
Abstract
Ependymoma is the second most common malignant brain tumor in children. The etiology is largely unknown and germline DNA sequencing studies focusing on childhood ependymoma are limited. We therefore performed germline whole-genome sequencing on a population-based cohort of children diagnosed with ependymoma in Denmark over the past 20 years (n = 43). Single nucleotide and structural germline variants in 457 cancer related genes and 2986 highly evolutionarily constrained genes were assessed in 37 children with normal tissue available for sequencing. Molecular ependymoma classification was performed using DNA methylation profiling for 39 children with available tumor tissue. Pathogenic germline variants in known cancer predisposition genes were detected in 11% (4/37; NF2, LZTR1, NF1 & TP53). However, DNA methylation profiling resulted in revision of the histopathological ependymoma diagnosis to non-ependymoma tumor types in 8% (3/39). This included the two children with pathogenic germline variants in TP53 and NF1 whose tumors were reclassified to a diffuse midline glioma and a rosette-forming glioneuronal tumor, respectively. Consequently, 50% (2/4) of children with pathogenic germline variants in fact had other tumor types. A meta-analysis combining our findings with pediatric pan-cancer germline sequencing studies showed an overall frequency of pathogenic germline variants of 3.4% (7/207) in children with ependymoma. In summary, less than 4% of childhood ependymoma is explained by genetic predisposition, virtually restricted to pathogenic variants in NF2 and NF1. For children with other cancer predisposition syndromes, diagnostic reconsideration is recommended for ependymomas without molecular classification. Additionally, LZTR1 is suggested as a novel putative ependymoma predisposition gene.
Similar content being viewed by others

Introduction
Ependymoma is the second most common malignant central nervous system (CNS) tumor in children and is associated with poor long-term survival [1, 2]. Apart from a very limited number of children with neurofibromatosis type-2 associated spinal ependymoma, the underlying causes of ependymoma remain unknown [3, 4]. Several factors indicate that genetic predisposition plays a role including increased population-based familial risk [5], reports of familial intracranial ependymoma [6, 7], genetic ancestry-based risk differences [8] and an absence of known environmental risk factors [9].
No systematic germline sequencing investigation of genetic predisposition specific to childhood ependymoma has been reported to date. Over the last decade, several large pediatric pan-cancer germline sequencing studies have been performed, with childhood ependymoma accounting for less than 5% (191/4833) of the combined sample size [10,11,12,13,14,15,16,17,18,19]. Taken together, these whole-exome/-genome sequencing (WES/WGS) studies report rare pathogenic germline variants in 4.7% (9/191) of children with ependymoma, although individual study estimates range from 0 to 21%. Lack of molecular tumor classification [10, 11, 13,14,15,16,17,18,19], small ependymoma sample sizes [11, 12, 14,15,16,17,18,19], restriction to gene panels [10,11,12,13,14,15,16,17,18,19] and lack of population-based study designs [10,11,12,13,14, 16,17,18,19] further complicate the delineation of the nature and extent of genetic predisposition in childhood ependymoma.
The aim of this population-based study was to investigate genetic predisposition in children with molecularly classified ependymoma due to rare pathogenic germline variants both in and outside known cancer genes. Moreover, we assessed the feasibility of performing germline WGS and tumor DNA methylation profiling in a combined retro-/prospective nationwide cohort spanning more than 20 years.
Material and methods
Retrospective cohort
Children (< 18 years) diagnosed with ependymoma from 2000 to 2016 in Denmark were identified through the Danish Childhood Cancer Registry (DCCR) [20]. Registry data on date of birth, gender, histopathology and tumor location was validated by cross-linkage with the National Pathology Registry. Living patients aged > 18 years at the time of the study were informed and offered inclusion both in writing and by telephone. For minors (< 18 years at the time of the study) and for deceased patients, parents or legal guardians were contacted. Detailed clinical and four-generational pedigrees were retrieved through patient health record review for included patients.
Prospective cohort
Since 2016, all children (< 18 years) diagnosed with cancer in Denmark have been offered germline WGS through the STAGING study, described in detail elsewhere [15, 21]. The prospective cohort consists of children with ependymoma included in STAGING from 2016 to 2021. Similarly to the retrospective cohort, data variables were retrieved through patient health record and histopathology report review.
Collection of tissue for germline DNA sequencing
Leukocyte DNA was isolated from peripheral blood samples drawn in parallel with clinical sampling when possible. For deceased patients, archived blood samples were collected from the Copenhagen Hospital Biobank (CHB) [22]. For those without obtainable blood samples, dissection of normal brain tissue was performed on formalin-fixed paraffin embedded (FFPE) tumor tissue samples.
Germline whole-genome and -exome sequencing
Germline WGS was performed on leukocyte DNA using the HiSeqX platform (Illumina, USA) with paired-end sequencing of 150 bp reads and target 30X average coverage. Germline WES of healthy brain tissue was performed using Novaseq 6000 (Illumina, USA). Exomes were sequenced as 2 × 150 bp paired-end reads to an average median coverage of 60X. Tissue handling, sequencing and bioinformatics procedures including variant filtering are further detailed in the Additional file 1: Methods.
Cancer gene panel analysis
For the gene panel analysis, WGS/WES data was limited to filtered SNVs and SV deletions identified in a predefined set of 457 genes. This panel consisted of 390 cancer related genes supplemented by 67 genes with either established or suggested roles in ependymoma tumorigenesis selected based on the scientific literature (Additional file 2: Tables S1 and S2). Variants were reviewed by a multidisciplinary team specialized in pediatric cancer predisposition. Variants were classified as either “benign”, “likely benign”, “likely pathogenic”, “pathogenic”, or as “variants of unknown significance” (VUS) in accordance with international standards [23]. In the context of this study, “likely pathogenic” and “pathogenic” variants are referred to simply as “pathogenic”.
Constrained gene analysis
For the constrained gene analysis, all rare, coding SNVs and SV deletions, were subsetted to variants predicted to cause loss-of-function (pLoF) in 2986 highly constrained genes. Based on metadata from 141,456 humans without serious childhood disease, evolutionarily constrained genes were defined by a LoF observed/expected upper bound fractions (LOEUF) score of ≤ 0.35 which is indicative of depletion of pLoF variation and in line with recent recommendations [24, 25]. Curation of resulting variants, including use of 586 in-house whole genome sequences from children with cancers other than ependymoma, is detailed in the Additional file 1: Methods and Additional file 2: Table S3.
Tumor DNA methylation profiling and molecular classification
Molecular tumor classification was performed using retrospectively collected iDAT files for patients with existing clinical DNA methylation profiles. For all others, archived FFPE or freshly frozen (FF) tumor samples were collected and underwent DNA methylation profiling using the Infinium MethylationEPIC BeadChip Kit (Illumina, USA). Archived tumor DNA was restored using the Infinium HD FFPE DNA Restore Kit (Illumina, USA) prior to methylation profiling. Tumor methylation class and subclass were predicted using a publicly available classifier tool [26]. The classifier version and employed cut-off scores are further detailed in Additional file 1: Methods. For an illustrative overview of the cohort and methods used, please see Fig. 1).

Graphic overview of the cohort (n = 43 children) and methods employed
Statistical analyses
Statistical analyses were performed using R v.3.6.1 and IBM SPSS Statistics v.25.
Ethical approvals
This study was approved by the Capital Region Scientific Ethical Committee (H-15016782, prospective cohort) and the Danish National Committee on Health Research Ethics (2000407). All patients and/or parents/legal guardians provided informed consent.
Results
Patient characteristics
A total of 43 children registered with an ependymoma diagnosis were included. Median age at diagnosis (5.3, SD 4.7), gender distribution (females 44.2%), histopathology diagnosis, and tumor location (Table 1) were in line with existing population-based reports [27,28,29]. The overall inclusion rate was 77% (43/56). For the retrospective cohort, in which both living and deceased patients were eligible for inclusion, a higher rate of inclusion was seen for deceased patients compared to living patients (91% vs. 66%, Fisher’s exact test, p = 0.067). The inclusion process, including main reasons for exclusion, is illustrated in Additional file 1: Fig. S3.
Molecular tumor classification
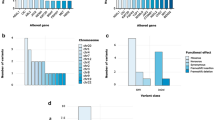
Molecular tumor (re-)classification based on DNA methylation profiling was possible for 90% (39/43) of patients. Distribution of original histopathological diagnosis and resulting tumor methylation class and subclass is listed in Table 1 and illustrated in Fig. 2, respectively.

Sankey plot illustrating original histopathological diagnosis (left) for 43 children registered with ependymoma and corresponding tumor methylation class (right). CPS, cancer predisposition syndrome; ANA-EPN, ependymoma WHO 3; EPN, ependymoma WHO 2; MYX-EPN, myxopapillary ependymoma WHO 2; GBM, atypical glioblastoma with several differential diagnoses considered; where clinical methylation profiling resulted in alteration of the diagnosis; EBLASTOMA, ependymoblastoma incorrectly registered in the Danish Childhood Cancer Registry as ependymoma; NA, not available
Ultimately, the reclassification rate for patients histopathologically diagnosed with ependymoma and with available tumor tissue was 7.7% (3/39). Initially, tumor methylation class prediction mandated amendment of the registered diagnosis to a non-ependymoma entity for four patients (Figs. 2 and 3). Two patients with histopathologically diagnosed WHO grade 3 ependymomas located in the pons and thalamus, respectively, were both reclassified as H3K27-mutant or EZHIP expressing diffuse midline gliomas (DMG_H3K27). Another tumor, extending through the aqueduct from the fourth ventricle also registered as ependymoma in the DCCR, was reclassified as a rosette-forming glioneuronal tumor (RGNT) based on DNA methylation profiling. Of note, the original histopathology report of this tumor revealed a discussion of several differential diagnoses. Finally, an ependymoblastoma incorrectly coded as ependymoma in the registry was specified as a C19mc-altered embryonal tumor with multilayered rosettes (ETMR). All reclassifications were supported by subsequent review of the original histopathology reports by a senior pediatric neuropathologist. For one of the molecularly classified ependymoma patients, a chart review revealed a previous alteration of the initial histopathological diagnosis of atypical glioblastoma to ependymoma based on clinical DNA methylation profiling (Fig. 2).

Overview of resulting molecular tumor classification for the four patients with detected pathogenic germline variants
Germline DNA sequencing
Tissue for germline DNA sequencing was available for 86% (37/43; new or prospectively collected blood samples n = 28, archived blood samples n = 6, normal brain tissue n = 3) of which 34 retained ependymoma status following molecular tumor classification. The six patients not undergoing germline sequencing were all deceased, part of the retrospective cohort, and without available archived blood samples or dissectible healthy brain tissue in the FFPE tumor samples.
Cancer panel analysis findings
Nine pathogenic variants (eight SNVs, one SV) in nine patients were detected across the 457 cancer panel genes. Five heterozygous loss-of-function variants in the recessive genes FANCM, ERCC3, and SBDS, along with relatively common risk allele variants in CHEK2 and BRIP1, were considered unrelated to ependymoma, but are further detailed in Additional file 2: Table S6.
Two of the four pathogenic variants at first assumed to be related to ependymoma were found in children where the histopathological diagnosis was subsequently altered following tumor DNA methylation profiling (Fig. 3). This included an NF1 nonsense variant (p.Arg1947Ter [c.5839C > T]) in a patient with a molecularly confirmed RGNT and a TP53 missense variant (p.Arg273Cys [c.817C > T]) in a child with a thalamic DMG_H3K27. Thus, the likelihood of diagnostic reclassification by DNA methylation profiling to a non-ependymoma tumor entity was significantly higher for children with detected pathogenic germline variants (2/4 vs. 0/29, Fisher’s exact test, p = 0.011, analysis limited to patients with ependymoma confirmed as initial histopathology diagnosis and both available tumor and germline tissue, n = 33, Additional file 2: Table S4).
A causative 364 bp NF2 deletion (chr22:30067648–30068012; p.Met334_Leu374del [c.1000-167_1122 + 75del]) was detected in a young child diagnosed with a WHO grade 2 ependymoma (methylation class spinal ependymoma (SP-EPN)) located at the cervicomedullary junction. The patient was initially treated with partial surgical resection followed by focal radiation and adjuvant chemotherapy, after which a minor contrast-enhancing tumor remnant has remained stable for more than 10 years. During follow-up, the patient developed bilateral vestibular schwannomas. Despite a family history with one third generation and several fourth generation relatives with clinically diagnosed neurofibromatosis type-2, the diagnosis had not been suspected until the patient debuted with ependymoma.
Finally, a pathogenic nonsense variant in LZTR1 (p.Gln762Ter [c.2284C > T]), a gene not formerly linked with ependymoma, was detected in an otherwise healthy child diagnosed with a WHO grade 3 ependymoma (methylation class posterior fossa group A (PF-EPN-A) ependymoma, subclass 1c), located in and around the foramen of Luschka. Of note, the only other LZTR1 variant observed in our cohort was a VUS (p.Asp703Asn [c.2107G > A]) in another child diagnosed with the same molecular ependymoma subclass (PF-EPN-A1c) in the same location.
No pathogenic variants were detected in the supplementary panel of 67 ependymoma related genes.
Constrained gene analysis findings
Sixteen pLoF variants (11 SNVs and five SVs) were observed in the same number of constrained genes in 12 patients. Both pLoF variants already known to cause ependymoma (in NF2 and NF1), were rediscovered. However, the nonsense NF1 variant was found in a patient for whom DNA methylation profiling amended the ependymoma diagnosis to RGNT (Additional file 2: Tables S3 and S5).
Following molecular reclassification, 14 constrained gene pLoF variants remained, and were located in the following genes (ordered according to rising LOEUF scores); CHD6, NF2, COL1A1, FGD5, BRWD1, UHRF2, ZNFX1, FOXO3, CDC42BPA, DHX37, DNAJC2, TRIM67, ZMYM2, VPS4A. No significant enrichments were detected using the String Database v.11 [30]. However, all but one (COL1A1) are expressed in normal brain tissue [31]. Interestingly, 6/7 of the constrained genes in which pLoF variants were found in patients with posterior fossa ependymoma show particularly high expression levels in cerebellar tissue (UHRF2, FOXO3, CDC42BPA, ZMYM2, CHD6 and DNAJC2) [32].
Other than a 3.4-fold enrichment of non-membrane-bounded organelles (false discovery rate 3.56e-3) the GO PANTHER Cellular Component Overrepresentation Test [33] did not reveal any other significant enrichments for the detected constrained genes when compared to all other genes.
Discussion
In this combined retro- and prospective study, we performed germline WGS/WES and tumor DNA methylation profiling of a population-based cohort diagnosed nationwide over a timespan of 21 years to determine the role of genetic predisposition in childhood ependymoma. Both known cancer genes and genes somatically or epigenetically associated with ependymoma were analyzed for pathogenic germline variants, as were evolutionarily constrained genes. Our findings establish ependymoma as a disease where germline pathogenic variants in known cancer genes only rarely play an underlying role, especially when precise molecular (re)classification is available. We also identify new putative ependymoma predisposition genes. Lastly, we highlight the essential role of including molecular tumor classification in ependymoma studies and the feasibility of using archived tumor samples for this purpose.
Pathogenic variants detected in known cancer genes
Of the 37 patients undergoing germline WGS, 11% (4/37) were found to harbor pathogenic variants in the cancer panel genes (NF1, NF2, TP53, and LZTR1)). By comparison, both the carrier frequency and the genes involved were similar to the findings of Zhang et al. from their pediatric pan-cancer germline study (n = 1120) which included 67 ependymoma patients (4/67 (6%), NF1, NF2, TP53) [10]. However, in our cohort, tumor DNA methylation profiling reclassified two of the patients with pathogenic germline variants in NF1 and TP53 to tumor types other than ependymoma. Consequently, only two pathogenic germline variants were detected among children with molecularly confirmed ependymoma (2/34, 5.9%). In this context, it is worth noting that the reclassification rate in our study (7.7%) is comparable to that reported by Capper et al. [26]. In their prospective cohort of 101 histopathologically diagnosed ependymoma samples, 6.0% (6/101) were reclassified based on tumor DNA methylation profiling to a non-ependymoma entity, including neuroepithelial tumors and two DMGs, similarly to our cohort.
As of this writing, ten large (n > 100) pediatric pan-cancer germline sequencing studies including children with ependymoma have been published (Table 2). Combined, these investigations report pathogenic germline variants in 4.7% (9/191) of children with histopathologically diagnosed ependymoma. Following exclusion of a likely benign TP53 variant (detailed below), three variants likely unrelated to ependymoma (incidental findings from the ACMG v2.0 [34]) and duplicate patients (detailed in Table 2), just 2.9% (5/173) of children with ependymoma are reported to harbor pathogenic germline variants in known cancer genes. Of these, all were in NF1 (n = 3) or NF2 (n = 2). This estimate is strikingly similar to our observations, especially when taking into consideration the low frequencies and sample size and the fact that the gene panels used in the majority of the previous studies did not include LZTR1.
Neurofibromatosis type-2 predisposes both to intraspinal and -cranial childhood ependymoma
The association between neurofibromatosis type-2 and spinal ependymoma is well established [35] and somatic NF2 variants are recurrently altered in ependymomas with intraspinal location [36]. Yet, several cases of intracranial ependymoma (especially located to the cervicomedullary junction) have been reported in children and young adults with neurofibromatosis type-2 [37,38,39,40,41]. Combined with our findings of a cervicomedullary located ependymoma in a child with a pathogenic germline NF2 variant, there is mounting evidence that germline NF2-related ependymomas may be located intracranially, as well as intraspinally. While the former will often represent SP-EPN located in or around the cervicomedullary junction, cases of PF-EPN-B ependymoma have also been reported [37].
Still, pathogenic germline NF2 variants are relatively rare in the overall pediatric ependymoma population and thus explain only a minority of cases: Among the 173 children with ependymoma included in the reviewed pan-childhood cancer germline sequencing studies [10,11,12,13,14,15,16,17,18,19], only two patients (1.2%) were reported to harbor pathogenic NF2 alterations [10, 16] (Table 2), for whom neither tumor location nor molecular subclass were described.
Questioning Li-Fraumeni Syndrome’s association with (molecularly classified) ependymoma
Both somatic and germline TP53 variants have been reported in other pediatric CNS tumors, yet such alterations are extremely rare in ependymoma tumor tissue [42]. Of all the children with ependymoma included in the aforementioned germline predisposition investigations, only one patient (0.6%, 1/173) was found to carry a TP53 variant characterized as pathogenic [10]. The variant (NM_000546:p.Tyr107His, c.319T > C), which was detected in a 10-year-old girl with an infratentorial ependymoma, has later been classified as benign in ClinVar [43] and was not reported as pathogenic by Gröbner et al., who included the same patient in their subsequent study [13]. Furthermore, the variant has been found in 0.1% of healthy adults that self-identified as African/African American [24]. Apart from the 173 children with ependymoma reviewed above, five cases of children with ependymoma and pathogenic germline TP53 variants have been reported in the literature [44,45,46]. Of note, molecular tumor classification was not performed in any of these cases. Were it not for DNA methylation profiling-based reclassification to DMG, the erroneous ependymoma phenotype in our cohort would have been reported as associated with the germline TP53 variant. This underscores the importance of molecular classification of ependymal tumors.
Pathogenic NF1 germline variants also appear to play a role in childhood ependymoma
Pathogenic NF1 germline variants are extremely rare among children with ependymoma. No such variants were detected among the 34 children with molecularly classified ependymoma following diagnostic revision to RGNT for the child with a nonsense variant in NF1. In comparison, three of the reported 173 germline sequenced children with ependymoma (1.7%) have been found to carry pathogenic NF1 variants (Table 2). These include two children with intracranial ependymoma reported by Zhang et al. [10] and one 6-year-old child with synchronous schwannoma and CNS ependymoma reported by Fiala et al. [16]. Only two additional cases of children with (clinically) diagnosed neurofibromatosis type-1 and intracranial ependymoma have been reported in the literature [47]. Diagnostic confirmation and tumor molecular subtyping by DNA methylation profiling was not reported for any of these patients. This may have inflated the reported NF1 carrier rate in patients with ependymoma. This phenomenon is illustrated by the diagnostic revision in both our cohort and others, where histopathologically diagnosed ependymomas were reclassified to pilocytic astrocytomas and neuroepithelial tumors based on DNA methylation profiling [26]. Importantly, both of these tumor types have a much higher rate of germline NF1 alterations [48, 49].
LZTR1 might represent a novel putative ependymoma predisposition gene
A likely pathogenic LZTR1 variant (p.Gln762Ter [c.2284C > T]), undetected among > 125,000 healthy adult in gnomAD [24], was found in a child diagnosed with a fourth ventricle PF-EPN-A1c ependymoma. Pathogenic germline variants in LZTR1 have not previously been reported in patients with ependymoma. The gene, which is centromeric to NF2 and SMARCB1 on chromosome 22q11.21, was recently uncovered as a germline predisposition gene in schwannomatosis [50]. Pathogenic LZTR1 germline variants have been reported in children with different cancer types, including high-grade glioma [13], but have not been evaluated in the majority of the existing large pan-childhood cancer germline sequencing studies [10, 11, 16,17,18]. Although monozygosity of 22q has been reported in ~ 40% of RELA-fusion positive supratentorial ependymoma (ST-EPN-RELA) [52], the rarity of pathogenic somatic NF2 variants in the majority of intracranial ependymoma suggests a different tumor suppressor gene to be located on chromosome 22 [51, 53, 54]. We therefore speculate that pathogenic germline LZTR1 variants may play a role in tumorigenesis for a limited subset of children with ependymoma, perhaps restricted to the PF-EPN-A1c molecular subtype.
Upon review of LZTR1 findings in our childhood (non-ependymoma) cancer control cohort, the LZTR1 missense VUS (p.Asp703Asn [c.2107G > A]) detected in another patient with PF-EPN-A1c was observed in a child with acute myeloid leukemia. Moreover, this variant has been reported in 5/26,128 (0.02%) Swedish individuals reported without serious childhood disease in gnomAD [24].
Less than 4% of childhood ependymoma is explained by pathogenic variants in known cancer genes
Based on the described meta-analysis, the current best estimate of germline predisposition in childhood ependymoma suggests that 3.4% (7/207) carry a causative pathogenic germline variant, mainly located in NF2 and NF1 (Fig. 2). This estimate indicates that germline predisposition is significantly less frequent than what is reported for pediatric brain and spinal cord tumors in general [3] (Fisher’s exact test, 7/207 vs. 147/1425, OR = 0.30 [0.11–0.66], p < 0.001). Consequently, one may question the need to perform extensive genetic testing in newly diagnosed children with ependymoma if no family history or other signs or symptoms of neurofibromatosis are present. Of course, the lack of germline findings in the majority of children with ependymoma may reflect limitations in our current knowledge of genetics. Or, perhaps more likely, other biological mechanisms including epigenetic dysregulation, which has been suggested as the main driver for the largest molecular subgroup, PF-EPN-A [55, 56].
Several factors may, however, have influenced the validity of the combined risk estimate; Opposite to our study, all but one of the germline investigations listed in Table 2 did not report molecular tumor classification [12]. Moreover, their lack of population-based study design may have introduced selection bias. As illustrated by the pathogenic NF2 deletion detected in our cohort, limiting bioinformatic analyses solely to SNVs, as done in one of the reviewed sequencing studies [18], may miss pathogenic alterations. Also affecting the generalizability of the combined estimate is the fact that the two cohorts contributing 65% (112/173) of the total ependymoma sample size were limited to intracranial ependymoma, likely resulting in underreporting of NF2-associated cases [10, 13].
Constrained gene analysis may explain additional genetic risk
Focusing on genes exhibiting evolutionary intolerance of inactivating alterations has recently emerged as a novel approach of investigating genetic predisposition to any state that limits reproduction, such as fatal childhood diseases [24]. We have previously detailed how a constrained gene approach may be useful in investigations of genetic predisposition to childhood (CNS) malignancies [21].
Constrained gene analysis of children with molecularly confirmed ependymoma rediscovered the NF2 deletion detected in our cancer gene panel analysis. Apart from NF2, none of the 14 constrained genes in which pLoF variants were detected have previously been linked with ependymoma. Interestingly, several are suggested to have tumor suppressor roles (FOXO3 [57], TRIM67 [58], UHRF2 [59, 60], CHD6 [61]).
As no single gene was found to harbor pLoF variants in more than one patient, further research of the concept is needed before a common or broader role for constrained genes in ependymoma predisposition can be ascertained. In our cohort, the lack of consistent constrained gene findings likely reflects the limited sample size and its subtype heterogeneity, or, alternatively, the growing notion that PF-EPN-A is an epigenetically driven disease. In this context, it is worth mentioning that two of the constrained genes, in which pLoF variants were detected in children with PF-EPN-A, affect epigenetic gene expression control (UHRF2 [60, 62] and DNAJC2 [63]). As neither the detected constrained genes nor LZTR1 have been analyzed in the majority of the aforementioned pediatric pan-cancer germline sequencing studies, their inclusion in future larger ependymoma cohorts will be important to confidently suggest any disease-related roles and indication for further study.
Strengths and limitations
Key strengths of this study include its population-based design, high inclusion rates and molecular tumor classification based on DNA methylation profiling. Moreover, our germline WGS-based SNV and SV and WES SNV analysis included not only 390 known cancer genes, but also 67 other genes with implied roles in ependymoma tumorigenesis and constrained gene analysis. The comprehensive literature review-based meta-analysis further strengthens the value of our investigation.
However, even with a nationwide inclusion period of more than 20 years, our sample size limits generalizability of the observed carrier frequencies. Tumor and germline tissue were unavailable for four and six patients, respectively. Finally, the use of a non-ependymoma childhood cancer control cohort in the filtering of germline variants might have affected variant filtration in a conservative direction. Optimally, an equal or larger control cohort of representative and ethnically comparable whole-genome sequenced children would have been available.
In summary
This population-based germline sequencing study of childhood ependymoma, including constrained gene analysis, establishes that genetic predisposition plays a role for less than 4% of patients. This is significantly lower than for pediatric CNS tumors in general. Moreover, we show that pathogenic germline variants in children with ependymoma are virtually restricted to NF2 and NF1. Our results emphasize the importance of molecular tumor classification, as the likelihood of diagnostic reclassification to a non-ependymoma tumor was significantly higher for children with detected pathogenic germline variants. We therefore advocate diagnostic reconsideration in children with non-molecularly classified ependymoma with cancer predisposition syndromes other than neurofibromatosis type-2. In addition, we present LZTR1 as a novel putative ependymoma predisposition gene.
Availability of data and materials
All data produced in the present work are contained in the manuscript with the exception of genetic sequencing data. Danish legal regulation does not permit uploading of raw sequencing data. Selected data may be made available upon reasonable request (dependent on required approvals from relevant scientific ethic boards) to the authors.
References
Ostrom QT, De Blank PM, Kruchko C et al (2014) Alex’s Lemonade stand foundation infant and childhood primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro-Oncology 16:x1–x35. https://doi.org/10.1093/neuonc/nou327
Marinoff AE, Ma C, Guo D et al (2017) Rethinking childhood ependymoma: a retrospective, multi-center analysis reveals poor long-term overall survival. J Neurooncol 135(1):201–211. https://doi.org/10.1007/s11060-017-2568-8
Muskens IS, Zhang C, de Smith AJ, Biegel JA, Walsh KM, Wiemels JL (2019) Germline genetic landscape of pediatric central nervous system tumors. Neuro-Oncology 21(11):1376–1388. https://doi.org/10.1093/neuonc/noz108
Zhang C, Ostrom QT, Semmes EC et al (2020) Genetic predisposition to longer telomere length and risk of childhood, adolescent and adult-onset ependymoma. Acta Neuropathol Commun 8(1):173. https://doi.org/10.1186/s40478-020-01038-w
Hemminki K, Tretli S, Sundquist J, Johannesen TB, Granström C (2009) Familial risks in nervous-system tumours: a histology-specific analysis from Sweden and Norway. Lancet Oncol 10(5):481–488. https://doi.org/10.1016/S1470-2045(09)70076-2
Chen BY, Praeger A, Christie M, Yuen T (2020) Familial intracranial ependymoma mimicking an extra-lesion: a case report and review of the literature. J Clin Neurosci 74:250–253. https://doi.org/10.1016/j.jocn.2020.01.051
Dimopoulos VG, Fountas KN, Robinson JS (2006) Familial intracranial ependymomas. Report of three cases in a family and review of the literature. Neurosurg Focus 20(1):E8. https://doi.org/10.3171/foc.2006.20.1.9
Zhang C, Ostrom QT, Hansen HM et al (2020) European genetic ancestry associated with risk of childhood ependymoma. Neuro-Oncology 22(11):1637–1646. https://doi.org/10.1093/neuonc/noaa130
Johnson KJ, Cullen J, Barnholtz-Sloan JS et al (2014) Childhood brain tumor epidemiology: a brain tumor epidemiology consortium review. Cancer Epidemiol Prev Biomark 23(12):2716–2736. https://doi.org/10.1158/1055-9965.EPI-14-0207
Zhang J, Walsh MF, Wu G et al (2015) Germline mutations in predisposition genes in pediatric cancer. N Engl J Med 373(24):2336–2346. https://doi.org/10.1056/NEJMoa1508054
Parsons DW, Roy A, Yang Y et al (2016) Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol 2(5):616–624. https://doi.org/10.1001/jamaoncol.2015.5699
Oberg JA, Glade Bender JL, Sulis ML et al (2016) Implementation of next generation sequencing into pediatric hematology-oncology practice: moving beyond actionable alterations. Genome Med 8:133. https://doi.org/10.1186/s13073-016-0389-6
Gröbner SN, Worst BC, Weischenfeldt J et al (2018) The landscape of genomic alterations across childhood cancers. Nature 555(7696):321–327. https://doi.org/10.1038/nature25480
Wong M, Mayoh C, Lau LMS et al (2020) Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat Med 26(11):1742–1753. https://doi.org/10.1038/s41591-020-1072-4
Byrjalsen A, Hansen TVO, Stoltze UK et al (2020) Nationwide germline whole genome sequencing of 198 consecutive pediatric cancer patients reveals a high frequency of cancer prone syndromes. PLoS Genet. https://doi.org/10.1371/JOURNAL.PGEN.1009231
Fiala EM, Jayakumaran G, Mauguen A et al (2021) Prospective pan-cancer germline testing using MSK-IMPACT informs clinical translation in 751 patients with pediatric solid tumors. Nat Cancer 2(3):357–365. https://doi.org/10.1038/s43018-021-00172-1
Newman S, Nakitandwe J, Kesserwan CA et al (2021) Genomes for kids: the scope of pathogenic mutations in pediatric cancer revealed by comprehensive DNA and RNA sequencing. Cancer Discov. https://doi.org/10.1158/2159-8290.CD-20-1631
von Stedingk K, Stjernfelt KJ, Kvist A et al (2021) Prevalence of germline pathogenic variants in 22 cancer susceptibility genes in Swedish pediatric cancer patients. Sci Rep 11(1):5307. https://doi.org/10.1038/s41598-021-84502-4
Wagener R, Taeubner J, Walter C et al (2021) Comprehensive germline-genomic and clinical profiling in 160 unselected children and adolescents with cancer. Eur J Hum Genet 29(8):1301–1311. https://doi.org/10.1038/s41431-021-00878-x
Schrøder H, Rechnitzer C, Wehner PS et al (2016) Danish childhood cancer registry. Clin Epidemiol 8:461–464. https://doi.org/10.2147/CLEP.S99508
Stoltze UK, Foss-Skiftesvik J, van Overeem Hansen T et al (2022) Genetic predisposition & evolutionary traces of pediatric cancer risk: a prospective 5-year population-based genome sequencing study of children with CNS tumors. Neuro-Oncology. https://doi.org/10.1093/neuonc/noac187
Sørensen E, Christiansen L, Wilkowski B et al (2021) Data resource profile: the Copenhagen Hospital Biobank (CHB). Int J Epidemiol 50(3):719–720e. https://doi.org/10.1093/ije/dyaa157
Richards S, Aziz N, Bale S et al (2015) Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. https://doi.org/10.1038/gim.2015.30
Karczewski KJ, Francioli LC, Tiao G et al (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581(7809):434–443. https://doi.org/10.1038/s41586-020-2308-7
Abramovs N, Brass A, Tassabehji M (2020) GeVIR is a continuous gene-level metric that uses variant distribution patterns to prioritize disease candidate genes. Nat Genet 52(1):35–39. https://doi.org/10.1038/s41588-019-0560-2
Capper D, Jones DTW, Sill M et al (2018) DNA methylation-based classification of central nervous system tumours. Nature. https://doi.org/10.1038/nature26000
Elsamadicy AA, Koo AB, David WB et al (2020) Comparison of epidemiology, treatments, and outcomes in pediatric versus adult ependymoma. Neuro-Oncol Adv 2(1):vdaa019. https://doi.org/10.1093/noajnl/vdaa019
Villano JL, Parker CK, Dolecek TA (2013) Descriptive epidemiology of ependymal tumours in the United States. Br J Cancer 108(11):2367–2371. https://doi.org/10.1038/bjc.2013.221
McGuire CS, Sainani KL, Fisher PG (2009) Incidence patterns for ependymoma: a surveillance, epidemiology, and end results study: clinical article. J Neurosurg 110(4):725–729. https://doi.org/10.3171/2008.9.JNS08117
Szklarczyk D, Gable AL, Lyon D et al (2019) STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47(D1):D607–D613. https://doi.org/10.1093/nar/gky1131
Fagerberg L, Hallström BM, Oksvold P et al (2014) Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics MCP 13(2):397–406. https://doi.org/10.1074/mcp.M113.035600
Duff MO, Olson S, Wei X et al (2015) Genome-wide identification of zero nucleotide recursive splicing in drosophila. Nature 521(7552):376–379. https://doi.org/10.1038/nature14475
Gene Ontology Consortium (2021) The gene ontology resource: enriching a GOld mine. Nucleic Acids Res 49(D1):D325–D334. https://doi.org/10.1093/nar/gkaa1113
Kalia SS, Adelman K, Bale SJ et al (2017) Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 19(2):249–255. https://doi.org/10.1038/gim.2016.190
Coy S, Rashid R, Stemmer-Rachamimov A, Santagata S (2020) An update on the CNS manifestations of neurofibromatosis type 2. Acta Neuropathol (Berl) 139(4):643–665. https://doi.org/10.1007/s00401-019-02029-5
Ebert C, von Haken M, Meyer-Puttlitz B et al (1999) Molecular genetic analysis of ependymal tumors. NF2 mutations and chromosome 22q loss occur preferentially in intramedullary spinal ependymomas. Am J Pathol. 155(2):627–632. https://doi.org/10.1016/S0002-9440(10)65158-9
Kresbach C, Dorostkar MM, Suwala AK et al (2021) Neurofibromatosis type 2 predisposes to ependymomas of various localization, histology, and molecular subtype. Acta Neuropathol (Berl) 141(6):971–974. https://doi.org/10.1007/s00401-021-02304-4
Ruggieri M, Praticò AD, Serra A et al (2016) Childhood neurofibromatosis type 2 (NF2) and related disorders: from bench to bedside and biologically targeted therapies. Acta Otorhinolaryngol Ital 36(5):345–367. https://doi.org/10.14639/0392-100X-1093
Wheeler A, Metrock K, Li R, Singh S (2022) Cystic meningioangiomatosis and cerebellar ependymoma in a child with neurofibromatosis type 2. Radiol Case Rep 17(4):1082–1087. https://doi.org/10.1016/j.radcr.2022.01.050
Halliday D, Emmanouil B, Vassallo G et al (2019) Trends in phenotype in the English paediatric neurofibromatosis type 2 cohort stratified by genetic severity. Clin Genet 96(2):151–162. https://doi.org/10.1111/cge.13551
Essayed WI, Bernard A, Kalamarides M (2015) Clinical response associated with radiographic regression of a cervicomedullary ependymoma in a NF2 patient treated by bevacizumab. J Neurooncol 125(2):445–446. https://doi.org/10.1007/s11060-015-1925-8
AACR Project GENIE (2017) Powering precision medicine through an International Consortium. Cancer Discov 7(8):818–831. https://doi.org/10.1158/2159-8290.CD-17-0151
Landrum MJ, Lee JM, Benson M et al (2018) ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46(D1):D1062–D1067. https://doi.org/10.1093/nar/gkx1153
Bougeard G, Renaux-Petel M, Flaman JM et al (2015) Revisiting Li-Fraumeni Syndrome from TP53 mutation carriers. J Clin Oncol 33(21):2345–2352. https://doi.org/10.1200/JCO.2014.59.5728
Metzger AK, Sheffield VC, Duyk G, Daneshvar L, Edwards MS, Cogen PH (1991) Identification of a germ-line mutation in the p53 gene in a patient with an intracranial ependymoma. Proc Natl Acad Sci U S A 88(17):7825–7829. https://doi.org/10.1073/pnas.88.17.7825
Hosoya T, Kambe A, Nishimura Y, Sakamoto M, Maegaki Y, Kurosaki M (2018) Pediatric case of Li-Fraumeni syndrome complicated with supratentorial anaplastic ependymoma. World Neurosurg 120:125–128. https://doi.org/10.1016/j.wneu.2018.08.203
Riffaud L, Vinchon M, Ragragui O, Delestret I, Ruchoux MM, Dhellemmes P (2002) Hemispheric cerebral gliomas in children with NF1: arguments for a long-term follow-up. Childs Nerv Syst 18(1–2):43–47. https://doi.org/10.1007/s00381-001-0534-3
Fisher MJ, Jones DTW, Li Y et al (2021) Integrated molecular and clinical analysis of low-grade gliomas in children with neurofibromatosis type 1 (NF1). Acta Neuropathol (Berl) 141(4):605–617. https://doi.org/10.1007/s00401-021-02276-5
Sievers P, Appay R, Schrimpf D et al (2019) Rosette-forming glioneuronal tumors share a distinct DNA methylation profile and mutations in FGFR1, with recurrent co-mutation of PIK3CA and NF1. Acta Neuropathol (Berl) 138(3):497–504. https://doi.org/10.1007/s00401-019-02038-4
Piotrowski A, Xie J, Liu YF et al (2014) Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat Genet 46(2):182–187. https://doi.org/10.1038/ng.2855
Ebert C, von Haken M, Meyer-Puttlitz B et al (1999) Molecular genetic analysis of ependymal tumors. Am J Pathol 155(2):627–632
Pajtler KW, Witt H, Sill M et al (2015) Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 27(5):728–743. https://doi.org/10.1016/j.ccell.2015.04.002
Singh PK, Gutmann DH, Fuller CE, Newsham IF, Perry A (2002) Differential involvement of protein 4.1 family members DAL-1 and NF2 in intracranial and intraspinal ependymomas. Mod Pathol Off J U S Can Acad Pathol Inc 15(5):526–531. https://doi.org/10.1038/modpathol.3880558
Slavc I, MacCollin MM, Dunn M et al (1995) Exon scanning for mutations of the NF2 gene in pediatric ependymomas, rhabdoid tumors and meningiomas. Int J Cancer 64(4):243–247. https://doi.org/10.1002/ijc.2910640406
Mack SC, Witt H, Piro RM et al (2014) Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506(7489):445–450. https://doi.org/10.1038/nature13108
Stuckert A, Bertrand KC, Wang P, Smith A, Mack SC (2020) Weighing ependymoma as an epigenetic disease. J Neurooncol 150(1):57–61. https://doi.org/10.1007/s11060-020-03562-0
Jiramongkol Y, Lam EWF (2020) FOXO transcription factor family in cancer and metastasis. Cancer Metastasis Rev 39(3):681–709. https://doi.org/10.1007/s10555-020-09883-w
Wang S, Zhang Y, Huang J et al (2019) TRIM67 activates p53 to suppress colorectal cancer initiation and progression. Cancer Res 79(16):4086–4098. https://doi.org/10.1158/0008-5472.CAN-18-3614
Lu H, Bhoopatiraju S, Wang H et al (2016) Loss of UHRF2 expression is associated with human neoplasia, promoter hypermethylation, decreased 5-hydroxymethylcytosine, and high proliferative activity. Oncotarget 7(46):76047–76061. https://doi.org/10.18632/oncotarget.12583
Mori T, Ikeda DD, Yamaguchi Y, Unoki M, NIRF Project (2012) NIRF/UHRF2 occupies a central position in the cell cycle network and allows coupling with the epigenetic landscape. FEBS Lett 586(11):1570–1583. https://doi.org/10.1016/j.febslet.2012.04.038
Egan CM, Nyman U, Skotte J et al (2013) CHD5 is required for neurogenesis and has a dual role in facilitating gene expression and polycomb gene repression. Dev Cell 26(3):223–236. https://doi.org/10.1016/j.devcel.2013.07.008
Pichler G, Wolf P, Schmidt CS et al (2011) Cooperative DNA and histone binding by Uhrf2 links the two major repressive epigenetic pathways. J Cell Biochem 112(9):2585–2593. https://doi.org/10.1002/jcb.23185
Richly H, Rocha-Viegas L, Ribeiro JD et al (2010) Transcriptional activation of polycomb-repressed genes by ZRF1. Nature 468(7327):1124–1128. https://doi.org/10.1038/nature09574
Acknowledgements
Jeanette Krogh Petersen, Marianne Schmidt, Henning Boldt and Benedicte Parm Ulhøi for retrieval of archived tumor tissue and iDAT files.
Funding
This work is part of Childhood Oncology Network Targeting Research, Organization & Life expectancy (CONTROL) and supported by the Danish Cancer Society (R-257-A14720), the Danish Childhood Cancer Foundation (2019-5934), the Danish Childhood Brain Tumor Foundation and the European Union’s Interregional Oresund–Kattegat–Skagerrak Grant.
Author information
Authors and Affiliations
Contributions
JF-S, UKS, RM, KW, JS-R & KS: conception, design. JF-S, UKS, SMAK, RM, JS-R, ES, SRY, AOL, LCM, DS, KW: patient inclusion, sample and/or data acquisition. JF-S, UKS, ES, SRO, TOvH, KW, KS, RM, LBA, LCM, DS, BWK, AOL: data interpretation. JF-S, UKS: drafting of manuscript. All authors: manuscript revision and approval. All authors have made substantial contributions to the manuscript and approved its final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Capital Region Scientific Ethical Committee (H-15016782, prospective cohort) and the Danish National Committee on Health Research Ethics (2000407). All patients and/or parents/legal guardians provided informed consent.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Redefining germline predisposition in children with molecularly characterized ependymoma: a population-based 20-year cohort. Figure S1. Flowchart illustrating the filtering of single nucleotide germline variants in 37 children with histopathologically diagnosed ependymoma. Figure S2. Flowchart illustrating the filtering of structural germline variants in 37 children with histopathologically diagnosed ependymoma. Figure S3. Overview of the inclusion process and germline tissue availability.
Additional file 2: Table S1
. Overview of the 390 genes associated with cancer included in the panel analysis. Table S2. Genes reported with potential germline/somatic role in ependymoma. Table S3. Constrained genes manual curation results. Table S4. Cohort overview: clinical, histopathological and molecular data. Table S5. Overview of pathogenic variants in known cancer associated genes and pLoF variants in evolutionarily constrained genes by molecular tumor type. Table S6. Five heterozygous loss-of-function variants in recessive genes considered unrelated to ependymoma.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Foss-Skiftesvik, J., Stoltze, U.K., van Overeem Hansen, T. et al. Redefining germline predisposition in children with molecularly characterized ependymoma: a population-based 20-year cohort. acta neuropathol commun 10, 123 (2022). https://doi.org/10.1186/s40478-022-01429-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-022-01429-1