
Abstract
Stress emanating from drought condition is one of the inevitable conditions that occurs in many environments and spread across borders and results in severe hindrance to the morphology, physiology, biochemistry and molecular attributes of plants. Highland barley drought tolerance has been demonstrated to be a polygenic related characteristic and genetic composition that can assist in dissecting the gene network(s) controlling the drought tolerance needs to be adequately understood. DNA affinity purification sequencing (DAP-seq) has been shown to contribute to ways of making pure some of the protein with rare sequence-specific DNA binding and can help understand the molecular dynamics in barley under varying exposure time to drought. The present study aimed at identifying novel transcription factors (TFs) in highland barley that are key in drought resistance through DAP-seq-based transcriptomic analysis. The experimental design included two hulless barley accessions; drought-resistant Sheera 10 (X) and drought-sensitive 5171–7 (W), that were both subjected to short-term (4 h) and long-term (48 h) exposure to drought (T1 and T5, respectively), with the control group (CK) involving not subjecting the two accessions to any treatment. Through transcriptome analysis, one candidate transcription factor (GATA family; (bHLH, MYB-related, GARP-G2-like, bZIP, HB-HD-ZIP, C2H2, SET, mTERF, AP2/ERF-ERF, ARID, NAC, GARP-ARR-B, C2C2-GATA, FAR1, Trihelix, NF-YB, B3 and AUX/IAA) was found. The motif obtained was found to be consistent with GATA transcription factor. The DAP-seq highlighted the differential expression target gene which were verified by RT-qPCR. The associated genes were found to be a cluster of structural gene IP_HOR_1, IP_HOR_2 and In_HOR locus. Through RT-qPCR, it was also proved that the gene expressions were indeed upregulated. The TF HOVUSG2784400 was confirmed to be responsible to drought stress under long-term exposure, which regulates the differential expression of the genes, thereby improving the drought resistance of barley.
Graphical Abstract

Similar content being viewed by others

Introduction
Stress that emanates from drought conditions is one of the inevitable conditions that occur in many environments and spread across borders without any clear warning, resulting to reduced plant biomass productivity, crop quality and the regulation of energy in an ecosystem [15, 31]. Inadequacy in soil moisture has been found to be the key significant environmental stress that emerges when there is change in temperature conditions, dynamics in light intensity and drop in rainfall intensity [42]. The impact that drought inflict on crop production may occur from a time of cumulative effects that are also multidimensional in nature. These complexities result in severe negative impact on the morphological, physiological, biochemical as well as molecular characteristics of plants, hindering their photosynthetic capacity [17]. For the survival mechanism, different plant species have continued to evolve in numerous ways to develop complex resistance as well as adaptation mechanisms that range from physiological and biochemical responses to transcriptome factor modulations [12, 43]. The sophisticated adaptation strategies and existing regularity network involved in the mechanism of water stress resilience and adaptation in most crops have continued to be discussed. For instance, the pattern of growth and structural dynamics, losses and reduction through evapo-transpiration occurring from stomatal conductance, as well as distribution, the mechanism of rolling of leaf, the changes in the ratio between the root and the shoot, alteration in the root length, accumulation of adaptable solutes, promotion of transpiration efficiency, osmotic status regulation, hormonal status regulation and controlled senescence time are the mechanisms that are pursued by plants when subjected to water inadequacy have been widely studied [15, 31]. Efforts to develop methods for mechanisms for drought tolerance are creating mechanisms that greatly focuses on plant molecules and genes levels, giving special consideration to the emerging science of omics hi-tech manipulations that greatly aim to promote the plants adaptation to stressful conditions [24, 29].
From the trend occurring within the change of environmental variables, water has been noted to be the major limiting factor that controls crop productivity a cross the glob, and generating climate-resilient crops has remained a significant requirement [22]. Efforts towards highland barley productivity improvement have involved studies that have explored the effects brought by freeze–thaw, extreme drought as well as artemisinin stress within the coverage of the arid parts of the Qinghai-Tibet Plateau [26]. Indeed, focus has also been placed on the strategies plants portray that enable them to respond to extremities of drought and salinity stress. Most plants have been recorded to apply numerous mechanisms that depend on their physiology and biochemistry structure, as well as soil administration design, crop inauguration, including other crop development factors that maintain enough amount of water on the leaves to help in maintaining the performance of osmotic and stomatal function [32]. Therefore, drought as a conglomerate natural occurrence, there is need to provide understanding on numerous structures at the levels of physiology, biochemistry, and molecule with the aim of generating crops that have better drought tolerance while avoiding yield penalties [22]. Studies have pointed out that improvement at molecular levels is required to promote the final yields under water inadequacy situations. Through this, new genetically engineered barley with a high level of resilience to drought are formed through breeding by undertaking breeding from highly drought-resilient genetically engineered groups and picking from among their progeny [32].
Some of the observed traits that depict enhanced productivity of the drought resilience barley types under water deficit conditions may include early developmental stages, elaborate root development at deeper depths, and high efficient water use portrayed by high water take up at post-anthesis stage [3]. Indeed, drought tolerance has been demonstrated to be a polygenic trait and genetic constitution that can greatly assist in dissecting the gene network(s) that play great role in controlling the drought tolerance [27, 32,33,34]. Other findings have also highlighted synergistic effects resulting from numerous stress factors occurring together under drought condition causing more serious effect on barley than when they occur singularly [5].
Under genetic engineering for improved barley productivity, genes such as HvMYB1 have been shown to produce protection against drought by serving as an arbiter of abscisic acid performance [1]. Moreover, drought resistance in barley has been shown to be under the regulation of numerous genes, accounting transcription factors (TF) that makes the plant to combat the harsh conditions. These TFs represent the major molecular shifts that orchestrate the control of plant developmental actions in reply to numerous unfavorable conditions [20]. Therefore, continued efforts on identification and validation of the highly appropriate reference genes for correction manipulation of gene expression within drought distress condition is encouraged [4]. Generally, plants have undergone development that has brought about a system of averagely complex distress action to adapt to water limitation. These response systems often make use of the gene encoding a specific TF together with its target gene which constitutes a regulon, which takes part in signal transduction to activate and or silence genes involved in response to drought. Through in-depth molecular and genomic studies, some five specific families of TFs (AP2/EREBP, bZIP, MYB/MYC, NAC and WRKY) (out of > 80 known families of TFs) have continued to attract increased consideration based on their useful role in drought resistant in many of the plants (Gahlaut et al., 2016; [30].
Going by the efforts for promoting barley productivity under drought condition, describing genes that high promote resistance is key. Indeed, through molecular studies, it has been demonstrated that young barley plants use complicated approaches to gain resilience ability at their early stages of development, with more in-depth functional confirmatory analysis indicating that the candidate genes require adequately research to avail sufficient knowledge on the genetic manipulation of drought resistance at early stages of development [39]. DNA affinity purification sequencing (DAP-seq) has been proven to avail mechanisms for purifying the rare sequence-specific DNA-binding proteins. Indeed, the DAP-seq, a TF-binding site (TFBs) combines the TFs for affinity purification with the genomic library under the next-generation sequencing [2]. Therefore, the present study focused at further identifying novel transcription factor in highland barley that are key in drought resistance through DNA affinity purification sequencing-based transcriptomic analysis. The study aimed at contributing to success in barley production under extreme drought conditions.
Materials and methods
Material
Two hulless barley accessions, Sheera 10 and 5171–7, referred to in this study as X and W, respectively, were used the experiment. The two were chosen from the 1700 germplasm resources, originally screened for drought stress resilience [44]. The X had displayed a drought-resistance ability and W displayed drought-sensitive ability. The two accessions were both subjected to two different drought simulation treatments durations (T1 and T5). T1 was a short-term treatment for 4 h, while T5 was long-term treatments for 48 h. The control group (CK) was obtained by not subjecting the two accessions to any drought simulation treatment. From the treatments described above, the groups were marked as (X_T1, X_T5 and X_CK) for drought-resistant variety, and (W_T1, W_T5 and W_CK) for drought-sensitive variety. All the treatment groups were done in triplicate and marked as 1, 2 and 3.
Experimental design
Transcriptome detection of differentially expressed genes
Transcriptome detection of differentially expressed genes (DEG) was conducted for X-T1 vs X-CK, X-T5 vs X-CK, W-T1 vs W-CK, W-T5 vs W-CK. DNA extraction, library preparation and sequencing were as described in [8]. Under this study, total DNA libraries from the all the 12 treatments (X-T1 vs X-CK in triplicate, (X-T5 vs X-CK in triplicate, W-T1 vs W-CK in triplicate and W-T5 vs W-CK in triplicate) were constructed. Bases exceeding 97.18% and relatively 94% from the more than 900 million raw reads showed a q-values of between ≥ 20 and ≥ 30 with error of probability of 0.02 to 0.025%, respectively. The GC-content value was found to be in the range of 55.67 and 57.76%. After undertaking filtering out of the reads with low-quality, an approximate total of 800 million clean reads were produced. Trinity was then applied to produce 144,806 transcripts with N50 of 1705 bp and N90 of 645 bp.
Gene screening and annotation and motif analysis
From the transcriptome results, the gene HOVUSG2784400 was screened and confirmed to be upregulated in X-T5 vs X-CK, and then was annotated. RT-qPCR verification was then performed for the transcription factor. The DAP-seq being considered a very high-throughput TF-binding region detection method was applied in the in vitro expression of TFs to interrogate the HOVUSG2784400 genomic DNA to establish the binding position (peak) and sequence motif. This was done through data preprocessing using Illumina HiSeqTM2000/ Miseq/BGISEQ-500 that involved the removal of connector sequence, the contamination sequence and the low mass base. From this the clean data sequence was obtained that was used in data analysis. The clean data were then localized to the reference genome to obtain the bam file. The detection was then done at the Peaks to obtain the enriched region information. This was followed by the determination of the distribution of energy elements, recent gene searches and motif predictions. Finally, the statistical Peak distribution, GO, KEGG function annotation and enrichment of Peak's recent genes and transcription factor prediction was undertaken.
Motif Enrichment Analysis (MEA) was done on the promoter region to establish the DNA-binding transcription factors that regulate the transcription of a group of genes by discovering enrichment of familiar binding motifs in the genes' control regions. The Peak sequences were then used in predicting motifs.
Results
Transcriptome detection of differentially expressed genes
To identify DEGs under the short-term (T1) and long-term (T5) treatment durations for the drought-resistant versus the control (X_T1 versus X_CK and X_T5 versus X_CK) and drought-sensitive versus the control (W_T1 versus W_CK, W_T5 versus W_CK) barley genotypes in return to low water, RNA-seq was undertaken through Illumina HiSeq 2000. The level of relationship between the biological replicates was found not to be low (R2 = 0.87–0.99) concurring with the reproducibility of the findings. Overall, approximately 767 million raw reads, and 758 million clean reads each 50 nucleotides long, were generated for each sample. Approximately 85–90% of the reads were aligned to the reference genome.
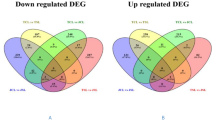
The calculation of transcript magnitude of every gene was computed as fragments per kilobase per million mapped reads (FPKM) (Figure a) and the quantified values were then applied in the determination of the DE as log2-fold change (FC) ratio among the control and the treatments for every time point and in every genotype. After employing the cut-off log2FC ≥ 2 for upregulated and ≤ − 2 for downregulated and the corrected q-value cut-off < 0.05, the DEGs were marked. The entire quantity of DE genes varied greatly among the genotypes and the control in relation to the various treatment times with the greatest quantities occurring under the long-term drought treatments (Fig. 1b). Within the DE genes, the quantities of upregulated genes were found to be less than the downregulated ones within all the treatments (Fig. 2), pin pointing that the barley genotypes were not similar in their reaction towards low water stress and the response towards this condition majorly associate up-regulation of some not many particular groups of drought-sensitive genes.

a Fragments contained in each kilobase per mapped reads in millions and b all sample reads for gene elements

Values of up- and down-regulated genes under W_T1 versus W_CK (a), W_T5 versus W_CK (b), X_T1 versus W_CK (c) and X_T5 versus X_CK (d)
Through GO Enrichment Analysis for DEGs, it was shown that under both the W_T1 versus W_CK and W_T5 versus W_CK, the cytoplasm and cytoplasmic vesicle gene sets were the most upregulated and downregulated, respectively. However, under the X_T1 versus W_CK, the heme binding and metal binding gene sets were, respectively, the most upregulated and downregulated, while under the X_T5 versus X_CK, binding transcription factor activity and cytoplasmic vesicle gene sets were, respectively, the most upregulated and downregulated, respectively.
Through KEGG Enrichment Analysis for DEGs, it was shown that under the W_T1 versus W_CK, phenylpropanoid biosynthesis and protein production within the endoplasmic reticulum were, respectively, the highly upregulated and downregulated KEGG pathways. Under W_T5 versus W_CK, phenylpropanoid biosynthesis and plant hormone signal transduction were, respectively, the most upregulated and downregulated KEGG pathways. Under the X_T1 versus X_CK, RNA transport and hormonal signal transduction in plant were the highly upregulated and downregulated KEGG pathways, respectively. Under the X_T5 versus X_CK, hormonal signal transduction in plant and biosynthesis of the phenylpropanoid were, respectively, the highly upregulated and downregulated KEGG pathways. The DEG annotation based on KEGG classification for the X_T5 versus X_CK is presented in Fig. 3.

KEGG classification for the X_T5 versus X_CK. Downregulated genes (a) and upregulated genes (b)
Gene screening and annotation
The transcriptome detection of differentially expressed genes was followed by the screening of HOVUSG2784400 gene and then upregulated in the X-T5 vs X-CK treatment group (Fig. 4). Based on that, the result for GO and KEGG enrichment is provided in Fig. 5. The study showed that iron-to-iron binding was the most expressed GO terms. Additionally, the metabolism of glycerolipid, starch as well as sucrose and plant hormone signal transduction portrayed high expression over expressed KEGG pathways. Through iTAK, the following GATA transcription factors were identified; bHLH (9), MYB-related (5), GARP-G2-like (5), bZIP (5), HB-HD-ZIP (4), C2H2 (4), SET (3), mTERF (3), AP2/ERF-ERF (3), ARID (3), NAC, GARP-ARR-B, C2C2-GATA (2), FAR1 (2), Trihelix (2), NF-YB (2), B3 (2), AUX/IAA (2) and other (26) (Additional file 1).

HOVUSG2784400 gene quantification across different treatments

GO enrichment (a) and KEGG enrichment for the HOVUSG2784400 gene
Functional elements and motif analysis
Through DAP-sequence analysis of HOVUSG2784400, the number of functional elements of each gene on the genome loci after statistical comparison of reads was plotted using the ChIPseeker/vennpie.R and presented in Fig. 6. Motif analysis was performed on the promoter region peak obtained by DAP-seq analysis, and the motif obtained found to be consistent with GATA transcription factor. The associated genes were found to be a cluster of structural gene IP_HOR_1, IP_HOR_2 and In_HOR locus. Through RT-qPCR, it was also proved that the gene expressions were indeed upregulated. The KEGG enrichment on the promoter region peak is provided in Fig. 7.

Functional elements of each gene on the genome loci a In_HOR, b IP_HOR_1, and c IP_HOR_2

KEGG enrichment analysis on the promoter region peak of structural gene loci a In_HOR, b IP_HOR_1 and c IP_HOR_2
Discussion
Seedling drought tolerance genetic architecture is a complex procedure that requires in-depth research. Through that, genetic engineered breeding for environmental resistant and hardy traits is key in ensuring improved crop productivity under extreme weather and climatic conditions. Indeed, the chosen genotypes can be more applicable in further improving drought resilience in barley with high productive potential [35]. Drought has remained a critical developmental factor for crop yield and its resilience has been shown to vary along genetical line, with little information on mediating molecular factors. More of the details for molecular and genetic modifications are useful factors in determining constraints on crop evolution, and therefore studies that provide further information on genes and their related natural alleles are required give knowledge of pleiotropy for new avenues in improvement [21]. Therefore, sufficient knowledge on the process of molecular action of the pleiotropic genetic mechanism and the phenotypically associated gene plasticity will give further knowledge into the resilience in local barley and avail fresh techniques required in promoting the crop productivity [21]. In the current study, it was shown that total quantity of DEGs varied between the genotypes and the control, which also occurred based on the contrasting duration of applying the treatment with the more quantity being observed under the long-term drought treatments. From this finding, it was demonstrated that resistant genes are expressed differently between the drought tolerant as well as the drought-resistant barley varieties and the variation is pegged on the time of exposure to environmental extreme. In fact, a previous study had demonstrated that the depicting of varying gene action to persistent unfavorable drought conditions pointed out the significant survival roles by gene regulations at different growth stages upon drought stress [14].
The existing advancement in drought tolerance crop research has been due to improvement in physiological, inter-breeding, and genetical aspects of studies. These three have given much emphasis on the physiology and biochemistry components of the metabolic networks that are applied by plants during the time they are subjected to the condition of drought-related stress. Therefore, alternative barley genotypic varieties with high level of drought resistance are being brought forward through genetic engineering of crosses from promising low water-resistant genotypes and choosing between such high-quality progeny. An additional identification of genes that make immense contribution to tolerance to low water conditions is extremely significant, and studies have shown tolerance to low water conditions as being a polygenic characteristics and genetic make-up is a useful process in dissecting the gene network(s) that take part in controlling the drought tolerance [33]. Specifically, in general, numerous polymorphisms of single nucleotide in nature were shown to be interconnected to several characteristics that are shared between numerous barley species, within which numerous genomic regions were shown to have a relation with candidate genes. Additionally, some of the previously documented quantitative trait loci (QTL) were associated to the survival of the barley l under low water conditions. Moreover, the determined QTL had similar colocalization with numerous genes with exclusive distribution on specific region of the chromosome. Within the DEGs, the quantity of upregulated genes were found to be lower than the downregulated under all the treatments, serving as an indication that the barley genotypes vary in their mode of action to low water stress and the their response under such conditions majorly entails up-regulation of few particular groups of drought-responsive genes [13].
Reports on genes like the ones from the AP2 and NAC families have indicated key TFs that are important in the regulation of drought-distressful actions in barley and have therefore been recommended as appropriate responsible genes for in-depth functional studies for promotion of barley productivity under low water condition [19]. From the present study, through GO Enrichment Analysis for DEGs, the cytoplasm, metal binding, binding transcription factor gene sets were the most upregulated. Indeed, TFs have remained useful points for gene manipulation for environmental stress resilience in plants and sequence-specific TFs control gene interpretation by binding to cis-regulatory factors in both the promoter and enhancer DNA [16]. A metal binding gene sets such as metallothionein, which are familiar in taking part in the process of metal homeostasis and detoxification and in reaction to oxidative stressful condition, their occurrence equally indicate a high aggregation of transcripts bounded to infected cells of nodules in reaction to low water conditions [7, 25]. The useful responsibilities of metallothionein genes in transgenic lines of chickpea in existence of varying stressful environmental conditions had also been associated with multi-stress tolerant, including conditions of extreme drought [23]. Moreover, transcriptome analysis has been widely used to mine drought tolerance genes in barley [41], but only a few studies have focused on wild barley from the China. In our study, the number of differentially expressed genes in drought-tolerant barley was significantly lower than that in drought-sensitive barley. These results support the conclusion of previous studies that, compared to drought-sensitive barley genotypes, drought-tolerant barley genotypes have more stable gene expression changes [18]. GO annotation analyses also revealed differences between the two barley varieties and treatment options. The tolerant variety showed strong energy metabolism (ATP metabolism GO:0042626; GO:0016887) and transport activity.
Through KEGG Enrichment Analysis for DEGs, phenylpropanoid biosynthesis, plant hormone signal transduction and RNA transport were found to be the largely upregulated pathways. Phenylpropanoid pathway is among the very significant metabolic pathways of plant secondary metabolism. Indeed, crops grown under challenging environments usually accumulate phenolic compounds to enhance plant tolerance [37]. Moreover, additional environmental stress factors like low water condition also stimulate the cell signaling mechanisms, bring about the transcriptional up-regulation of phenylpropanoid pathway. The development in resistant ability of most crops to low water availability has been shown to be connected with the multiple activities of polyphenols. Moreover, polyphenols might take part in other important environmental functions during unfavorable conditions, functioning for instance as info-chemicals that in most plants. Therefore, there is increased need to increase, for example, the work of specially designed polyphenols as a feedback to certain unfavorable environmental conditions and to support the operations intimal system which tend to change from main metabolism to the up-regulation of phenylpropanoid pathway as a cross response reaction to several unfavorable abiotic conditions [6, 9]. According to the KOG annotation, the successfully annotated unigenes were classified based on the 26 KOG groups, and among these, more were classified as translation, ribosomal structure, and biogenesis, which accounted for the highest proportion, followed by posttranslational modification, protein turnover, and chaperones. After the KO annotations of the unigenes, the biochemical pathways regulated were identified by KEGG metabolic pathway analysis. Under this numerous unigenes were also annotated and divided into KEGG pathways. Furthermore, the major TF families with the largest number of differentially regulated genes indicated that the families were widely involved in the regulation of the responses to drought stress. Overall, TFs clearly play crucial roles in the responses of barley to drought stress.
Plant hormone signal transduction contains ionic and osmotic homeostasis signaling pathways, detoxification and pathways for growth regulation. Genes have been found to initiate hormonal metabolism, and their cross-talk have as well been found to facilitate resilience to low water conditions in plants [38], and the current study therefore highlighted the regulatory circuits of phytohormones in drought tolerance mechanism. Furthermore, some miRNAs are practically conserved within different plant groups and are controlled by low water stress conditions, and as confirmed by the findings of this study, the regulation of the RNA transport pathways enhanced the tolerance to drought possibly by elimination of metabolites that can greatly influence the metabolism in the course of low water stressing conditions [36]. GATA TFs are type IV zinc-finger proteins that play significant role in the development as well as growth of plants [10], profiles for expressions showed that all the GATA genes were expressed in the barley under simulated drought condition and most were induced by the long-term treatment. Research studies have continued to recommend GATA transcription factor in improving the adaptation of sweet potato and other plants to abiotic stress [11, 39, 40].
The expressed GATA TFs (bHLH, MYB-related, GARP-G2-like, bZIP, HB-HD-ZIP, C2H2, SET, mTERF, AP2/ERF-ERF, ARID, NAC, GARP-ARR-B, C2C2-GATA, FAR1, Trihelix, NF-YB, B3 and AUX/IAA) could therefore be part of the barley bio-molecule reaction that promotes the plant tolerance to drought, especially under long-term tolerance. Indeed, the Motif analysis on the promoter region peak obtained by DAP-seq analysis consistency with GATA transcription factor also pointed to the same understanding. More so, the associated gene clusters IP_HOR_1, IP_HOR_2 and In HOR locus could also be a proof that that the gene expressions were indeed upregulated. In fact, the HOR-2 gene family encoding the B-hordeins has been shown to harbor 15–30 copies. The Hor2 locus of barley, which specifies B hordein, is a complex group of genes that arise from the multiplication of a single ancestral gene [28], and needs to be studied for increased knowledge on its role in environmental tolerance.
Conclusion
Through the transcriptome results under different treatments of two materials (anti-vs no drought resistance), a candidate transcription factor (GATA family, HOVUSG2784400) was found, and then DAP-seq performed identified the DNA bound by the transcription factor. The DAP-seq highlighted the differential expression target gene which were verified by RT-qPCR as the differential expression. Finally, the transcription factor HOVUSG2784400 was confirmed to be in response to low water stressful condition, which controls the differential expression of the genes, thereby improving the drought resistance of barley.
Availability of data and materials
The primary research data from this research are available and can be given on request application through the corresponding author.
References
Alexander RD, Wendelboe-Nelson C, Morris PC. The barley transcription factor HvMYB1 is a positive regulator of drought tolerance. Plant Physiol Biochem. 2019;142:246–53. https://doi.org/10.1016/J.PLAPHY.2019.07.014.
Bartlett A, O’Malley RC, Huang SSC, Galli M, Nery JR, Gallavotti A, Ecker JR. Mapping genome-wide transcription-factor binding sites using DAP-seq. Nat Protoc. 2017. https://doi.org/10.1038/nprot.2017.055.
Carter AY, Hawes MC, Ottman MJ. Drought-tolerant barley I field observations of growth and development. Agronomy. 2019. https://doi.org/10.3390/AGRONOMY9050221.
Cassol D, Cruz FP, Espindola K, Mangeon A, Müller C, Loureiro ME, Corrêa RL, Sachetto-Martins G. Identification of reference genes for quantitative RT-PCR analysis of microRNAs and mRNAs in castor bean (Ricinus communis L.) under drought stress. Plant Physiol Biochem PPB. 2016;106:101–7. https://doi.org/10.1016/J.PLAPHY.2016.02.024.
Chang Y, Zhang J, Bao G, Yan B, Qu Y, Zhang M, Tang W. Physiological responses of highland barley seedlings to NaCl, drought, and freeze-thaw stress. J Plant Growth Regul. 2020;40(1):154–61. https://doi.org/10.1007/S00344-020-10085-5.
Chen Y, Huang L, Liang X, Dai P, Zhang Y, Li B, Lin X, Sun C. Enhancement of polyphenolic metabolism as an adaptive response of lettuce (Lactuca sativa) roots to aluminum stress. Environ Poll. 2020. https://doi.org/10.1016/j.envpol.2020.114230.
Clement M, Lambert A, Herouart D, Boncompagni E. Identification of new up-regulated genes under drought stress in soybean nodules. Gene. 2008;426(1–2):15–22. https://doi.org/10.1016/J.GENE.2008.08.016.
Dudhate A, Shinde H, Tsugama D, Liu S, Takano T. Transcriptomic analysis reveals the differentially expressed genes and pathways involved in drought tolerance in pearl millet [Pennisetum glaucum (L) R Br]. PLoS ONE. 2018;13(4):0195908. https://doi.org/10.1371/JOURNAL.PONE.0195908.
Grace SG, Logan BA. Energy dissipation and radical scavenging by the plant phenylpropanoid pathway. Philosophical Trans Royal Soc Biol Sci. 2000;355(1402):1499–510. https://doi.org/10.1098/rstb.2000.0710.
Guo J, Bai X, Dai K, Yuan X, Guo P, Zhou M, Shi W, Hao C. Identification of GATA transcription factors in Brachypodium distachyon and functional characterization of BdGATA13 in drought tolerance and response to Gibberellins. Front Plant Sci. 2021;12:2386. https://doi.org/10.3389/FPLS.2021.763665/BIBTEX.
Guo J, Bai X, Dai K, Yuan X, Guo P, Zhou M, Shi W, Hao C. Identification of GATA transcription factors in brachypodium distachyon and functional characterization of BdGATA13 in drought tolerance and response to gibberellins. Frontiers Plant Sci. 2021. https://doi.org/10.3389/FPLS.2021.763665.
Gupta A, Rico-Medina A, Caño-Delgado AI. The physiology of plant responses to drought. Science. 2020;368(6488):266–9. https://doi.org/10.1126/SCIENCE.AAZ7614.
Gürel F, Öztürk NZ, Uçarlı C. Transcriptomic responses of barley (Hordeum vulgare L) to drought and salinity. Plant Omics Trends Appl. 2016. https://doi.org/10.1007/978-3-319-31703-8_7/COVER.
He C, Du Y, Fu J, Zeng E, Park S, White F, Zheng J, Liu S. Early drought responsive genes are variable and relevant to drought tolerance. Genes Genomes Genet. 2020. https://doi.org/10.1534/G3.120.401199.
Hossain MA, Wani SH, Bhattacharjee S, Burritl DJ, Tran LSP. Drought stress tolerance in plants vol 1 Physiology and biochemistry. Drought Stress Tolerance Plants Physiol Biochem. 2016. https://doi.org/10.1007/978-3-319-28899-4.
Inukai S, Kock KH, Bulyk ML. Transcription factor-DNA binding: beyond binding site motifs. Curr Opin Genet Dev. 2017;43:110–9. https://doi.org/10.1016/J.GDE.2017.02.007.
Iqbal MS, Singh AK, Ansari MI. Effect of drought stress on crop production. New Frontiers Stress Manage Durable Agric. 2020. https://doi.org/10.1007/978-981-15-1322-0_3/COVER.
Janiak A, Kwasniewski M, Sowa M, Gajek K, Żmuda K, Kościelniak J, Szarejko I. No time to waste: transcriptome study reveals that drought tolerance in Barley may be attributed to stressed-like expression patterns that exist before the occurrence of stress. Frontiers Plant Sci. 2018. https://doi.org/10.3389/FPLS.2017.02212.
Javadi SM, Shobbar ZS, Ebrahimi A, Shahbazi M. New insights on key genes involved in drought stress response of barley: gene networks reconstruction, hub, and promoter analysis. J Genet Eng Biotechnol. 2021;19(1):1–12. https://doi.org/10.1186/S43141-020-00104-Z/TABLES/4.
Joshi R, Wani SH, Singh B, Bohra A, Dar ZA, Lone AA, Pareek A, Singla-Pareek SL. Transcription factors and plants response to drought stress: current understanding and future directions. Frontiers Plant Sci. 2016. https://doi.org/10.3389/FPLS.2016.01029.
Juenger TE. Natural variation and genetic constraints on drought tolerance. Curr Opin Plant Biol. 2013;16(3):274–81. https://doi.org/10.1016/J.PBI.2013.02.001.
Kishor PBK, Rajesh K, Reddy PS, Seiler C, Sreenivasulu N. Drought stress tolerance mechanisms in barley and its relevance to cereals. Biotechnol Agric For. 2014;69:161–79. https://doi.org/10.1007/978-3-662-44406-1_9/COVER.
Kumar S, Yadav A, Verma R, Dubey AK, Narayan S, Pandey A, Sahu A, Srivastava S, Sanyal I. Metallothionein (MT1): a molecular stress marker in chickpea enhances drought and heavy metal stress adaptive efficacy in transgenic plants. Environ Exp Bot. 2022. https://doi.org/10.1016/J.ENVEXPBOT.2022.104871.
Li T, Wang YH, Liu JX, Feng K, Xu ZS, Xiong AS. Advances in genomic, transcriptomic, proteomic, and metabolomic approaches to study biotic stress in fruit crops. Crit Rev Biotechnol. 2019;39(5):680–92. https://doi.org/10.1080/07388551.2019.1608153.
Linh TM, Mai NC, Hoe PT, Lien LQ, Ban NK, Hien LTT, Chau NH, Van NT. Metal-based nanoparticles enhance drought Tolerance in Soybean. J Nanomater. 2020. https://doi.org/10.1155/2020/4056563.
Liu H, Bao G, Dou Z, Liu H, Bai J, Chen Y, Yuan Y, Zhang X, Xi J. Response characteristics of highland barley under freeze-thaw, drought and artemisinin stresses. BMC Plant Biol. 2022;22(1):1–12. https://doi.org/10.1186/S12870-022-03520-0/TABLES/1.
Nevo E, Chen G. Drought and salt tolerances in wild relatives for wheat and barley improvement. Plant, Cell Environ. 2010;33(4):670–85. https://doi.org/10.1111/j.1365-3040.2009.02107.x.
Pedersen C, Linde-Laursen I. The relationship between physical and genetic distances at the Hor1 and Hor2 loci of barley estimated by two-colour fluorescent in situ hybridization. Theor Appl Genet Theor Und Angewandte Genetik TAG. 1995. https://doi.org/10.1007/BF00223904.
Razzaq MK, Aleem M, Mansoor S, Khan MA, Rauf S, Iqbal S, Siddique KHM. Omics and crispr-cas9 approaches for molecular insight, functional gene analysis, and stress tolerance development in crops. Int J Mol Sci. 2021;22(3):1–13. https://doi.org/10.3390/ijms22031292.
Reddy BM, Anthony Johnson AM, Jagadeesh Kumar N, Venkatesh B, Jayamma N, Pandurangaiah M, Sudhakar C. De novo Transcriptome analysis of drought-adapted cluster bean (Cultivar RGC-1025) reveals the wax regulatory genes involved in drought resistance. Frontiers Plant Sci. 2022. https://doi.org/10.3389/FPLS.2022.868142.
Salehi-Lisar SY, Bakhshayeshan-Agdam H. Drought stress in plants causes, consequences, and tolerance. Drought Stress Tolerance Plants Physiol Biochem. 2016. https://doi.org/10.1007/978-3-319-28899-4_1/COVER.
Sallam A, Alqudah AM, Dawood MFA, Baenziger PS, Börner A. Drought stress tolerance in wheat and barley advances in physiology breeding and genetics research. Int J Mol Sci. 2019. https://doi.org/10.3390/IJMS20133137.
Sallam A, Alqudah AM, Dawood MFA, Baenziger PS, Börner A. Drought stress tolerance in wheat and barley: advances in physiology breeding and genetics research. Int J Mol Sci. 2019. https://doi.org/10.3390/IJMS20133137.
Sallam A, Amro A, Elakhdar A, Dawood MFA, Moursi YS, Baenziger PS. Marker–trait association for grain weight of spring barley in well-watered and drought environments. Mol Biol Rep. 2019. https://doi.org/10.1007/s11033-019-04750-6.
Sallam A, Mourad AMI, Hussain W, Stephen Baenziger P. Genetic variation in drought tolerance at seedling stage and grain yield in low rainfall environments in wheat (Triticum aestivum L). Euphytica. 2018. https://doi.org/10.1007/S10681-018-2245-9.
Shanker AK, Maheswari M. Small RNA and drought tolerance in crop plants. Indian J Plant Physiol. 2017. https://doi.org/10.1007/S40502-017-0335-7.
Sharma A, Shahzad B, Rehman A, Bhardwaj R, Landi M, Zheng B. Response of phenylpropanoid pathway and the role of polyphenols in plants under abiotic stress. Molecules. 2019. https://doi.org/10.3390/MOLECULES24132452.
Singh PK, Srivastava D, Tiwari P, Tiwari M, Verma G, Chakrabarty D. Drought tolerance in plants: molecular mechanism and regulation of signaling molecules. Plant Signal Mol Role Regul under Stressful Environ. 2019. https://doi.org/10.1016/B978-0-12-816451-8.00006-X.
Thabet SG, Moursi YS, Karam MA, Graner A, Alqudah AM. Genetic basis of drought tolerance during seed germination in barley. PLoS ONE. 2018;13(11):e0206682. https://doi.org/10.1371/JOURNAL.PONE.0206682.
Gahlaut V, Jaiswal V, Kumar A, Gupta PK. Transcription factors involved in drought tolerance and their possible role in developing drought tolerant cultivars with emphasis on wheat (Triticum aestivum L). TAG Theor Appl Genet Theor Und Angewandte Genetik. 2016. https://doi.org/10.1007/S00122-016-2794-Z.
Wu Y, Shi H, Yu H, Ma Y, Hu H, Han Z, Zhang Y, Zhen Z, Yi L, Hou J. Combined GWAS and transcriptome analyses provide new insights into the response mechanisms of sunflower against drought stress. Frontiers Plant Sci. 2022;13:847435. https://doi.org/10.3389/fpls.2022.847435.
Xu M, Liu Q, Wu D, Wang T, Espoire M, Chai Q. Characterization of spatiotemporal patterns of soil water stable isotopes at an agricultural field. Sci Total Environ. 2022;828:154538. https://doi.org/10.1016/J.SCITOTENV.2022.154538.
Yang S, Chu N, Zhou H, Li J, Feng N, Su J, Deng Z, Shen X, Zheng D. Integrated analysis of transcriptome and metabolome reveals the regulation of chitooligosaccharide on drought tolerance in sugarcane (Saccharum spp hybrid) under drought stress. Int J Mol Sci. 2022;23(17):9737. https://doi.org/10.3390/ijms23179737.
Yuan H, Zeng X, Shi J, Xu Q, Wang Y, Jabu D, Sang Z, Nyima T. Time-course comparative metabolite profiling under osmotic stress in tolerant and sensitive tibetan hulless barley. BioMed Res Int. 2018. https://doi.org/10.1155/2018/9415409.
Acknowledgements
Wuhan Igenebook Biotechnology Company Ltd, China, is acknowledged for the molecular and biotechnology work on this study.
Funding
This work was funded by the Central Government Guides Local Projects (XZ202001YD0027C), Tibet Autonomous Region Financial Special Fund (XZNKY-2018-C-021) and National Barley Industry Technology System (CARS-05-01A-08).
Author information
Authors and Affiliations
Contributions
All authors made equal contribution on study proposal writing, data collection, data analysis and manuscript production.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No ethical approval was required in this study.
Consent to publication
This manuscript has been approved by all authors for publication.
Competing interests
There is no competing interest between the authors or institutions.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Additional identified GATA transcription factors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, Y., Li, H., Zhao, C. et al. Identification of a novel transcription factor under long-term drought resistance in highland barley: a DNA affinity purification sequencing-based transcriptomic analysis. Chem. Biol. Technol. Agric. 10, 1 (2023). https://doi.org/10.1186/s40538-022-00376-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40538-022-00376-2