire H, Beers MF (2008) Surfactant protein D protects against acute hyperoxic lung injury. Am J Respir Crit Care Med 178(8):805–813. https://doi.org/10.1164/rccm.200804-582OC
Bridges JP, Davis HW, Damodarasamy M, Kuroki Y, Howles G, Hui DY et al (2000) Pulmonary surfactant proteins A and D are potent endogenous inhibitors of lipid peroxidation and oxidative cellular injury. J Biol Chem 275(49):38848–38855. https://doi.org/10.1074/jbc.M005322200
LeVine AM, Elliott J, Whitsett JA, Srikiatkhachorn A, Crouch E, DeSilva N et al (2004) Surfactant protein-d enhances phagocytosis and pulmonary clearance of respiratory syncytial virus. Am J Respir Cell Mol Biol 31(2):193–199. https://doi.org/10.1165/rcmb.2003-0107OC
Salez L, Balloy V, van Rooijen N, Lebastard M, Touqui L, McCormack FX et al (2001) Surfactant protein a suppresses lipopolysaccharide-induced IL-10 production by murine macrophages. J Immunol 166(10):6376–6382
Jiang Z, Zhou Q, Gu C, Li D, Zhu L (2017) Depletion of circulating monocytes suppresses IL-17 and HMGB1 expression in mice with LPS-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol 312(2):L231–LL42. https://doi.org/10.1152/ajplung.00389.2016
Luan ZG, Zhang XJ, Yin XH, Ma XC, Zhang H, Zhang C et al (2013) Downregulation of HMGB1 protects against the development of acute lung injury after severe acute pancreatitis. Immunobiology 218(10):1261–1270. https://doi.org/10.1016/j.imbio.2013.04.013
Campos KK, Manso RG, Goncalves EG, Silva ME, de Lima WG, Menezes CA et al (2013) Temporal analysis of oxidative effects on the pulmonary inflammatory response in mice exposed to cigarette smoke. Cell Immunol 284(1–2):29–36. https://doi.org/10.1016/j.cellimm.2013.07.002
Marklund S, Marklund G (1974) Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem 47(3):469–474
Buege JA, Aust SD (1978) Microsomal lipid peroxidation. Methods Enzymol 52:302–310
Levine RL, Williams JA, Stadtman ER, Shacter E (1994) Carbonyl assays for determination of oxidatively modified proteins. Methods Enzymol 233:346–357
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Mandarim-de-Lacerda CA (2003) Stereological tools in biomedical research. An Acad Bras Cienc 75(4):469–486
de Souza ABF, Chirico MTT, Cartelle CT, de Paula Costa G, Talvani A, Cangussu SD et al (2018) High-fat diet increases HMGB1 expression and promotes lung inflammation in mice subjected to mechanical ventilation. Oxidative Med Cell Longev 2018:7457054. https://doi.org/10.1155/2018/7457054
Ramos CO, Campos KKD, Costa GP, Cangussu SD, Talvani A, Bezerra FS (2018) Taurine treatment decreases inflammation and oxidative stress in lungs of adult mice exposed to cigarette smoke. Regul Toxicol Pharmacol 98:50–57. https://doi.org/10.1016/j.yrtph.2018.07.008
Petrache I, Choi ME, Otterbein LE, Chin BY, Mantell LL, Horowitz S et al (1999) Mitogen-activated protein kinase pathway mediates hyperoxia-induced apoptosis in cultured macrophage cells. Am J Phys 277(3):L589–L595. https://doi.org/10.1152/ajplung.1999.277.3.L589
Bakogiannis C, Sachse M, Stamatelopoulos K, Stellos K (2017) Platelet-derived chemokines in inflammation and atherosclerosis. Cytokine. https://doi.org/10.1016/j.cyto.2017.09.013
Thepen T, Van Rooijen N, Kraal G (1989) Alveolar macrophage elimination in vivo is associated with an increase in pulmonary immune response in mice. J Exp Med 170(2):499–509
Winterbourn CC, Kettle AJ, Hampton MB (2016) Reactive oxygen species and neutrophil function. Annu Rev Biochem 85:765–792. https://doi.org/10.1146/annurev-biochem-060815-014442
Nagato AC, Bezerra FS, Talvani A, Aarestrup BJ, Aarestrup FM (2015) Hyperoxia promotes polarization of the immune response in ovalbumin-induced airway inflammation, leading to a TH17 cell phenotype. Immun Inflamm Dis 3(3):321–337. https://doi.org/10.1002/iid3.71
Yan C, Naltner A, Martin M, Naltner M, Fangman JM, Gurel O (2002) Transcriptional stimulation of the surfactant protein B gene by STAT3 in respiratory epithelial cells. J Biol Chem 277(13):10967–10972. https://doi.org/10.1074/jbc.M109986200
Korn T, Bettelli E, Oukka M, Kuchroo VK (2009) IL-17 and Th17 cells. Annu Rev Immunol 27:485–517. https://doi.org/10.1146/annurev.immunol.021908.132710
Shi Y, Sandoghchian Shotorbani S, Su Z, Liu Y, Tong J, Zheng D et al (2012) Enhanced HMGB1 expression may contribute to Th17 cells activation in rheumatoid arthritis. Clin Dev Immunol 2012:295081. https://doi.org/10.1155/2012/295081
Hu FY, Chi CF, Wang B, Deng SG (2015) Two novel antioxidant nonapeptides from protein hydrolysate of skate (Raja porosa) muscle. Mar Drugs 13(4):1993–2009. https://doi.org/10.3390/md13041993
Giorgio M, Trinei M, Migliaccio E, Pelicci PG (2007) Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol 8(9):722–728. https://doi.org/10.1038/nrm2240
Thannickal VJ, Fanburg BL (2000) Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol 279(6):L1005–L1028. https://doi.org/10.1152/ajplung.2000.279.6.L1005
Hensley K, Robinson KA, Gabbita SP, Salsman S, Floyd RA (2000) Reactive oxygen species, cell signaling, and cell injury. Free Radic Biol Med 28(10):1456–1462
Andersson S, Kheiter A, Merritt TA (1999) Oxidative inactivation of surfactants. Lung 177(3):179–189
Kuzmenko AI, Wu H, Bridges JP, McCormack FX (2004) Surfactant lipid peroxidation damages surfactant protein a and inhibits interactions with phospholipid vesicles. J Lipid Res 45(6):1061–1068. https://doi.org/10.1194/jlr.M300360-JLR200
Filoche M, Tai CF, Grotberg JB (2015) Three-dimensional model of surfactant replacement therapy. Proc Natl Acad Sci U S A 112(30):9287–9292. https://doi.org/10.1073/pnas.1504025112
Spragg RG, Lewis JF, Walmrath HD, Johannigman J, Bellingan G, Laterre PF et al (2004) Effect of recombinant surfactant protein C-based surfactant on the acute respiratory distress syndrome. N Engl J Med 351(9):884–892. https://doi.org/10.1056/NEJMoa033181
Spragg RG, Taut FJ, Lewis JF, Schenk P, Ruppert C, Dean N et al (2011) Recombinant surfactant protein C-based surfactant for patients with severe direct lung injury. Am J Respir Crit Care Med 183(8):1055–1061. https://doi.org/10.1164/rccm.201009-1424OC
Ganguly S, Moolchandani V, Roche JA, Shapiro PS, Somaraju S, Eddington ND et al (2008) Phospholipid-induced in vivo particle migration to enhance pulmonary deposition. J Aerosol Med Pulm Drug Deliv 21(4):343–350. https://doi.org/10.1089/jamp.2007.0675
Acknowledgements
The authors would like to express their gratitude to the Federal University of Ouro Preto and this work was supported by the Federal University of Ouro Preto (# 23109.003267/2017-01 and # 23109.003268/2017-47) and Coordenação de Aperfeiçoamento de Pessoal de Ensino Superior (CAPES) [Bolsista CAPES - PVEX - Process # 88881.172437/2018-01] to FSB. AT credits CNPq for the fellowship of research productivity. FSB is doing a Post-Doctoral Fellow in the Interdepartmental Division of Critical Care Medicine, St. Michael’s Hospital, University of Toronto, Toronto, ON, Canada.
Funding
This study was supported by Coordenação de Aperfeiçoamento de Pessoal de Ensino Superior (CAPES) and Federal University of Ouro Preto (UFOP).
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Contributions
FSB, COR, TFC, and NPSA participated in the design of the study, carried out the experiments, performed data analyses, and drafted the manuscript. ABFS, ACBB, CTC, and SDC performed the histological analyses and helped draft the manuscript. GPC and AT carried out the immunoassay analyses and contributed to the manuscript. LB and ACN helped write the manuscript, contributed to the study design, supervised the experimental work and statistical analysis, and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
All experiments were approved by Animal Ethics Committee at the Federal University of Ouro Preto, according to the protocol number 2015/14.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bezerra, F.S., Ramos, C.d., Castro, T.d. et al. Exogenous surfactant prevents hyperoxia-induced lung injury in adult mice. ICMx 7, 19 (2019). https://doi.org/10.1186/s40635-019-0233-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40635-019-0233-6