
Abstract
Mild traumatic brain injury (mTBI) results in broad neurological symptoms and an increased risk of being diagnosed with a neurodegenerative disease later in life. While the immediate oxidative stress response and post-mortem pathology of the injured brain has been well studied, it remains unclear how early pathogenic changes may drive persistent symptoms and confer susceptibility to neurodegeneration. In this study we have used a mouse model of repeated mTBI (rmTBI) to identify early gene expression changes at 24 h or 7 days post-injury (7 dpi). At 24 h post-injury, gene expression of rmTBI mice shows activation of the DNA damage response (DDR) towards double strand DNA breaks, altered calcium and cell–cell signalling, and inhibition of cell death pathways. By 7 dpi, rmTBI mice had a gene expression signature consistent with induction of cellular senescence, activation of neurodegenerative processes, and inhibition of the DDR. At both timepoints gliosis, microgliosis, and axonal damage were evident in the absence of any gross lesion, and by 7 dpi rmTBI also mice had elevated levels of IL1β, p21, 53BP1, DNA2, and p53, supportive of DNA damage-induced cellular senescence. These gene expression changes reflect establishment of processes usually linked to brain aging and suggests that cellular senescence occurs early and most likely prior to the accumulation of toxic proteins. These molecular changes were accompanied by spatial learning and memory deficits in the Morris water maze. To conclude, we have identified DNA damage-induced cellular senescence as a repercussion of repeated mild traumatic brain injury which correlates with cognitive impairment. Pathways involved in senescence may represent viable treatment targets of post-concussive syndrome. Senescence has been proposed to promote neurodegeneration and appears as an effective target to prevent long-term complications of mTBI, such as chronic traumatic encephalopathy and other related neurodegenerative pathologies.
Similar content being viewed by others


Introduction
Mild traumatic brain injury (mTBI) is common and can cause a broad range of debilitating symptoms, including headaches, fatigue, irritability, sleep disturbances, depression and anxiety, and attention deficits [61, 94, 101]. While some individuals recover within days to weeks after injury, others go on to have persistent long-term effects [147]. In particular, repeated mTBI (rmTBI) has been identified as a major risk factor for neurodegenerative diseases including Alzheimer’s disease (AD) [100], Parkinson’s disease (PD) [68] amyotrophic lateral sclerosis (ALS) [21, 2], fronto-temporal dementia (FTD) [126, 124], and chronic traumatic encephalopathy (CTE) [95].
Brain changes following mTBI are still under investigation, however, it is established that immediately following mTBI there is a significant increase of oxidative stress [52, 140]. This can lead to widespread DNA damage [28]. Indeed, accumulation of DNA damage has been reported in various models of mTBI [3, 123, 151] and our lab has previously shown evidence of double-strand breaks (DSBs) in human brains with mTBI history [131, 132]. Although cells throughout the body regularly encounter DNA damage from endogenous sources, such as metabolic reactive oxygen species (ROS), the cell’s DNA damage response (DDR) can become overwhelmed when facing pathological levels of damage. The DDR is a dynamic pathway which functions to restore DNA integrity following various types of lesions, involving signalling molecules, transcription factors, and repair enzymes. If the DDR becomes persistently activated in the face of persistent DNA damage, cellular senescence pathways may be activated. Cellular senescence is characterized by cell-cycle arrest, chronic inflammation via the production of senescence-associated secretory phenotype (SASP) factors, morphological abnormalities, and anti-apoptotic gene expression signatures. Senescent cells accumulate with age [23, 157, 175], neurodegenerative disease and cognitive decline [15], neuropsychiatric disorders [33], and have recently been discovered to accumulate after TBI [3, 123, 131, 132, 151].
In this paper, we use a mouse model of rmTBI to identify gene expression changes at 24 h and 7 days post-injury. We suggest that activation of the DDR occurs in the 24 h period following mTBI, which drives changes consistent with cellular senescence and early neurodegeneration by 7d post-injury.
Materials and methods
Animals
All experiments were approved by the Centre for Phenogenomics (TCP) Animal Care Committee. Adult (7–9 week old) male C57BL/6 mice (TCP in-house colony), housed randomly, were used in this study. Mice were kept under standard laboratory conditions with access to food and water ad libitum. Separate groups of mice were used for neurobehavioural testing including righting reflex and Morris water maze (n = 8 mice per group), NanoString gene expression analysis and histology (n = 3 mice per group), and molecular analysis with qPCR and Western Blot (n = 3 mice per group).
Repeated mild traumatic brain injury model
Mice were randomly assigned to receive rmTBI or sham procedures. The brain injury model used in this study is a closed-skull controlled impact model. Mice received pre-operative subcutaneous injections of sustained release buprenorphine (1.2 mg/kg) and were then anesthetized with isoflurane (induced at 4% and maintained at 2.5%). Once anesthetized, mice were injected subcutaneously with lactated ringers with 5% dextrose (0.75 ml), and under the scalp with equal parts xylocaine (with epinephrine) and bupivacaine (total injection volume of 0.1 ml). Mice were placed in a stereotactic frame, preventing movement of the head, where a midline incision was made to expose the skull. The desired location (2.5 mm right of Bregma, over the right somatosensory cortex) was identified and an impact of 200 m/s was administered with an electromagnetically driven Impact One Stereotaxic Impactor (Leica, Buffalo Grove, IL) using a 5 mm metal tip, to a depth of 1.5 mm, and with a dwell time of 200 ms.
Neurobehavioural testing
Following the impact, mice were placed in a clean recovery cage and righting reflex was assessed by placing mice on their backs and measuring the time for mice to “right” by turning over onto all four paws. This procedure was repeated for a total of three impacts with a 24-h inter-impact interval. Sham animals received all procedures without the impact. One week following the final injury, mice were subjected to neurobehavioural testing with the Morris water maze task over 4 days. Each testing day starts with the mice being left in their home cages inside of the testing room or in an anteroom for at least 30 min prior to testing, to help them acclimate to the testing environment. The maze is positioned centrally under a ceiling camera and roughly 1 m away from surrounding walls with four visual cues around the pool. The pool is filled with water to approximately 10 cm from the edge (providing mice with a clear view of external cues), heated to 25 °C, and coloured with non-toxic white paint to make the platform, placed in the centre of a target quadrant, invisible. Days 1–3 make up the training days, in which each mouse undergoes four blocks of three trials (total of 12 trials per day per mouse) with ~ 15 min of rest between trial blocks. On day 4 the platform was removed, and the time spent in the platform zone was measured.
Statistical analysis of neurobehavioural tasks
Raw data from Ethovision (Noldus) was obtained and analyzed using the Rtrack in R, a package for reproducible and automated water maze analysis using a machine learning classifying strategy [116]. Metrics obtained from this package were latency to platform, time in goal zone, and search strategy classification.
Animal sacrifice
Following the three impacts or sham surgeries, mice for tissue analysis were sacrificed at 1 day or 1 week after the final injury, and mice used for behavioural testing (not included in our tissue analyses) were sacrificed on the final day of behavioural testing. For NanoString analysis and histology, mice were sacrificed via transcardial perfusion, first with PBS and heparin then with 4% paraformaldehyde, under ketamine (150 mg/kg) and xylazine (10 mg/kg). Following perfusion, the brain in the skull was removed and post-fixed overnight in 4% PFA. Following post-fixation, the hemispheres were separated, and each brain half was processed and embedded in paraffin blocks. For molecular analysis, mice were sacrificed via transcardial perfusion under ketamine and xylazine with PBS and heparin. The brains from these mice were immediately dissected out of the skull and frozen in a bath of isopentane in liquid nitrogen.
Histology
Formalin-fixed paraffin embedded (FFPE) blocks from the ipsilateral side of each mouse brain were used for immunohistochemistry. Sagittal blocks were cut into six micron sections and mounted on glass slides. Sections were stained with hematoxylin and eosin (H&E), glial fibrillary acidic protein (GFAP, Dako), Ionized calcium binding adaptor molecule 1 (IBA1, Abcam), amyloid precursor protein (APP, EMD Millipore 1:5000), and Lamin A/C (Santa Cruz, 1:200).
RNA extraction and isolation
FFPE blocks from the ipsilateral side of each mouse brain were used for gene expression. Shavings from each block (containing structures spanning the entire ipsilateral hemisphere excluding cerebellum) were taken, and total RNA was extracted and isolated using the RNeasy FFPE Kit by Qiagen (Qiagen Inc., Toronto, ON, Canada) with no changes to the manufacturer’s protocol. Total RNA was quantified using the Nanodrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). Approximately 200 ng RNA was used from each sample for expression profiling.
Nanostring nCounter mouse panel
Gene expression was assessed with a NanoString nCounter gene expression panel (Nanostring Technologies, Seattle, WA, USA). This panel allows multiplex gene expression analysis in fixed tissue of 770 genes broadly involved in pathways involved in inflammation, neurobiology, neuropathology, and metabolism and stress. For data normalization processes, this panel includes 13 internal references genes. A list of all target genes used in this study can be found in Additional files 1 and 2, where ratio data can also be found.
Statistical analysis of nanostring data
Raw data was collected and assessed using NanoString Technologies nSolver Analysis Software 4.0. Briefly, quality control was performed using default settings, normalization was performed using the geometric mean of positive control counts and housekeeping normalization. Fold change (FC) ratios were calculated between rmTBI and shams and a two-tailed t-test was performed to determine FC ratio significance. All fold change data for 24 h post-injury can be found in Additional file 1, and for 7d post-injury can be found in Additional file 2. Heat map clusters were generated using Euclidian distance and average linkage methods and all gene expression figures were generated using nSolver. Gene ontology analysis was performed using the g:Profiler open-source g:GOSt software. Specifically, functional terms were retrieved from the Gene Ontology (GO) database containing terms for biological process, molecular function, and cellular component. Pathway analysis used the Kyoto Encyclopedia of Genes and Genomes (KEGG), Reactome, and WikiPathways (WP) databases. Terms unrelated to the brain were omitted, and the top 20 results from each group (i.e. from each biological process, molecular function, cellular component, and pathway groups). Statistical analysis was performed using the g:SCS method in g:Profiler, a default method for multiple testing corrections. Statistical threshold was set to adjusted p ≤ 0.05.
Western Blot and quantitative real-time PCR
For Western Blot (WB) the ipsilateral hemisphere, excluding cerebellum and brainstem, was homogenized in RIPA buffer (Millipore) containing a protease and phosphatase inhibitor cocktail (Thermofisher). The supernatants were harvested after centrifugating at 12,000g for 20 min. Protein concentration was measured using a protein assay kit (Biorad). Protein samples were loaded in 10% polyacrylamide gels (Biorad) and transferred to a nitrocellulose membrane (Biorad). Membranes were blocked in 5% BSA of tris buffered saline with 0.05% tween-20 (TBS-T) for 2 h and incubated with primary antibodies for 2 h at room temperature or overnight at 4 °C. HRP conjugated secondary antibodies [Sigma, anti-mouse (A9044, 1:10,000) and rabbit (A0545, 1:20,000)] were employed. Membranes were visualized by the Odyssey Fc imaging system (LI-COR). Primary antibodies were beta-tubulin (Sigma, T7816, 1:20,000), 53BP1 (Novus biologicals, NB100-304, 1:10,000), DNA2 (Thermofisher, PA5-68167, 1:1000), Bcl-w (Cell signaling technology, #2724, 1:1000), and p53 (Santa cruz biotechnology, sc-126, 1:1000).
For qPCR: the ipsilateral hemisphere, excluding cerebellum and brainstem, was homogenized in Trizol (Invitrogen) for total RNA isolation. RNA quality was determined using a Nanodrop one (Thermofisher). 1–2 µg of total RNA was reverse-transcribed using the cDNA synthesis kit (SuperScript VILO (Invitrogen)) according to the manufacturer’s protocol. qPCR was performed by QuantaStudio 3 system (Applied Biosystems) using SYBR select master mix (Thermofisher). The information of primers is in Additional file 3: Table S1. Statistical analysis for both WB and qPCR data was performed using unpaired student t-test.
Results
rmTBI mice show loss of righting reflex indicative of a mild injury
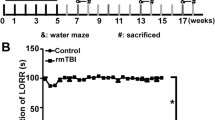
Loss of righting reflex was evident in the rmTBI mice (Fig. 1). rmTBI mice took an average of 186.0 s longer to right after the first impact (p = 0.001), 61.7 s longer after the second impact (p = 0.02), and 63.1 s longer after the third impact (p = 0.01). The longest righting reflex recorded remained under 15 min (900 s), which is indicative of a mild injury [106].

The duration of loss of righting reflex in rmTBI was significantly longer than in sham-treated mice after each impact (p < 0.001 main effect of injury status, repeated measures ANOVA), and decreased significantly with increased impact number (p < 0.001, interaction between injury status and impact number, repeated measures ANOVA). Righting reflex was under 15 min for all mice, supportive of a mild injury
Spatial learning and memory impairment 1 week after rmTBI
rmTBI mice and sham mice differed slightly in their average swim speed, with rmTBI mice swimming 1.8 cm/s faster compared to shams on average (Fig. 2a). Across three training days, rmTBI mice spent significantly longer searching for the hidden platform (latency to goal) compared to shams at 1 week post-injury with a main effect of injury (p = 0.0001, repeated measures ANOVA), a main effect of training day (p = 0.047, repeated measures ANOVA) and a significant interaction between injury status and training day (p = 0.001, repeated measures ANOVA) indicating that while the shams improved their performance over time whereas the rmTBI mice did not (Fig. 2b). In the probe test rmTBI mice spent less time in the goal zone compared to shams, which was almost significant (p = 0.55, Mann–Whitney U Test) (Fig. 2c).

rmTBI mice show evidence of spatial learning and memory impairment at 1 week post-injury. a rmTBI mice swam an average of 1.8 cm/s faster than sham mice. b Across three training days rmTBI mice had a significantly increased latency to find the platform compared to shams (p < 0.001, main effect of injury status, repeated measures ANOVA). c In the probe test rmTBI mice spent less time in the platform zone compared to shams (p = 0.55, Mann–Whitney U Test). Analysis of search strategies with Rtrack revealed no significant differences one training day one, however rmTBI mice used significantly different search strategies by training days two (χ2 = 6.2, df = 2, p = 0.05) and three (χ2 = 33.9, df = 2, p < 0.0001) with rmTBI mice utilizing less allocentric strategies than shams by the end of training
Search strategy use was analyzed using a machine learning classifier package in R called RTrack [116] (Fig. 2d). Three main categories of search strategies were assessed. Non-goal-oriented strategies include thigmotaxis, circling, and random paths, indicating that the mouse is not attempting to reach the goal but is instead motivated to escape the pool. Procedural strategies, including scanning and chaining, indicating that the mouse is aware of a goal, but is using egocentric measures to find it. Last, allocentric strategies including direct searches, corrected searches, and direct paths indicate that the mouse is orienting itself based on cues around the room to find the goal.
On the first training day the use of search strategies was not significantly different between rmTBIs and shams (χ2 = 3.4, df = 2, p = 0.18). However, rmTBI and sham mice utilized significantly different search strategies on training day two (χ2 = 6.2, df = 2, p = 0.05) and three (χ2 = 33.9, df = 2, p < 0.0001). Across the trials from training day one to training day three rmTBI mice only increased the use of allocentric strategies by 4.2% by the end of training, whereas shams had increased the use of allocentric strategies by 36.9%. Similarly, rmTBI mice remained stagnant using 65.6% non-goal oriented and procedural strategies by training day three, whereas shams only used these strategies 26.2% of the time. These results suggest that rmTBI mice have significant deficits in their spatial learning and memory retention, specifically for the ability to learn and use spatial cues and rely instead on mostly egocentric measures to complete the task.
Histological assessment of brains
rmTBI brains showed normal gross morphology, with an absence of any visible lesion at the region of impact and did not visibly differ from sham brains (Fig. 3). Parasagittal histological section of the brain stained with Hematoxylin and eosin show absence of structural damage such as contusion or hemorrhage in ipsilateral cortex and underlying structures.

Gross morphology of the mouse brain following Sham (a) and rmTBI (b) procedures. H&E staining of the rmTBI brain (c) reveals no visible lesion induced by the injury model, indicating a mild injury has been elicited
Gliosis, microgliosis, axonal damage
Histology revealed increased expression of GFAP and Iba1 at 1 day post-injury (Fig. 4b, e) and 1 week post-injury (Fig. 4c, f) on the side receiving the impact compared to shams (Fig. 4a, d). This suggests the presence of incremental gliosis and microglial activation, two well-researched and reproducible repercussions of the mild injury model used in this study [9]. Similarly, evidence of axonal damage after injury by increased APP staining was present at 7 days (Fig. 5b) after injury. These findings, along with a lack of gross morphology and the loss of righting reflex in injured animals, are supportive of eliciting a mild injury. Additionally, Lamin A/C (Fig. 5) immunohistochemistry shows loss of expression on the nuclear membrane of a subset of glial cells in the cortex and decreased expression in the dentate nucleus of the hippocampus. Loss of lamin A/C is associated with the induction of cellular senescence through DNA-damage sensing pathways [45].

rmTBI brains have increased expression of GFAP at 1 day (b) and 7 days (c) post-injury, compared to shams (a) in the impact region of the isocortex, indicating gliosis. Microglial activation, shown with Iba1 staining, can be seen in rmTBI brains at both 1 day (e) and 7 days (f) post-injury compared to shams (d). At 7 days post-injury rmTBI mice showed evidence of axonal damage by the presence of APP positive spheroids in the subcortical white matter (h), which was not present in shams (i, low power view in g). Scale bars: 0.3 mm in a–f, 0.1 mm in g, 0.05 mm in h, 0.08 mm in i

Lamin A/C expression on the nuclear membrane of glial cells was reduced at 1 day post injury in the cortex (b) and in neurons of the dentate (e), and this loss was exacerbated by 1 week post-injury in the cortex (c) and dentate (f) compared to shams (a, d). Arrows in b and c point to glial cells that have lost lamin expression. Scale bar: 150 μm in a–c, 300 μm in d, 120 μm in e, f
Activation of the DNA damage response 24 h after rmTBI
24 h following the final injury, there were a total of 15 genes significantly changed between rmTBI mice and shams in the ipsilateral hemisphere (Fig. 6). Of these changes, 4 genes were significantly upregulated (Fig. 7). Three of these four genes encode for DNA damage repair proteins in the DDR: ATG7 (FC = 1.09, p = 0.03), DNA2 (FC = 1.11, p = 0.04), and NBN (FC = 1.09, p = 0.02). ATG7 is an autophagy-related protein [136] which can modulate p53-dependent cell cycle pathways during prolonged stress [78] and plays a critical role in homology-directed repair (HDR) of DSBs [46]. Similarly, DNA2 encodes a HDR DNA repair protein for repair of DSBs [62] and NBN encodes a non-homologous end joining (NHEJ) DNA repair protein for repair of DSBs [158]. Together, the significant upregulation of these three genes in rmTBI mice compared to shams indicates the presence of DSBs at this timepoint and the activation of the DDR. The fourth gene found to be upregulated 24 h post-injury was CD163 (FC = 1.22, p = 0.04), which encodes a cell surface marker of activated M2 phenotype macrophages [71], suggesting an increase in the presence and activation of macrophages 24 h post-injury.

Overview of significantly (p ≤ 0.05) differentially expressed genes between rmTBI and sham mice at 24 h post-injury

Significantly (p ≤ 0.05) upregulated genes in rmTBI mice compared to shams at 24 h post-injury. Data are expressed as normalized RNA count number for shams and rmTBI. The median value is represented by the horizontal line within the box, defined by the first and third quartiles, and tails represent 1.5 × the interquartile range
Gene ontology of genes significantly upregulated 24 h post-injury revealed enrichment of 22 GO terms (Fig. 8a). Five molecular terms were enriched: DNA helicase activity, helicase activity, catalytic activity acting on DNA, ATG8 activating enzyme, and ATG12 activating enzyme. These suggest enrichment of DNA repair pathways and autophagy signalling. Sixteen biological process terms were enriched, including autophagy, cell-cycle checkpoint, chromosome organization, DNA replication, integrity, elongation, and unwinding, response to stress, and telomere maintenance. These terms support the activation of the DDR at this timepoint, particularly the induction of cell-cycle arrest and DNA regulating and maintenance pathways to begin repair processes. One cell component term was enriched, gamma DNA polymerase complex, further supporting activation of DNA repair processes at this timepoint.

Gene set enrichment analysis of genes significantly (p ≤ 0.05) upregulated 24 h post-injury. a Gene ontology (GO) functions significantly enriched include DNA damage response pathways and b pathways significantly enriched include the DNA damage response and cell-cycle checkpoints
Pathway analysis of upregulated genes at 24 h post-injury revealed enrichment of 20 terms (Fig. 7b). Two terms from the WP database were enriched: homologous recombination and macrophage markers. Sixteen terms from the Reactome database were enriched, encompassing DNA repair (including DNA double-strand break repair, homology-directed repair, and resolution of D-loop structures) and cell-cycle checkpoints. Lastly, one term from the KEGG database, autophagy-other, was enriched. These terms support the activation of the DNA damage response, particularly towards DSBs, at this timepoint.
11 genes were significantly downregulated in the rmTBI ipsilateral hemisphere 24 h post-injury (Fig. 9), representing changes in major signalling pathways including DNA methylation, apoptosis, autophagy, TNF-associated signalling, and calcium signalling.

Significantly (p ≤ 0.05) downregulated genes in rmTBI mice compared to shams at 24 h post-injury. Data are expressed as normalized RNA count number for shams and rmTBI. The median value is represented by the horizontal line within the box, defined by the first and third quartiles, and tails represent 1.5 × the interquartile range
AGO4 (FC = −1.63, p = 0.04) plays a critical role in the RNA-directed DNA methylation complex [19], supporting altered DNA methylation patterns 24 h post-injury. CASP9 (FC = −1.28, p = 0.026) is an initiator in the apoptotic pathway, and so its downregulation is both anti-apoptotic and protective against TNF-receptor-activated apoptosis [93]. EPG5 (FC = −1.12, p = 0.05) is an autophagy-related gene which is essential for the formation of degradative autolysosomes [162]. EPG5 is reduced in rodent models of ischemia [177] and mediates neuronal survival and normal functioning [179]. Furthermore, 7 single-nucleotide polymorphisms (SNPs) in EPG5 have been associated with AD, and 8 SNPs have been associated with an earlier age of onset of AD [161], suggesting that alterations in EPG5’s expression may confer susceptibility to neurodegeneration. HAT1 (FC = −1.12, p = 0.05) is a cytoplasmic histone deacetylase, for which loss is associated with early onset ageing and mitochondrial defects [105], and for which expression is reduced under treatment with hypoxic agents [159]. MAP1LC3A (FC = −1.34, p = 0.04) is a microtubule associated protein which mediates the interaction between microtubules and the cytoskeleton [22] and is a key protein in the autophagy pathway. PPP3R2 (FC = −1.25, p = 0.02) is a subunit of calcineurin, a critical calcium and calmodulin-dependent serine/threonine protein phosphatase involved in a wide variety of brain functions including neurotransmitter release, receptor functioning, signal transduction systems, cell death pathways, and gene expression changes [41, 49]. It has been previously reported that calcineurin subunit 2 expression decreases following TBI [165, 166] and its decrease may confer susceptibility to AD [120, 156]. Indeed, calcineurin is hypothesized to be a modulator of tau in the presence of amyloid deposition and its downregulation may accelerate AD progression [120, 156]. TRADD (FC = −1.26, p = 0.02) encodes TNFRSF1A-associated death domain and is a mediator of the TNF-receptor-activated apoptosis [20]. In addition, deficiency of TRADD has been shown to result in the accumulation of DSB foci [75]. TRAF3 (FC = −1.12, p = 0.03) is a TNF-receptor-associated factor which is critical for NF-kappaB activation and cell death pathways [60]. TRAF3 has been shown to reduce expression with age [82]. Lastly, 2 significantly downregulated genes at this timepoint, ADAMTS16 (FC = −1.35, p = 0.006) and TMEM64 (FC = −1.09, p = 0.05), do not yet have established functions in the brain. Taken together, this expression profile suggests that 24 h post-injury there is significant reduction in expression of major cell death signalling pathways, possibly conferring vulnerability to subsequent brain damage and future neurodegenerative pathologies. C6 (FC = −1.84, p = 0.04) encodes complement factor 6, a late complement component which forms the membrane attack complex. Deficiency in this component in small mammals has been shown to cause a progressive neurological syndrome characterized by multifocal motor neuropathy and severe axonal degeneration [44].
Gene ontology analysis of genes significantly downregulated 24 h post-injury revealed enrichment of 17 GO terms (Fig. 10a). Five molecular function terms were enriched: protein binding, tumor necrosis factor receptor superfamily binding, catalytic activity acting on a protein, tumor necrosis factor receptor binding, and binding. Eleven biological process terms were enriched, including response to stimulus, regulation of cell death, response to stress, and regulation of apoptotic process. One cellular component term, protein-containing complex, was enriched. Enrichment of these terms in the downregulated genes at 24 h post-injury suggests that TNF-receptor-mediated activity, cell death including apoptosis, and the cellular stress response are reduced at this timepoint.

Gene set enrichment analysis of genes significantly (p ≤ 0.05) upregulated 24 h post-injury. a Gene ontology (GO) functions and b pathway analysis suggest inhibition of cell death pathways and altered metabolic function
Pathway analysis of downregulated genes at 24 h post-injury revealed enrichment of 19 terms (Fig. 10b). Seven terms from the WP database were enriched: histone modifications, TNF-alpha-NF-kB signalling, EGFR1 signalling, MAPK signalling, Alzheimer’s disease, oxidative damage, and apoptosis. Three terms from the Reactome database were enriched: programmed cell death, apoptosis, and caspase activation via extrinsic apoptotic signalling. Lastly, ten terms from the KEGG database were enriched including ALS, AD, MAPK signalling, apoptosis, TNF signalling, NF-kappa-B signalling, IL-17 signalling, RIG-I-like receptor signalling, VEGF signalling, and Prion disease. This analysis suggests inhibition of cell death and major energy-dependent signalling pathways 24 h post-injury, consistent with analyses of up-regulated genes at this timepoint showing activation of the DNA damage response.
Cellular senescence, neurodegeneration, and signalling alterations 7 days after rmTBI
7 days after rmTBI there were a total of 32 genes significantly changed between rmTBI mice and shams in the ipsilateral hemisphere (Fig. 11). 19 genes had significantly increased expression levels (Fig. 12), suggesting the induction of cellular senescence, activation of neurodegenerative pathways, and changes in large signalling pathways, particularly to cell death pathways, mitophagy, autophagy, metabolism, and oxidation and reduction reactions.

Overview of significantly (p ≤ 0.05) differentially expressed genes between rmTBI and sham mice at 7d post-injury

Significantly (p ≤ 0.05) upregulated genes in rmTBI mice compared to shams at 7d post-injury. Data are expressed as normalized RNA count number for shams and rmTBI. The median value is represented by the horizontal line within the box, defined by the first and third quartiles, and tails represent 1.5 × the interquartile range
AGO4 (FC = 1.33, p = 0.04) is a member of the Argonaute family which plays a role in DNA methylation and miRNA maintenance [19]. Although relatively little is known about the role of AGO4 compared to the other known members of this protein family, for example AGO2 regulates gene expression silencing in the context of cellular senescence [6, 8], it is thought to be specific for gene expression silencing [97]. It is unclear what role AGO4 plays in the brain, but it likely plays a role in gene expression signatures perhaps in senescent cells. CASP3 (FC = 1.11, p = 0.04) encodes caspase 3, an executioner in the apoptosis pathway when it is proteolytically cleaved from its pro- form. CASP3 was significantly increased 7d post-injury, suggesting some level of apoptosis occurring at this timepoint. Although caspase 3 is thought to be inhibited by cellular senescence [117], it paradoxically increases expression with ageing and has even been suggested as an initiating factor for cellular senescence [152].
Several genes within the BCL2 pro-senescence signalling pathway were increased at this timepoint. BCL2L2 (FC = 1.11, p = 0.04) encodes the anti-apoptotic and pro-senescence BCL-W protein [173]. Increased expression of BCL2L2 is associated with neurofibrillary tangle pathology in AD [182], and it promotes the expression of senescence-associated beta-galactosidase, p53, p21, and p16 which are all hallmark features of cellular senescence [24, 54]. Inhibition of BCL2L2 results in direct elimination of senescent cells [173], supporting the role of this protein in the induction and maintenance of cellular senescence. In relation to BCL2L2, BNIP3 (FC = 1.17, p = 0.04) encodes BCL2 interacting protein 3, which is increased in hypoxic-ischemic-encephalopathy [18] and physiological ageing [31], and has been shown to increase in association with cellular senescence especially in cells over-expressing p21 and p16 [16]. Similarly, BNIP3L (FC = 1.05, p = 0.04) encodes BCL2 interacting protein 3 like. Although BNIP3L is typically thought of as pro-apoptotic [181], anti-apoptotic roles have been recently discovered, particularly in reactive astrocytes during ALS disease progression [35]. Similarly, BNIP3L has been shown to increase in senescent cells to direct mitochondria to autophagosomes for degradation [174]. RAD51 (FC = 1.2, p = 0.05) encodes a DNA repair enzyme which associate with gH2AX foci [103]. Together, these four genes suggest increased expression of cellular senescence pathways 7d post-injury.
In addition to the above changes reflecting cellular senescence, several of the significantly increased genes at this timepoint have been associated with neurodegenerative phenotypes and diseases. CHST8 (FC = 1.1, p = 0.005) encodes the enzyme carbohydrate sulfotransferase 8. This enzyme has been suggested to be enriched in AD pathology progression [144] and increased in the hippocampus of AD brains [64]. CLEC7A (FC = 2.02, p = 0.02) is a disease-associated microglia gene which has been shown to increase after TBI in mice [19], in a mouse model of AD [38, 56], and in aged microglia associated with neurodegenerative disease [135]. DAPK1 (FC = 1.08, p = 0.05) is a calcium-dependent serine/threonine kinase that is highly expressed in the cortex in AD [167], after ischemic damage [154], and in physiological ageing where it has been suggested to play a role in the decay of learning and memory with age [51]. DAPK1 has been found to phosphorylate tau protein [118], suggesting that it possibly plays a functional role in the progression of AD-related pathology. GPR34 (FC = 1.36, p = 0.002) encodes an inhibitory G-protein coupled receptor found on microglia which is overexpressed in regions of the brain affected by neurodegeneration in both humans and mice [10]. Although its functional importance remains unclear, GPR34 is also found to increase in aged human brains and has been postulated as a possible risk factor for AD [110]. MAFB (FC = 1.29, p = 0.03) is a transcriptional regulator of mature microglia which is upregulated during multiple sclerosis disease progression [81] and found to inhibit apoptosis of macrophages [89]. MAFB positively regulates the expression of MSR1 (FC = 1.45, p = 0.002) [137], which was also significantly increased at this timepoint. MSR1 encodes a macrophage scavenger receptor, and it is increased in mouse models of AD [128] where it plays a role in amyloid beta uptake [138]. OLFML3 (FC = 1.18, p = 0.01) encodes a secreted extracellular matrix glycoprotein which is highly linked to AD pathogenesis in brain tissues where it correlates with amyloid beta levels, and in the cerebrospinal fluid (CSF) where it is considered a potential biomarker of AD [160]. OLFML3 has also been identified with single cell RNA sequencing as a senescence-associated microglia gene [72]. SUMO1 (FC = 1.10, p = 0.042) encodes a smell ubiquitin-like modifier protein that has been shown to increase in models of AD [107] and of PD and ageing [164]. Functionally, increased levels of SUMO1 leads to hippocampal dysfunction, decreased cell proliferation, and reduced neuroblast differentiation [172]. In the context of neurodegenerative pathologies, SUMO1 overexpression leads to increased amyloid plaque density, increased dendritic spine loss, and more severe cognitive deficits in a mouse model of AD [73] and impairment of synaptic function associated with memory impairment [91]. Last in this group, TYROBP (FC = 1.15, p = 0.04) encodes a TREM2 adaptor protein and has been shown to be increased in AD patients and mouse models [17,55] as well as after TBI [18]. Overexpression of TYROBP can exacerbate tau-mediated neurodegeneration [133] and targeting of TYROBP in AD models is therapeutic, resulting in less severe neuritic dystrophy and attenuation of learning and behavioural impairment [85]. Together, these 10 upregulated genes suggest neurodegenerative-like changes in the brain 7d post-injury.
Three genes with increased expression levels suggest changes to largescale signalling pathways which likely have vast repercussions on brain function and health. MAPK10 (FC = 1.09, p = 0.05) encodes mitogen-activated protein kinase 10, which is a neuron-specific map kinase enzyme in the JNK signalling pathway. MAPK10 has been identified as a possible target and biomarker of neurodegenerative diseases [104]. Indeed, MAPK10 is associated with processes of ageing and neurological deficits [168] and enhances both amyloid beta production through the phosphorylation of amyloid precursor protein (APP) [74] and the maturation and development of neurofibrillary tangles [170]. MAPK10 has also been shown to mediate the death of dopaminergic neurons in PD [25]. RAB6B (FC = 1.14, p = 0.017) is also regulated by the JNK signalling pathway [178] and was significantly increased at 7d post-injury. RAB6B encodes a novel GTPase localized to microglia, pericytes, and Purkinje cells [113] where its role in the brain is not well established aside from its role in retrograde transport of cargo in neuronal cells [163]. PIK3CD (FC = 1.26, p = 0.005) encodes the phosphoinositide 3-kinase catalytic subunit which is a kinase in the PKA pathway associated with schizophrenia [63] and neurologically compromised brains [168]. PIK3CD is related to processes of ageing, and gain of function mutations in this gene result in increased number of senescent cells [88] and similarly inhibition of this PKA subunit is senolytic [183]. RB1CC1 (FC = 1.06, p = 0.041) encodes an autophagy-related protein for which expression increases in aged glial cells [139]. RB1CC1 activates the expression of the senescent factor p16 (or CDKN2A) [109], but is also an upstream regulator of autophagy [153]. Importantly, loss of autophagy signalling has been shown to cause deficiency in the DDR, particularly towards DSBs [86]. Together, these 3 upregulated genes indicate changes to large-scale signalling pathways including the JNK pathway, PKA pathway, and autophagy.
Thirteen genes had significantly decreased expression in the ipsilateral hemisphere 7d post-injury (Fig. 13). Several of these expression signatures are consistent with reports on cellular senescence, neurodegenerative disease, and neuroinflammation resulting in barrier dysfunction.

Significantly (p ≤ 0.05) downregulated genes in rmTBI mice compared to shams at 7d post-injury. Data are expressed as normalized RNA count number for shams and rmTBI. The median value is represented by the horizontal line within the box, defined by the first and third quartiles, and tails represent 1.5 × the interquartile range
BAX (FC = −1.07, p = 0.05) encodes a protein in the BCL2 protein family which regulates susceptibility to apoptosis [122, 169], being anti-apoptotic under low expression conditions, and which is known to decrease in senescent cells [37, 155]. In support of this anti-apoptotic mechanism, the apoptosis inducer CIDEA (FC = −1.38, p = 0.05) was also significantly decreased at this timepoint. Base-excision repair DNA repair and senescence-associated gene EXO1 (FC = −1.81, p = 0.04) was significantly decreased 7d post-injury. It has been reported that repression of EXO1 induces cellular senescence and, vice versa, that the activation of the p53 senescence pathway is sufficient to repress DNA repair activity [27]. SNPs in EXO1 have been associated with cognitive ageing [83], supporting its role in cellular senescence. MPG (FC = −1.34, p = 0.04) is also a DNA repair gene in the base-excision repair pathway. Its inhibition results in increased markers of senescence [142] and, similarly, its expression is decreased in senescent cells [4]. MERTK (FC = −1.38, p = 0.024) encodes a tyrosine kinase which is known to decrease in senescent cells and ageing [127], where it results in increased blood brain barrier permeability [98]. MERTK plays multiple roles in the cell, including mediating synapse remodelling and neural circuit refinement [26] and its dysregulation in AD has been suggested to contribute to chronic inflammation in AD pathology [69]. Inhibition of MERTK leads to cellular senescence in glioblastoma cells [146]. These 5 genes together suggest increased levels of cellular senescence at 7d post-injury.
Several downregulated genes at this timepoint reflect neurodegenerative changes. CYP27A1 (FC = −1.33, p = 0.03) encodes a cholesterol metabolizing enzyme in the brain, which when reduced leads to increased levels of 27-hydroxycholesterol, a feature of AD disease progression [148, 149] and learning and memory impairment [176]. 27-hydroxycholesterol has also been associated with impaired glucose metabolism in neurons [66]. CYP27A1 expression is reduced in neurons and around amyloid plaques in AD [13] and it is significantly reduced with physiological ageing [84]. CYP7B1 (FC = −1.10, p = 0.03) is another cholesterol metabolizing enzyme in the brain, for which loss results in increased oxysterol substrate accumulation and transport across the blood brain barrier [57]. CYP7B1 decreases with age [168] and its loss is associated with impaired spatial memory in rats [171]. RELN (FC = −1.16, p = 0.05) encodes the secreted extracellular matrix protein reelin, which plays important roles in controlling cell–cell interactions in the brain. RELN expression decreases early in AD prior to the deposition of amyloid pathology [59], and its loss is associated with cognitive deficits, several neuropsychiatric disorders, and loss of synapse maintenance [65]. Indeed, RELN expression is reduced in autism spectrum disorder [184] and schizophrenia and bipolar disorder where it associates with reduced dendritic spine density [50]. In addition, RELN plays a role in protecting against amyloid beta toxicity [76]. TTR (FC = −1.79, p = 0.04) encodes a carrier protein secreted by the choroid plexus endothelial cells into the CSF [180]. Reduced levels of TTR are seen in AD CSF and brain tissue [143]. Indeed, TTR plays a role in the clearance of amyloid peptides from the brain by binding to amyloid peptides and crossing the blood brain barrier specifically in the brain-to-blood direction exclusively to transport to the liver [1].
Consistent with some of the above changes, two downregulated genes suggest the presence of astrogliosis and possible dysfunction of critical brain barriers including the blood–brain barrier and the ependymal epithelial lining of the ventricles. CD44 (FC = −1.40, p = 0.03) is a marker of pro-inflammatory astrocytes, for which reduced expression is found in aged mice and in mice with neurological deficits [145]. In addition, CD44 deficient mice have significantly increased blood–brain barrier permeability which severely worsens the progression of multiple sclerosis [39]. DAB2 (FC = −1.22, p = 0.04) encodes a mitogen-responsive protein which plays a role in establishing epithelial cell polarity [99]. Notably, specialized epithelial cells in the ventricular lining of the brain called ependymal cells require maintenance of their polarity to regulate CSF dynamics [112, 141, 166].
At this timepoint two additional genes were significantly reduced: CD163 (FC = −1.26, p = 0.01), a macrophage marker, and ATP6V0E (FC = −1.18, p = 0.02), an ATPase transporter subunit for which its role in the brain is not well established.
Gene ontology analysis of upregulated genes 7d post-injury revealed enrichment of 20 GO terms, all of which were biological process terms (Fig. 14a). These terms encompass regulation of cell death pathways, regulation of membrane permeability, regulation of the mitochondrial membrane, and the cellular response to stress. This GO enrichment analysis indicates that 7d post-injury the stress-response remains activated, and that significant regulation (both positive and negative) of cell death pathways is occurring. Similarly, pathway analysis of upregulated genes 7d post-injury (Fig. 15b) revealed enrichment of two terms from the WP database, apoptosis modulation by HSP70 and apoptosis, and four terms from the KEGG database, TNF signalling pathway, AGE-RAGE signalling pathway, mitophagy, and autophagy. This pathway analysis supports the dysregulation of cell death at this timepoint, with negative regulation suggested by apoptosis modulation by HSP70 and positive regulation suggested by apoptosis. Furthermore, this analysis suggests activation of the hypoxia-induced AGE-RAGE pathway (related to multiple age-related pathologies), and increased TNF, mitophagy, and autophagy signalling. These changes are consistent with the induction of cellular senescence at this timepoint.

Gene ontology (a) and pathway (b) enrichment analyses of genes significantly (p ≤ 0.05) upregulated 7d post-injury

Gene ontology (a) and pathway (b) enrichment analyses of genes significantly (p ≤ 0.05) downregulated 7d post-injury
Concomitantly, gene ontology analysis of downregulated genes 7d post-injury revealed enrichment of 40 terms (Fig. 15a), including 16 MF, 19 BP, and 5 CC terms. Enriched molecular function terms related primarily to the activity of various DNA glycosylases, cholesterol metabolizing enzymes, DNA exonucleases, and oxidoreductase and HSP70 activity. This analysis indicates a significant impairment of the DNA damage response, particularly the reduction of DNA damage repair enzymes, and impaired hypoxia response at 7d post-injury, which are consistent with the induction of cellular senescence. The enriched biological process terms encompassed regulation of cell death pathways, locomotion and metabolism, cell motility and migration, and the response to stress, suggesting significant changes in basic biological processes of the cell, perhaps due to the morphological changes associated with cellular senescence. Enriched cell component terms were the BAX complex, cytoplasm, intracellular anatomical structure, membrane, and cellular anatomical entity. Pathway analysis at 7d post-injury revealed enrichment of 12 terms (Fig. 15b). From the WP database, macrophage markers, mismatch repair, metapathway biotransformation, and oxidation by cytochrome P450 were significantly enriched. Similarly, from the Reactome database activation, translocation, and oligomerization of BAX, metabolism of steroids and lipids, functionalization of compounds, cyrochrome P450, and endogenous sterols were enriched. Lastly, ECM-receptor interaction was enriched from the KEGG database. Together, the pathway analysis at this timepoint supports reduction of the DDR, impaired oxidation reduction reactions, and impeded metabolism at this timepoint. These pathway changes support the roles of individual genes detailed above and suggests the induction of cellular senescence and neurodegenerative changes 7d post-injury.
qPCR and Western Blot validation of targets
We validated our findings using six qPCR and four WB targets related to DNA damage-induced cellular senescence. Using qPCR (Fig. 16a), we found elevation of IL1α at 1 day post-injury, which is a DNA damage sensing agent [125] and an upstream regulator of cellular senescence [114]. By 1 week post-injury, qPCR revealed significant elevation of senescence-associated interleukins IL1β (p = 0.04) and IL10 (p = 0.05), which promote senescence-mediated inflammation [77] and senescence induction through p53 and p21 [134], respectively. At 1 week post-injury qPCR also revealed elevation of the senescence marker p21, the DNA damage marker 53BP1, and EXO1, a DNA repair gene for which expression is reduced in senescent cells [27]. WB analysis supported these findings (Fig. 16b), showing elevation of the DNA damage-induced cellular senescence markers 53BP1 (p = 0.03), DNA2 (p = 0.01), and p53 (p = 0.03). These findings support and validate our gene expression data from NanoString, showing activation of the p21 and p53-dependent DNA damage-induced senescence pathways.

a qPCR and b Western Blot validation. Statistical analysis done in R using unpaired student t-test vs sham
Discussion
mTBI is associated with a long list of acute symptoms including, but not limited to, headaches, fatigue, anxiety, depression, attention deficits, short-term memory problems, insomnia, confusion, and irritability [90]. In addition to these acute changes, rmTBI can cause persistent symptoms for longer than 3 months, known as post-concussion syndrome [61], and an increased risk for several neurodegenerative diseases later in life [43]. There has been a focus in recent years on the characterization of the neuropathology of the injured brain, with CTE suggested as the pathological signature and driver of symptoms after mTBI [96]. While the risk of developing neurological disorders, including CTE, after repeated head injury is undeniable, it remains unclear what early pathophysiological mechanisms after mTBI may drive early symptoms and ultimately the toxic protein accumulation and neurodegeneration later in life. Furthermore, it also remains elusive which molecular changes drive the broad symptoms associated with mTBI both in the acute and chronic periods following injury.
Here we have presented a murine model of repeated mTBI. Histological analysis indicates absence of any gross visible lesion, yet progressively increased levels of gliosis and microglial activation at 1 day and 7 days post-injury and axonal damage at 7 days post-injury. These changes were accompanied by significant loss of righting reflex after each impact and impairment in the Morris water maze at 1 week post-injury. These changes are consistent with other studies using a closed skull model of mTBI, in which gliosis, microglial activation, and axonal damage are evident alongside neurobehavioural deficits [9].
Additionally, there are significant changes in numerous genes that point to DNA damage and early neurodegeneration as early as 24 h and ramping up after 7 days. 24 h following repeated injury, rmTBI mice had significantly increased expression of DSB repair enzymes in the DDR (ATG7, DNA2, NBN) and a marker of M2 macrophages (CD163), indicating the presence of neuroinflammation and DNA damage in the form of DSBs. At this timepoint rmTBI mice showed decreased expression of several cell death pathways including the membrane attack complex (C6), TNF-associated cell death (CASP9, TRADD, TRAF3), autophagy-mediated cell death (EPG5, MAPILC3A), and DNA methylation patterns (AGO4). At 24 h post-injury, therefore, the DDR and inhibition of cell death are evident, suggesting that cells are attempting to repair damage inflicted by the mTBI.
By 7d post-injury, many components of the DDR were significantly reduced (EXO1, MPG) with additional downregulation of cholesterol metabolizing enzymes (CUP27A, CYP7B1), loss of toxic protein clearance and other protective mechanisms against neurodegenerative mechanisms (MERTK, RELN, TTR, CD44, DAB2) and reduction of apoptosis (BAX, CIDEA). Up-regulated pathways at this timepoint include cellular senescence (AGO4, BCL2L2, BNIP3, BNIP3L, RAD51, RB1CC1), PKA and JNK signalling pathways (MAPK10, RAB6B, PIK3CD), and early neurodegenerative processes (CHST8, CLEC7A, DAPK1, GPR34, MAFB, MSR1, OLFML3, SUMO1, TYROBP). These results suggest that by 7d post-injury signs of brain senescence and neurodegeneration are detectable. These findings were supported with qPCR and WBs, directly showing elevation of IL1β, IL10, p21, 53BP1, DNA2, and reduction in EXO1—all of which are molecular signatures of cellular senescence induced by genotoxic stress.
Many studies using rodent models have been published characterizing the diverse secondary injury cascade that occurs in the acute period following mTBI. Neuroinflammation has been identified as a hallmark response in the acute (24 h to 7 days) period post-injury [87]. Indeed, various rodent models of mTBI have shown increased levels of infiltrating immune cells [34], reactive astrogliosis [12], microgliosis [12, 124], and increased levels of pro-inflammatory cytokines [131]. This acute period of neuroinflammation after injury has been shown to prime neurons towards a neurodegenerative phenotype in the long-term [102]. In addition to neuroinflammation, disruption of the blood brain barrier resulting in its increased permeability and changes in CSF dynamics has been reported in the acute period after mTBI in rodents [7, 47, 166]. These changes are frequently accompanied by behavioural changes suggestive of anxiety [12], depression [80], and cognitive impairment [12, 80]. Evidence of neuronal loss [14], diffuse axonal injury [36], and atrophy [53] are also evident in this acute period. Indeed, in this study we identified gliosis, microgliosis, and axonal damage at both 1 and 7 days post-injury. The gene expression changes found in this study support and reflect the findings presented above, but we have further identified DNA damage-induced cellular senescence as activated in the acute period following mTBI and a possible driver of these changes and of long-term effects of injury.
The results of this study are consistent with recently published mouse models of mTBI in which DNA damage has been reported in the early (24 h) period [3, 123, 151]. Our own studies on human autopsy cases showed strong evidence of DSBs in brains of athletes with a history of rmTBI [131, 132]. Activation of the DDR in response to DNA damage is essential for the maintenance of genome integrity [67]. Detection of the DDR is often lost once cellular senescence is induced [5, 40]. Concomitantly, the DDR is lost in ageing in association with the accumulation of senescent cells and “inflammaging” [111, 150]. IL1α was found to be elevated 1 day post-injury, an interleukin which senses DNA damage and is a regulator of the cellular senescence response to such genotoxic stress—yet by 1 week post-injury IL1α returns to baseline and signatures of senescence (namely elevated p21, p53, and reduced EXO1) are evident. Indeed, this mechanism is consistent with the gene expression and gene ontology analysis data presented here suggesting that by 7d post-injury the gene expression signature of rmTBI brains reflect cellular senescence and processes consistent with the aged brain.
A key finding in this study is the activation of neurodegenerative processes as early as 7d post-injury. Several significantly changed genes at this timepoint have been associated with AD-like pathology and disease progression (BCL2L2, CHST8, CLEC7A, CYP27A, CYP7B1, DAPK1, GPR34, MERTK, OLFML3, SUMO1, TTR, TYROBP), cognitive impairment (DAPK1, EXO1, RELN, SUMO1, TYROBP), PD progression (SUMO1, MAPK10), and the accumulation of toxic proteins (DAPK1, MAPK10, MSR1, OLFML3, SUMO1, TYROBP). In addition, several significantly changed genes at this timepoint play roles in the maintenance of critical brain barriers including the blood–brain barrier (CYP7B1, MERTK, CD44) and synapse maintenance in neuronal networks (MERTK, RELN), and associate with neurodegenerative disease pathogenesis in early stages prior to protein accumulation. Gene ontology and pathway analyses with GProfiler support these alterations. We therefore suggest that very early changes, namely DNA damage-induced cell senescence, after mTBI may predispose the brain to abnormal protein accumulation and neurodegenerative processes, due to a combination of increased pathogenic processes and a loss of protective mechanisms.
Gene expression changes reflective of early neurodegenerative processes in the acute period following rmTBI suggests that these processes may occur early and even prior to the accumulation of any abnormal neurotoxic protein species, such as hyperphosphorylated tau or amyloid beta. While these protein species are indisputably neurotoxic, we believe that they represent end-stage pathology and perhaps may not be the most optimal treatment target for preventing long-term symptoms after mTBI. Indeed, we suggest that the emergence of proteinopathy may be a result of some or all of these early pathogenic processes and loss of protective mechanisms and, because these changes occur very early, they may represent viable treatment targets for the prevention of long-term neurological sequalae after mTBI.
Decreased expression of critical DDR factors at 7d post-injury suggest that the brain may be more at risk in general to DNA damage in the acute period following mTBI. This poses an increased vulnerability for individuals who are at risk of repeated mTBI, such as professional athletes, military, or victims of domestic violence [2, 42, 121], who may face DNA damage from a subsequent brain injuries days or weeks apart and repeated many times over years. If the DDR is overwhelmed and consequently downregulated, subsequent damage from these injuries could hasten the progression of neurodegenerative conditions in these individuals. This may be why professional athletes, who experience head injuries frequently [29], commonly present with CTE or a combination of neurodegenerative pathologies at a significantly younger than average age [130]. Indeed, accumulation of DNA damage and defects in DNA repair machinery are linked to neurodegenerative mechanisms and severe neurological dysfunction. For example, mutations in the upstream DNA damage sensor protein ATM result in movement and coordination dysfunction, cerebellar atrophy, and speech impairments [8]. Similarly, mutations in the DNA repair enzyme Ligase 4 result in microcephaly [108, 115] and inactivation of the DNA repair factor PNKP has been shown to cause seizures, microcephaly, oligodendrocyte dysfunction, and mutations in this gene lead to cognitive impairments and even severe dementia [11]. Although a better understanding of the mechanisms linking DNA damage with neurological dysfunction and neurodegeneration are needed, there is sufficient evidence to suggest that the accumulation of DNA damage and, by extension, senescent cells, is sufficient to cause neurological symptoms and chronic pathology. Indeed, DNA damage-induced cellular senescence can lead to the induction of the SASP, in which pro-inflammatory secreted molecules contribute to chronic levels of neuroinflammation.
The gene expression signature at 7 days post-injury indeed suggests activation of DNA damage-induced cellular senescence pathways. This was validated on the tissue using immunohistochemistry for Lamin A/C, a nuclear lamin protein located adjacent to the inner nuclear membrane. Lamin A/C is a scaffolding protein important for the maintenance of genomic integrity through its chromatin stabilization role [70]. Loss of lamin A/C is associated with impaired DNA damage repair mechanisms [45] and induces cellular senescence characterized by defective DNA repair [119]. Hundreds of mutations in LMNA, the gene encoding Lamin A/C, have been identified as causing degenerative disorders, collectively called laminopathies, an example of which is Hutchinson–Gilford Progeria Syndrome (HGPS). Molecularly, HGPS dysregulated gene expression, DNA repair defect, and premature accumulation of senescent cells [48, 148] Clinically, patients with HGPS exhibit signs of accelerated aging with a significantly reduced lifespan (mid-teens), severe atherosclerosis and cardiovascular pathology, hair loss, and reduced bone density [148]. Reduced expression of Lamin A/C in glial cells following rmTBI is therefore concerning, and suggestive of DNA damage-associated cellular senescence, perhaps contributing to neurological dysfunction.
It is important to note that not all individuals who experience repetitive brain injury have long-term symptoms nor are diagnosed with a neurodegenerative disease [58]. Indeed, the presentation of mTBI is extremely heterogeneous between individuals [92] and between sexes, where women tend to experience more severe symptoms [30]. We hypothesize that individual differences in reversing the DNA damage and related consequences may drive long term outcomes of brain injury. As an example, naturally occurring polymorphisms in DDR genes may confer different susceptibilities to mTBI. Consistent with this hypothesis, polymorphisms in the DNA repair gene EXO1, have been shown in humans to associate with cognitive ageing [83]. Susceptibility or vulnerability towards mTBI-induced DNA damage may therefore determine a patient’s long-term outcome following mTBI. In the context of sex differences, women have additional physiological levels of DNA damage in the brain, due to estrogen and estrogen metabolites [129], which may lower the threshold of what the DDR can handle before it is overwhelmed. Estrogen-induced DNA damage has further been associated with cognitive impairment [79], suggesting it plays a functional role in brain health. DNA repair pathways have previously been suggested as personalized medicine approaches for targeted cancer therapy, as several cancers present with defects in DNA repair machinery [32]. More research is warranted into the inter-individual and sex heterogeneity of the DDR in response to mTBI, as this represents a viable personalized medicine approach to early mTBI intervention.
Conclusions
In this study, using a murine model of rmTBI we have shown evidence of a response to DNA damage in the brain 24 h following repeated brain injury, and progression to cellular senescence, impaired DNA damage responses, and brain ageing by 7 days. We suggest that these pathogenic processes are critical early events which may drive early symptoms as well as late onset neurodegenerative processes after mTBI they represent viable treatment targets for early intervention strategies. Within this strategy, personalized medicine approaches may be foreseeable due to naturally occurring polymorphisms and alterations in the DDR or hypoxia response which may underlie individual and sex differences.
Availability of data and materials
The data supporting the conclusions of this article are included within the article’s additional files.
Abbreviations
- mTBI:
-
Mild traumatic brain injury
- rmTBI:
-
Repeated mild traumatic brain injury
- AD:
-
Alzheimer’s disease
- PD:
-
Parkinson’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- FTD:
-
Fronto-temporal dementia
- CTE:
-
Chronic traumatic encephalopathy
- DSB:
-
Double strand break
- ROS:
-
Reactive oxygen species
- DDR:
-
DNA damage response
- SASP:
-
Senescence-associated secretory phenotype
- TCP:
-
Centre for phenogenomics
- FFPE:
-
Formalin-fixed paraffin-embedded
- FC:
-
Fold change
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of genes and genomes
- WP:
-
WikiPathways
- HDR:
-
Homology-directed repair
- NHEJ:
-
Non-homologous end joining
- SNP:
-
Single nucleotide polymorphism
- CSF:
-
Cerebrospinal fluid
- APP:
-
Amyloid precursor protein
References
Alemi M, Gaiteiro C, Ribeiro CA, Santos LM, Gomes JR, Oliveira SM et al (2016) Transthyretin participates in beta-amyloid transport from the brain to the liver–involvement of the low-density lipoprotein receptor-related protein 1? Sci Rep 6:20164
Armistead-Jehle P, Soble JR, Cooper DB, Belanger HG (2017) Unique aspects of traumatic brain injury in military and veteran populations. Phys Med Rehabil Clin N Am 28(2):323–337
Arun P, Rossetti F, Wilder DM, Sajja S, Van Albert SA, Wang Y et al (2020) Blast exposure leads to accelerated cellular senescence in the rat brain. Front Neurol 11:438
Atamna H, Nguyen A, Schultz C, Boyle K, Newberry J, Kato H et al (2008) Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J 22(3):703
Bakkenist CJ, Drissi R, Wu J, Kastan MB, Dome JS (2004) Disappearance of the telomere dysfunction-induced stress response in fully senescent cells. Cancer Res 64(11):3748
Benhamed M, Herbig U, Ye T, Dejean A, Bischof O (2012) Senescence is an endogenous trigger for microRNA-directed transcriptional gene silencing in human cells. Nat Cell Biol 14(3):226
Bharadwaj VN, Nguyen DT, Kodibagkar VD, Stabenfeldt SE (2018) Nanoparticle-based therapies for brain injury. Adv Healthc Mater 7(1):1700668
Biton S, Barzilai A, Shiloh Y (2008) The neurological phenotype of ataxia-telangiectasia: solving a persistent puzzle. DNA Repair (Amst) 7(7):1028
Bolton AN, Saatman KE (2014) Regional neurodegeneration and gliosis are amplified by mild traumatic brain injury repeated at 24-hour intervals. J Neuropathol Exp Neurol 73(10):933–947
Bonham LW, Sirkis DW, Yokoyama JS (2019) The transcriptional landscape of microglial genes in aging and neurodegenerative disease. Front Immunol 10:1170
Bras J, Alonso I, Barbot C, Costa MM, Darwent L, Orme T et al (2015) Mutations in PNKP cause recessive ataxia with oculomotor apraxia type 4. Am J Hum Genet 96(3):474
Broussard JI, Acion L, De Jesus-Cortes H, Yin T, Britt JK, Salas R et al (2018) Repeated mild traumatic brain injury produces neuroinflammation, anxiety-like behaviour and impaired spatial memory in mice. Brain Inj 32(1):113
Brown J 3rd, Theisler C, Silberman S, Magnuson D, Gottardi-Littell N, Lee JM et al (2004) Differential expression of cholesterol hydroxylases in Alzheimer’s disease. J Biol Chem 279(33):34674
Bu W, Ren H, Deng Y, Del Mar N, Guley NM, Moore BM et al (2016) Mild traumatic brain injury produces neuron loss that can be rescued by modulating microglial activation using a cb2 receptor inverse agonist. Front Neurosci 10:449
Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ (2018) Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562(7728):578
Capparelli C, Chiavarina B, Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J et al (2012) CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumolir growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle 11(19):3599
Castranio EL, Mounier A, Wolfe CM, Nam KN, Fitz NF, Letronne F et al (2017) Gene co-expression networks identify Trem2 and Tryobp as major hubs in human APOE expressing mice following traumatic brain injury. Neurobiol Dis 105:1
Castranio EL, Wolfe CM, Nam KN, Letronne F, Fitz NF, Lefterov I et al (2018) ABCA1 haplodeficiency affects the brain transcriptome following traumatic brain injury in mice expressing human APOE isoforms. Acta Neuropathol Commun 6(1):69
Chalertpet K, Pin-on P, Aporntewan C, Patchsung M, Ingrungruanglert P, Israsena N et al (2019) Argonaute 4 as an effector protein in RNA-directed DNA methylation in human cells. Front Genet 10:645
Chang X, Wang L, Wang Z, Wu S, Zhu X, Hu S et al (2017) TRADD mediates the tumor necrosis factor-induced apoptosis of L929 cells in the absence of RIP3. Sci Rep 7:16111
Chen H, Richard M, Sandler DP, UmbachKamel DMFA (2007) Head injury and amyotrophic lateral sclerosis. J Epidemiol 166(7):810–816
Cheng X, Wang Y, Gong Y, Li F, Guo Y, Hu S et al (2016) Structural basis of FYCO1 and MAP1LC3A interaction reveals a novel binding mode for Atg8-family proteins. Autophagy 12(8):1330
Chinta SJ, Woods G, Rane A, Demaria M, Campisi J, Andersen JK (2015) Cellular senescence and the ageing brain. Exp Gerontol 68:3–7
Choi JY, Shin HJ, Bae IH (2018) mir-93-5p suppresses cellular senescence by directly targeting bcl-w and p21. Biochem Biophys Res Commun 505(4):1134
Choi WS, Abel G, Klintworth H, Flavell RA, Xia Z (2010) JNK3 mediates paraquat- and rotenone-induced dopaminergic neuron death. J Neuropathol Exp Neruol 69(5):511
Chung WS, Clarke LE, Wang GX, Stafford BJ, Sher A, Chakraborty C et al (2014) Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504(7480):394
Collin G, Huna A, Warnier M, Flaman JM, Bernard D (2018) Transcriptional repression of DNA repair genes is a hallmark and a cause of cellular senescence. Cell Death Dis 9:259z
Czarny P, Kwiatkowski D, Kacperska D, Kawczynska D, Talarowska M, Orzechowska A et al (2015) Elevated levels of DNA damage and impaired repair of oxidative DNA damage in patients with recurrent depressive disorder. Med Sci Monit 21:412–418
Daneshvar DH, Nowinski CJ, McKee AC, Cantu RC (2011) The epidemiology of sport-related concussion. Clin Sports Med 30(1):1
Davis-Hayes C, Gossett JD, Levine WN, Shams T, Harada J, Mitnick J, Noble J (2017) Sex specific outcomes and predictors of concussion recovery. J Am Acad Orthop Surg 25(12):818–828
Dethlefsen MM, Halling JF, Moller HD, Plomgaard P, Regenberg B, Ringhold S et al (2018) Regulation of apoptosis and autophagy in mouse and human skeletal muscle with aging and lifelong exercise training. Exp Gerontol 111:141
Dietlein F, Thelen L, Reinhardt HC (2014) Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet 30(8):326
Diniz BS, Reynolds CF, Sibille E, Lin CW, Tseng G, Lotrich F et al (2017) Enhanced molecular aging in late-life depression: the senescent associated secretory phenotype. Am J Geriatr Psychiatry 25(1):64–72
Doran SJ, Henry RJ, Shirey KA, Barrett JP, Ritzel RM, Lai W et al (2020) Early or late bacterial lung infection increases mortality after traumatic brain injury in male mice and chronically impairs monocyte innate immune function. Crit Care Med 48(5):418
Duval N, Sumner WA, Andrianakos AG, Gray JJ, Bouchard RJ, Wilkins HM et al (2018) The bcl-2 homology-3 domain (bh3)-only proteins, bid, dp5/hrk, and bnip3l, are upregulated in reactive astrocytes of end-stage mutant sod1 mouse spinal cord. Front Cell Neurosci 12:15
Ekmark-Lewen S, Flygt J, Kiwanuka O, Meyerson BJ, Lewen A, Hillered L et al (2013) Traumatic axonal injury in the mouse is accompanied by a dynamic inflammatory response, astroglial reactivity and complex behavioral changes. J Neuroinflamm 10:44
Fielder E, von Zglinicki T, Jurk D (2017) The DNA damage response in neurons: die by apoptosis or survive in a senescence-like state? J Alzheimers Dis 60(s1):S107–S131
Fitz NF, Wolfe CM, Playso BE, Biedrzycki RJ, Lu Y, Nam KN et al (2020) Trem2 deficiency differentially affects phenotype and transcriptome of human APOE3 and APOE4 mice. Mol Neurodegen 15(1):41
Flynn KM, Michaud M, Madri JA (2013) CD44 deficiency contributes to enhanced experimental autoimmune encephalomyelitis: a role in immune cells and vascular cells of the blood–brain barrier. Am J Pathol 182(4):1322
Fumagalli M, Rossiello F, Mondello C, d’Adda di Fagagna F (2014) Stable cellular senescence is associated with persistent DDR activation. PLoS ONE 9(10):e110969
Furman JL, Norris CM (2014) Calcineurin and glial signaling: neuroinflammation and beyond. J Neuroinflammation 11:158
Gagnon KL, DePrince AP (2017) Head injury screening and intimate partner violence: a brief report. J Trauma Dissociation 18(4):635–644
Gardner RC, Yaffe K (2015) Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol Cel Neurosci 66(Pt B):75–80
Giannini C, Monaco S, Kirschfink M, Rother KO, Lorbacher de Ruiz H, Nardelli E et al (1992) Inherited neuroaxonal dystrophy in C6 deficient rabbits. J Neuropathol Exp Neruol 51(5):514
Gibbs-Seymour I, Markiewicz E, Bekker-Jensen S, Mailand N, Hutchison CJ (2015) Lamin A/C-dependent interaction with 53BP1 promotes cellular responses to DNA damage. Aging Cell 14:162–169
Gillespie DA, Ryan KM (2015) Autophagy is critically required for DNA repair by homologous recombination. Mol Cell Oncol 3(1):e1030538
Glushakova OY, Johnson D, Hayes RL (2014) Delayed increases in microvascular pathology after experimental traumatic brain injury are associated with prolonged inflammation, blood–brain barrier disruption, and progressive white matter damage. J Neurotrauma 31(13):1180
Gonzalo S, Kreienkamp R, Askjaer P (2017) Hutchinson–Gilford Progeria Syndrome: a premature aging disease caused by LMNA gene mutations. Ageing Res Rev 33:18–29
Groth RD, Dunbar RL, Mermelstein PG (2003) Calcineurin regulation of neuronal plasticity. Biochem Biophys Res Commun 311(4):1159
Guidotti A, Grayson DR, Caruncho HJ (2016) epigenetic RELN dysfunction in schizophrenia and related neuropsychiatric disorders. Front Cell Neurosci 10:89
Guo Y, Li H, Ke X, Deng M, Qu Z, Cai Y et al (2019) Degradation of caytaxin causes learning and memory deficits via activation of DAPK1 in aging. Mol Neurobiol 56(5):3368
Halstrom A, MacDonald E, Neil C, Arendts G, Fatovich D, Fitzgerald M (2017) Elevation of oxidative stress indicators in a pilot study of plasma following traumatic brain injury. J Clin Neurosci 35:104–108
Harris TC, de Rooij R, Kuhl E (2019) The shrinking brain: cerebral atrophy following traumatic brain injury. Ann Biomed Eng 47(9):1941–1959
Hartman ML, Czyz M (2020) BCL-w: apoptotic and non-apoptotic role in health and disease. Cell Death Dis 11:260
Haure-Mirande JV, Audrain M, Fanutza T, Kim SH, Klein WL, Glabe C et al (2017) Deficiency of TYROBP, an adapter protein for TREM2 and CR3 receptors, is neuroprotective in a mouse model of early Alzheimer’s pathology. Acta Neuropathol 134(5):769
Haure-Mirande JV, Wang M, Audrain M, Fanutza T, Kim SH, Heja S et al (2018) Integrative approach to sporadic Alzheimer’s disease: deficiency of TYROBP in cerebral Aβ amyloidosis mouse normalizes clinical phenotype and complement subnetwork molecular pathology without reducing Aβ burden. Mol Psychiatry 24(3):431
Hauser S, Poenisch M, Schelling Y, Hoflinger P, Schuster S, Teegler A et al (2019) mRNA as a novel treatment strategy for hereditary spastic paraplegia type 5. Mol Ther Methods Clin Dev 15:359
Hazrati LN, Tartaglia MC, Diamandi P, Davis KD, Green RE, Wennberg R et al (2013) Absence of chronic traumatic encephalopathy in retired football players with multiple concussions and neurological symptomatology. Front Hum Neurosci 7:222
Herrin A, Donath A, Steiner KM, Widera MP, Hamzehian S, Kanakis D et al (2012) Reelin depletion is an early phenomenon of Alzheimer’s pathology. J Alzheimers Dis 30(4):963
Hildebrand JM, Yi Z, Buchta CM, Poovassery J, Stunz LL, Bishop GA (2011) Roles of tumor necrosis factor receptor associated factor 3 (TRAF3) and TRAF5 in immune cell functions. Immunol Rev 244(1):55
Hiploylee C, Dufort PA, Davis HS, Wennberg RA, Tartaglia MC, Mikulis D, Hazrati LN, Tator CH (2017) Longitudinal study of postconcussion syndrome: Not everyone recovers. J Neurotrauma 34(8):1511–1523
Hoa NN, Kobayashi J, Omura M, Hirakawa M, Yang SH, Komatsu K et al (2015) BRCA1 and CtIP are both required to recruit DNA2 at double-strand breaks in homologous recombination. PLoS ONE 10(4):e0124495
Hood VL, Berger R, Freedman R, Law AJ (2019) Transcription of PIK3CD in human brain and schizophrenia: regulation by proinflammatory cytokines. Hum Mol Genet 28(19):3188
Huynh MB, Ouidja MO, Chantepie S, Carpentier G, Maiza A, Zhang G et al (2019) Glycosaminoglycans from Alzheimer’s disease hippocampus have altered capacities to bind and regulate growth factors activities and to bind tau. PLoS ONE 14(1):e0209573
Ishii K, Kubo KI, Nakajima K (2016) Reelin and neuropsychiatric disorders. Front Cell Neurosci 10:229
Ismail M, Mateos L, Maioli S, Merino-Serrais P, Ali Z, Lodeiro M et al (2017) 27-Hydroxycholesterol impairs neuronal glucose uptake through an IRAP/GLUT4 system dysregulation. J Exp Med 214(3):699
Jackson SP, Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461(7267):1071
Jafari S, Etminan M, Aminzadeh F, Samii A (2013) Head injury and risk of Parkinson disease: a systematic review and meta-analysis. Mov Disord 28(9):1222–1229
Jhang KA, Park JS, Kim HS, Chong YH (2018) Sulforaphane rescues amyloid-β peptide-mediated decrease in MerTK expression through its anti-inflammatory effect in human THP-1 macrophages. J Neuroinflammation 15(1):75
Jung HJ, Lee JM, Yang SH, Young SG, Fong LG (2013) Nuclear lamins in the brain - new insights into function and regulation. Mol Neurobiol 47(1):290–301
Kim WK, Alvarez X, Fisher J, Bronfin B, Westmoreland S, McLaurin J et al (2006) CD163 identifies perivascular macrophages in normal and viral encephalitic brains and potential precursors to perivascular macrophages in blood. Am J Pathol 168(3):822
Kiss T, Nyul-Toth A, Balasubramanian P, Tarantini S, Ahire C, DelFavero J et al (2020) Single-cell RNA sequencing identifies senescent cerebromicrovascular endothelial cells in the aged mouse brain. Geroscience 42(2):429
Knock E, Matsuzaki S, Takamura H, Satoh K, Rooke G, Han K et al (2018) SUMO1 impact on Alzheimer disease pathology in an amyloid-depositing mouse model. Neurobiol Dis 110:154
Kolbe K, Bukhari H, Loosse C, Leonhardt G, Glotzbach A, Pawlas M et al (2016) Extensive nuclear sphere generation in the human Alzheimer’s brain. Neurobiol Aging 48:103
Koo GB, Ji JH, Cho H, Morgan MJ, Kim YS (2017) Nuclear TRADD prevents DNA damage-mediated death by facilitating non-homologous end-joining repair. Sci Rep 7(1):3332
Lane-Donovan C, Philips GT, Wasser CR, Durakoglugil MS, Masiulis I, Upadhaya A et al (2015) Reelin protects against amyloid β toxicity in vivo. Sci Signal 8(384):ra67
Lau L, Porciuncula A, Yu A, Iwakura Y, David G (2019) Uncoupling the senescence-associated secretory phenotype from cell cycle exit via Interleukin-1 inactivation unveils its protumorigenic role. Mol Cell Biol 39(12):e00586-e618
Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJR, Motoyama N et al (2012) Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 336(6078):225
Leffa DD, Damiani AP, Damazio DD, Guerra NP, Moretti M, de Brito GG et al (2014) Longterm effects of ageing and ovariectomy on aversive and recognition memory and DNA damage in the hippocampus of female rats. Acta Neuropsychiatr 26(3):161–169
Lesniak A, Leszczynski P, Magdalena B-Z, Pick CG, Sacharczuk M (2017) Naloxone exacerbates memory impairments and depressive-like behavior after mild traumatic brain injury (mTBI) in mice with upregulated opioid system activity. Behav Brain Res 326:209
Lewis ND, Hill JD, Juchem KW, Stefanopoulos DE, Modis LK (2014) RNA sequencing of microglia and monocyte-derived macrophages from mice with experimental autoimmune encephalomyelitis illustrates a changing phenotype with disease course. J Neueroimmunol 277(1–2):26
Li J, Ayoub A, Xiu Y, Yin X, Sanders JO, Mesfin A et al (2019) (2019) TGFβ-induced degradation of TRAF3 in mesenchymal progenitor cells causes age-related osteoporosis. Nat Commun 10(1):2795
Lin E, Kuo PH, Liu YL, Yang AC, Tsai SJ (2019) Polymorphisms of the DNA repair gene EXO1 modulate cognitive aging in old adults in a Taiwanese population. DNA Repair (Amst) 78:1
Lin JB, Sene A, Santeford A, Fujiwara H, Sidhu R, Ligon MM et al (2018) Oxysterol signatures distinguish age-related macular degeneration from physiologic aging. EBioMedicine 32:9
Linnartz-Gerlach B, Bodea LG, Klaus C, Ginolhac A, Halder R, Sinkkonen L et al (2019) TREM2 triggers microglial density and age-related neuronal loss. Glia 67(3):539
Liu EY, Xu N, O’Prey J, Lao LY, Joshi S, Long JS et al (2015) Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc Natl Acad Sci USA 112(3):773
Lozano D, Gonzales-Portillo GS, Acosta S, de la Pena I, Tajiri N et al (2015) Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr Dis Treat 11:97
Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U et al (2014) Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol 15(1):88
Machiya JI, Shibata Y, Yamauchi K, Hirama N, Wada T, Inoue S et al (2007) Enhanced expression of MafB inhibits macrophage apoptosis induced by cigarette smoke exposure. Am J Respir Cell Mol Biol 36(4):418
Marshall S, Bayley M, McCullagh S, Velikonja D, Verrigan L, Ouchterlony D et al (2015) Updated clinical practice guidelines for concussion/mild traumatic brain injury and persistent symptoms. Brain Inj 29(6):688
Matsuzaki S, Lee L, Knock E, Srikumar T, Sakurai M, Hazrati LN et al (2015) SUMO1 affects synaptic function. Spine Density Mem Sci Rep 5:10730
Mayer AR, Quinn DK, Master CL (2017) The spectrum of mild traumatic brain injury: a review. Neurology 89(6):623
McDonnell MA, Wang D, Khan SM, Vander Heiden MG, Kelekar A (2003) Caspase-9 is activated in a cytochrome c-independent manner early during TNFalpha-induced apoptosis in murine cells. Cell Death Differ 10(9):1005
McInnes K, Friesen CL, MacKenzie DE, Westwood DA, Boe SG (2017) Mild traumatic brain injury and chronic cognitive impairment: a scoping review. PLoS ONE 12(4):e0174847
McKee AC, Alosco ML, Huber BR (2016) Repetitive head impacts and chronic traumatic encephalopathy. Neurosurg Clin N Am 27(4):529–535
McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I et al (2016) The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 131(1):75–86
Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T (2004) Human argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell 15(2):185
Montgomery RR (2017) Age-related alterations in immune responses to West Nile virus infection. Clin Exp Immunol 187(1):26
Moore R, Cai KQ, Tao W, Smith ER, Xu XX (2013) Differential requirement for Dab2 in the development of embryonic and extra-embryonic tissues. BC Dev Biol 13:39
Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A et al (1991) Head trauma as a risk factor for Alzheimer’s disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol 20(Suppl 2):S28-35
Mounce LT, Williams WH, Jones JM, Harris A, Haslam SA, Jetten J (2013) Neurogenic and psychogenic acute postconcussion symptoms can be identified after mild traumatic brain injury. J Head Trauma Rehabil 28(5):397–405
Mrak RE, Griffin WS (2005) Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging 26(3):349
Munch S, Weidtkamp-Peters S, Klement K, Grigaravicius P, Monajembashi S, Salomoni P et al (2014) The tumor suppressor PML specifically accumulates at RPA/RAD51-containing DNA damage repair foci but is nonessential for DNA damage-induced fibroblast senescence. Mol Cell Biol 34(10):1733
Musi CA, Agro G, Santarella F, Iervasi E, Borsello T (2020) JNK3 as therapeutic target and biomarker in neurodegenerative and neurodevelopmental brain diseases. Cells 9(10):2190
Nagarajan P, Agudelo Garcia PA, Iyer CC, Popova LV, Arnold WD, Parthun MR (2019) Early-onset aging and mitochondrial defects associated with loss of histone acetyltransferase 1 (Hat1). Aging Cell 18(5):e12992
Namjoshi DR, Cheng WH, McInnes KA et al (2014) Merging pathology with biomechanics using CHIMERA (Closed-Head Impact Model of Engineered Rotational Acceleration): a novel, surgery-free model of traumatic brain injury. Mol Neurodegener 9:55
Nistico R, Ferraina C, Marconi V, Blandini F, Negri L, Egebjerg J et al (2014) Age-related changes of protein SUMOylation balance in the AβPP Tg2576 mouse model of Alzheimer’s disease. Front Pharmacol 5:63
O’Driscoll M, Cerosaletti KM, Girard PM, Stumm M, Kysela B, Hirsch B et al (2001) DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol Cell 8(6):1175
Ochi Y, Chano Y, Ikebuchi K, Inoue H, Isono T, Arai A, Tameno H et al (2011) Rb1CC1 activates the p16 promoter through the interaction with hSNF5. Oncol Rep 26(4):805
Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ et al (2020) Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat Commun 11:6129
Olivieri F, Albertini MC, Orciani M, Ceka A, Cricca M, Procopio AD et al (2015) DNA damage response (DDR) and senescence: shuttled inflamma-miRNAs on the stage of inflamm-aging. Oncotarget 6(34):35509
Olstad EW, Ringers C, Hansen JN, Wens A, Brandt C, Wachten D et al (2019) Ciliary beating compartmentalizes cerebrospinal fluid flow in the brain and regulates ventricular development. Curr Biol 29(2):229
Opdam FJ, Echard A, Croes HJ, van den Hurk JA, van den Vorstenbosch RA, Ginsel LA et al (2000) The small GTPase Rab6B, a novel Rab6 subfamily member, is cell-type specifically expressed and localised to the Golgi apparatus. J Cell Sci 113(Pt. 15):2725
Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J (2009) Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A 106(40):17031–17036
Ou HL, Schumacher B (2018) DNA damage response and p53 in the aging process. Blood 131(5):488
Overall RW, Zocher S, Garthe A, Kempermann G (2020) Rtrack: a software package for reproducible automated water maze analysis. bioRxiv. https://doi.org/10.1101/2020.02.27.967372
Panneer Selvam S, Roth BM, Nganga R, Kim J, Cooley MA, Helke K et al (2018) Balance between senescence and apoptosis is regulated by telomere damage-induced association between p16 and caspase-3. J Biol Chem 293(25):9784
Pei L, Wang S, Jin H, Bi L, Wei N, Yan H et al (2015) A novel mechanism of spine damages in stroke via DAPk1 and tau. Cereb Cortex 25(11):4559
Pekovic V, Gibbs-Seymour I, Markiewicz E et al (2011) Conserved cysteine residues in the mammalian lamin A tail are essential for cellular responses to ROS generation. Aging Cell 10(6):1067–1079
Peterson D, Munger C, Crowley J, Corcoran C, Cruchaga C, Goate AM et al (2014) Variants in PPP3R1 and MAPT are associated with more rapid functional decline in Alzheimer’s disease: the Cache County Dementia Progression Study. Alzheimers Dement 10(3):366
Pfister T, Pfister K, Hagel B, Ghali WA, Ronksley PE (2016) The incidence of concussion in youth sports: a systematicm review and meta-analysis. Br J Sports Med 50(5):292–297
Raisova M, Hossini AM, Eberle J, Riebeling C, Wieder T, Sturm I et al (2001) The Bax/Bcl-2 ratio determines the susceptibility of human melanoma cells to CD95/Fas-mediated apoptosis. J Invest Dermatol 117(2):333
Ritzel RM, Doran SJ, Glaser EP, Meadows VE, Faden AI, Stoica BA et al (2019) Old age increases microglial senescence, exacerbates secondary neuroinflammation, and worsens neurological outcomes after acute traumatic brain injury in mice. Neurobiol Aging 77:194–206
Robinson S, Berglass JB, Denson JL, Berkner J, Anstine CV, Winer JL et al (2017) Microstructural and microglial changes after repetitive mild traumatic brain injury in mice. J Neurosci Res 95(4):1025
Rodier F, Coppé JP, Patil CK et al (2009) Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 11(10):1272
Rosso SM, Landweer EJ, Houterman M, Donker Kaat L, van Duijn CM, van Swieten JC (2003) Medical and environmental risk factors for sporadic frontotemporal dementia: a retrospective case-control study. J Neurol Neurosurg Psychiatry 74(11):1574–1576
Rymut N, Heinz J, Sadhu S, Hosseini Z, Riley CO, Marinello M et al (2020) Resolvin D1 promotes efferocytosis in aging by limiting senescent cell-induced MerTK cleavage. FASEB J 34(1):597
Sandoval K, Umbaugh D, House A, Crider A, Witt K (2019) Somatostatin receptor subtype-4 regulates mrna expression of amyloid-beta degrading enzymes and microglia mediators of phagocytosis in brains of 3xtg-ad mice. Neurochem Res 44(11):2670
Savage KI, Matchett KB, Barros EM, Cooper KM, Irwin GW, Gorski JJ, Orr KS et al (2014) BRCA1 deficiency exacerbates estrogen-induced DNA damage and genomic instability. Cancer Res 74(10):2773–2784
Schaffert J, LoBue C, White CL, Chiang HS, Didehbani N, Lacritz L et al (2018) Traumatic brain injury history is associated with an earlier age of dementia onset in autopsy-confirmed alzheimer’s disease. Neuropsychol 32(4):410
Schwab N, Grenier K, Hazrati LN (2019) DNA repair deficiency and senescence in concussed professional athletes involved in contact sports. Acta Neuropathol Commun 7(1):182
Schwab N, Tator C, Hazrati LN (2019) DNA damage as a marker of brain damage in individuals with history of concussions. Lab Invest 99(7):1008
Sekiya M, Wang M, Fujisaki N, Sakakibara Y, Quan X et al (2018) Integrated biology approach reveals molecular and pathological interactions among Alzheimer’s Aβ42, Tau, TREM2, and TYROBP in Drosophila models. Genome Med 10(1):26
Shang D, Hong Y, Xie W, Tu Z, Xu J (2020) Interleukin-1β drives cellular senescence of rat astrocytes induced by oligomerized amyloid β peptide and oxidative stress. Front Neurol 11:929
Shi L, Rocha M, Zhang W, Jian M, Li S, Ye Q et al (2020) Genome-wide transcriptomic analysis of microglia reveals impaired responses in aged mice after cerebral ischemia. J Cereb Blood Flor Metab 40(1_suppl):S49
Shi W, Liu W, Ma J, Lu J, Yang X, Wang J et al (2020) The role of Atg7-mediated autophagy in ionizing radiation-induced neural stem cell damage. Gene 738:144485
Shicita T, Ito M, Morita R, Komai K, Noguchi Y, Ooboshi H et al (2017) MAFB prevents excess inflammation after ischemic stroke by accelerating clearance of damage signals through MSR1. Nat Med 23(6):723
Sikorska K, Gradzka I, Wasyk I, Brzoska K, Stepkowski TM, Czerwinska M et al (2020) The impact of ag nanoparticles and cdte quantum dots on expression and function of receptors involved in amyloid-β uptake by bv-2 microglial cells. Materials (Basel) 13(14):3227
Simin S, Aixiao L, Jiadong L, Candy W, Patrizia CB (2009) Epigenetic memory loss in aging oligodendrocytes in the corpus callosum. Neurobiol Aging 29(3):452
Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED (2006) Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J Cereb Blood Flow Metab 26(11):1407–1418
Siyahhan B, Knobloch V, de Zelicourt D, Asgari M, Daners MS, Poulikakos D et al (2014) Flow induced by ependymal cilia dominates near-wall cerebrospinal fluid dynamics in the lateral ventricles. J R Soc Interface 11(94):20131189
Song S, Xing G, Yuan L, Wang J, Wang S, Yin Y et al (2012) N-methylpurine DNA glycosylase inhibits p53-mediated cell cycle arrest and coordinates with p53 to determine sensitivity to alkylating agents. Cell Res 22(8):1285
Sousa JC, Cardoso I, Marques F, Saraiva MJ, Palha JA (2007) Transthyretin and Alzheimer’s disease: where in the brain? Neurobiol Aging 28(5):713
Stefanova NA, Ershov NI, Kolosova NG (2019) Suppression of Alzheimer’s disease-like pathology progression by mitochondria-targeted antioxidant SkQ1: a transcriptome profiling study. Oxid Med Cell Longev 2019:3984906
Su W, Foster SC, Xing R, Feistel K, Olsen RHJ, Acevedo SF et al (2017) CD44 transmembrane receptor and hyaluronan regulate adult hippocampal neural stem cell quiescence and differentiation. J Biol Chem 292(11):4434
Sufit A, Lee-Sherik AB, DeRyckere D, Rupji M, Dwivedi B, Varella-Garcia M et al (2016) MERTK inhibition induces polyploidy and promotes cell death and cellular senescence in glioblastoma multiforme. PLoS ONE 11(1):e0165107
Tator CH, Davis HS, Duford PA, Tartaglia MC, Davis KD, Ebraheem A et al (2016) Postconcussion syndrome: demographics and predictors in 221 patients. J Neurosurg 125(5):1206
Testa G, Gamba P, Badilli U, Gargiulo S, Maina M, Guina T et al (2014) Loading into nanoparticles improves quercetin’s efficacy in preventing neuroinflammation induced by oxysterols. PLoS ONE 9(5):e96795
Testa G, Staurenghi E, Zerbinati C, Gargiulo S, Iuliano L, Giaccone G et al (2016) Changes in brain oxysterols at different stages of Alzheimer’s disease: Their involvement in neuroinflammation. Redox Biol 10:24
Tiwari V, Wilson DM (2019) DNA damage and associated DNA repair defects in disease and premature aging. Am J Hum Genet 105(2):237
Tominaga T, Shimada R, Okada Y, Kawamata T, Kibayashi K (2019) Senescence-associated-β-galactosidase staining following traumatic brain injury in the mouse cerebrum. PLoS ONE 14(3):e0213673
Tong Q, Zhang M, Cao X, Xu S, Wang D, Zhao Y (2017) Expression and activation of Daphnia pulex caspase-3 are involved in regulation of aging. Gene 634:37
Towers CG, Wodetzki D, Thorburn A (2020) Autophagy-dependent cancer cells circumvent loss of the upstream regulator RB1CC1/FIP200 and loss of LC3 conjugation by similar mechanisms. Autophagy 16(7):1332
Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM et al (2010) DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell 140(2):222
Uraoka M, Ikeda K, Kurimoto-Nakano R, Nakagawa Y, Koide M, Akakabe Y et al (2011) Loss of bcl-2 during the senescence exacerbates the impaired angiogenic functions in endothelial cells by deteriorating the mitochondrial redox state. Hypertension 58(2):254
Vacher M, Porter T, Villemagne VL, Milicic L, Peretti M, Fowler C et al (2019) Validation of a priori candidate Alzheimer’s disease SNPs with brain amyloid-beta deposition. Sci Rep 9(1):17069
van Deursen JM (2014) The role of senescent cells in ageing. Nature 509(7501):439–446
Varon R, Vissinga C, Platzer M, Cerosaletti KM, Chrzanowska KH, Saar K et al (1998) Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 93(3):467
Vrtacnik P, Zupan J, Mlakar V, Kranjc T, Marc J, Kerm B et al (2018) Epigenetic enzymes influenced by oxidative stress and hypoxia mimetic in osteoblasts are differentially expressed in patients with osteoporosis and osteoarthritis. Sci Rep 8:16215
Wang H, Dey KK, Chen PC, Li Y, Niu M, Cho JH et al (2020) Integrated analysis of ultra-deep proteomes in cortex, cerebrospinal fluid and serum reveals a mitochondrial signature in Alzheimer’s disease. Mol Neurodegener 15(1):43
Wang KS, Liu X, Xie C, Liu Y, Xu C (2016) Non-parametric survival analysis of EPG5 gene with age at onset of Alzheimer’s disease. J Mol Neurosci 60(4):436
Wang Z, Miao G, Xue X, Guo X, Yuan C, Wang Z et al (2016) The Vici syndrome protein EPG5 is a Rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol Cell 63(5):781
Wanschers BFJ, van de Vorstenbosch R, Schlager MA, Splinter D, Akhmanova A, Hoogenraad CC et al (2007) A role for the Rab6B Bicaudal-D1 interaction in retrograde transport in neuronal cells. Exp Cell Res 313(16):3408
Weetman J, Wong MB, Sharry S, Rcom-H’cheo-Gauthier A, Gai WP, Meedeniya A et al (2013) Increased SUMO-1 expression in the unilateral rotenone-lesioned mouse model of Parkinson’s disease. Neurosci Lett 544:119
Wu P, Zhao Y, Haidacher SJ, Wang E, Parsley MO, Gao J et al (2013) Detection of structural and metabolic changes in traumatically injured hippocampus by quantitative differential proteomics. J Neurotrauma 30(9):775
Xiong G, Elkind JA, Kundu S, Smith CJ, Antunes MC, Tamashiro E et al (2014) Traumatic brain injury-induced ependymal ciliary loss decreases cerebral spinal fluid flow. J Neurotrauma 31(16):1396
Xu LZ, Li BQ, Jia JP (2019) DAPK1: A novel pathology and treatment target for Alzheimer’s disease. Mol Neurobiol 56(4):2838
Xu SF, Hu AL, Xie L, Liu JJ, Qu Q, Liu J (2019) Age-associated changes of cytochrome P450 and related phase-2 gene/proteins in livers of rats. PeerJ 7:e74929
Yadav R, Srivastava P (2018) Clustering, pathway enrichment, and protein-protein interaction analysis of gene expression in neurodevelopmental disorders. Adv Pharmacol Sci 3632159
Yarza R, Vela S, Solas M, Ramirez MJ (2016) c-Jun N-terminal kinase (jnk) signaling as a therapeutic target for Alzheimer’s disease. Front Pharmacol 6:321
Yau JLW, Rasmuson S, Andrew R, Graham M, Noble J, Olsson T et al (2003) Dehydroepiandrosterone 7-hydroxylase CYP7B: predominant expression in primate hippocampus and reduced expression in Alzheimer’s disease. Neuroscience 121(2):307
Yoo DY, Kim DW, Kwon HJ, Jung HJ, Nam SM, Kim JW et al (2017) Chronic administration of SUMO1 has negative effects on novel object recognition memory as well as cell proliferation and neuroblast differentiation in the mouse dentate gyrus. Mol Med Rep 16(3):3427
Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S et al (2016) Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun 7:11190
Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JFJ et al (2009) Autophagy mediates the mitotic senescence transition. Genes Dev 23(7):798
Yousefzadeh MJ, Zhao J, Bukata C, Wade EA, McGowan SJ, Angelini LA, Bank MP et al (2020) Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell 19(3):e13094
Zhang X, Lv C, An Y, Liu Q, Rong H, Tao L et al (2018) Increased levels of 27-hydroxycholesterol induced by dietary cholesterol in brain contribute to learning and memory impairment in rats. Mol Nutr Foods Res 62(3):1700531
Zhang X, Wang X, Khurm M, Zhan G, Zhang H, Ito Y et al (2020) Alterations of brain quantitative proteomics profiling revealed the molecular mechanism of diosgenin against cerebral ischemia reperfusion effects. J Proteome Res 19(3):1154
Zhao L, Xue M, Zhang L, Guo B, Qin Y, Jiang Q et al (2020) MicroRNA-4268 inhibits cell proliferation via AKT/JNK signalling pathways by targeting Rab6B in human gastric cancer. Cancer Gene Ther 27(6):4461
Zhao YG, Zhao H, Sun H, Zhang H (2013) Role of EPG5 in selective neurodegeneration and Vici syndrome. Autophagy 9(8):1258
Zheng W, Blaner WS, Zhao Q (1999) Inhibition by lead of production and secretion of transthyretin in the choroid plexus: its relation to thyroxine transport at blood-CSF barrier. Toxicol Appl Pharmacol 155(1):24
Zhu L, Qi B, Hou D (2019) Roles of HIF1α- and HIF2 α-regulated BNIP3 in hypoxia-induced injury of neurons. Pathol Res Pract 215(4):822
Zhu X, Wang Y, Ogawa O, Lee H, Raina AK, Siedlak SL et al (2004) Neuroprotective properties of Bcl-w in Alzheimer disease. J Neurochem 89(5):1233
Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N et al (2015) The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14(4):644
Zhubi A, Chen Y, Guidotti A, Grayson DR (2017) Epigenetic regulation of RELN and GAD1 in the frontal cortex (FC) of autism spectrum disorder (ASD) subjects. Int J Dev Neurosci 62:63
Funding
This study was funded by the Canadian Institute of Health Research (CIHR).
Author information
Authors and Affiliations
Contributions
NS performed surgeries, collected, analyzed, and interpreted data presented in this paper and wrote the manuscript. LNH played a major role in design of experiments, data analysis and writing of the manuscript. YJ performed technical work including qPCR, WB, and statistical analysis of molecular work. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
NanoString data for 1-day post-injury group.
Additional file 2.
NanoString data for 1-week post-injury group.
Additional file 3: Table S1.
List of qPCR primers used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Schwab, N., Ju, Y. & Hazrati, LN. Early onset senescence and cognitive impairment in a murine model of repeated mTBI. acta neuropathol commun 9, 82 (2021). https://doi.org/10.1186/s40478-021-01190-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-021-01190-x